

Abstract
Mutations in voltage-gated Na+ channels have been linked to several channelopathies leading to a wide variety of diseases including cardiac arrhythmias, epilepsy, and myotonia. We have previously demonstrated that voltage-gated Na+ channel (Nav)1.5 trafficking-deficient mutant channels could lead to a dominant negative effect by impairing trafficking of the wild-type (WT) channel. We also reported that voltage-gated Na+ channels associate as dimers with coupled gating properties. Here, we hypothesized that the dominant negative effect of mutant Na+ channels could also occur through coupled gating. This was tested using cell surface biotinylation and single channel recordings to measure the gating probability and coupled gating of the dimers. As previously reported, coexpression of Nav1.5-L325R with WT channels led to a dominant negative effect, as reflected by a 75% reduction in current density. Surprisingly, cell surface biotinylation showed that Nav1.5-L325R mutant is capable of trafficking, with 40% of Nav1.5-L325R reaching the cell surface when expressed alone. Importantly, even though a dominant negative effect on the Na+ current is observed when WT and Nav1.5-L325R are expressed together, the total Nav channel cell surface expression was not significantly altered compared with WT channels alone. Thus, the trafficking deficiency could not explain the 75% decrease in inward Na+ current. Interestingly, single channel recordings showed that Nav1.5-L325R exerted a dominant negative effect on the WT channel at the gating level. Both coupled gating and gating probability of WT:L325R dimers were drastically impaired. We conclude that dominant negative suppression exerted by Nav1.5 mutants can also be caused by impairing the WT gating probability, a mechanism resulting from the dimerization and coupled gating of voltage-gated Na+ channel α-subunits.
NEW & NOTEWORTHY The presence of dominant negative mutations in the Na+ channel gene leading to Brugada syndrome was supported by our recent findings that Na+ channel α-subunits form dimers. Up until now, the dominant negative effect was thought to be caused by the interaction of the wild-type Na+ channel with trafficking-deficient mutant channels. However, the present study demonstrates that coupled gating of voltage-gated Na+ channels can also be responsible for the dominant negative effect leading to arrhythmias.
Keywords: arrhythmia, Brugada syndrome, channelopathies, SCN5A, sodium channel
INTRODUCTION
Voltage-gated Na+ channels are responsible for the rapid upstroke of the action potential and its propagation through excitable tissues. Mutations in voltage-gated ion channel genes are responsible for cardiac, muscular, and neuronal channelopathies. In the heart, hundreds of mutations in SCN5A, the gene encoding cardiac voltage-gated Na+ channel Nav1.5, lead to a wide variety of cardiac arrhythmias: Brugada syndrome (BrS), long QT syndrome type 3 (LQT3), cardiac conduction defect, or sick sinus syndrome. Unlike voltage-gated K+ channel genes, which encode for one domain protein assembling as a tetramer to form a functional channel, voltage-gated Na+ channel genes SCNXA encode for the entire functional four-domain channel protein (9). For this reason, the presence of Na+ channel mutants producing a dominant negative (DN) effect was unexpected. However, we and others have reported Na+ channel mutations able to exert a DN effect (3, 5, 7, 10, 11). Furthermore, we demonstrated that Nav1.5 α-subunits could interact with each other and that a trafficking-deficient channel was able to produce DN suppression by impairing trafficking of the wild-type (WT) channel (3). Moreover, we have also demonstrated that a trafficking-deficient channel coexpressed with a gating-deficient channel (both nonconductive mutant channels) were capable of transcomplementation. This led to a partial rescue of inward Na+ current (INa) when these two channels were expressed together through a mechanism by which the gating-deficient channel rescued the trafficking-deficient channel from the endoplasmic reticulum (ER) toward the cell surface due to their physical interaction (3). Finally, we (4) have also reported the presence of atypical BrS mutations that do not present defects when expressed alone but lead to reduced current amplitudes when coexpressed with WT channels due to trafficking deficiency of the interacting dimers. Altogether, DN effect and transcomplementation seem to be mainly driven by the interaction of the channels trafficking together toward the cell surface. This interaction between Na+ channel α-subunits and the presence of DN effects or transcomplementation led us to question the stoichiometry of voltage-gated Na+ channels. Interestingly, we demonstrated in a previous study that voltage-gated Na+ channel α-subunits form dimers through an interaction site located in the domain I–II linker (2). Importantly, we identified that the channels not only physically interact as dimers but also present with coupled gating properties (2).
Considering that we have now established that Na+ channels can form dimers with coupled gating (2), we investigated in the present study whether coupled gating of a mutant channel with the WT channel could also be a mechanism for the DN suppression observed by some Nav1.5 mutant channels. To do so, we used the BrS DN mutant Nav1.5-L325R. In human embryonic kidney (HEK)-293 cells, this mutation, located in the pore, results in very little current (5), suggesting that only a few mutant channels are able to reach the cell surface. In fact, we demonstrated here, through cell surface biotinylation assays, that 40% of the Nav1.5-L325R mutant channels are able to reach the cell surface compared with WT channels expressed alone. However, when both WT and Nav1.5-L325R channels were coexpressed together, the cell surface expression of total Nav channels was not significantly different from WT channels expressed alone. This suggests that another mechanism other than trafficking deficiency is needed to explain the drastic 75% loss of INa observed at the heterozygous state (WT + Nav1.5-L325R). Since we recently reported that Nav1.5 displays coupled gating properties (2), we hypothesized that the Nav1.5-L325R channel could exert a DN effect by impairing WT gating. Indeed, single channel recordings showed that the presence of the mutation reduced the open probability of the WT channel and its coupling coefficient. Additionally, we observed that the patterns of channel openings presenting with coupled or uncoupled openings were drastically different compared with WT channels alone. In fact, the number of channels displaying coupled openings was almost abolished. These findings suggest that the presence of Nav1.5-L325R in the dimer impairs WT gating. Therefore, this study suggests that DN mechanisms not only involve trafficking deficiencies but can also arise from coupled gating properties of voltage-gated sodium channels.
MATERIAL AND METHODS
SCN5A mutagenesis
Mutations were introduced in the pIRES-GFP-SCN5A plasmid using a QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions and verified by sequencing.
HEK-293 cell culture and transfection.
HEK-293 cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin. All transfections were done with polyfect from Qiagen according to the manufacturer’s instructions. HEK-293 cells were transfected in 35-mm dishes with WT or mutant or WT + mutant channels to mimic the heterozygous state of the patient with a total of 0.3 µg (for patch-clamp recordings) or 0.6 µg (for cell surface biotinylation assays) of plasmids per dish.
Patch-clamp solutions.
Thirty-six hours after transfection, HEK-293 cells were trypsinized and seeded to a density that enabled single cells to be identified. Green positive cells were chosen for patch-clamp experiments. For whole cell current recordings, HEK-293 cells were bathed in extracellular Tyrode solution containing (in mM) 150 NaCl, 2 KCl, 1 MgCl2, 1.5 CaCl2, 1 NaH2PO4, 10 glucose, and 10 HEPES (pH 7.4; NaOH). The patch pipette medium was (in mM) 35 NaCl, 105 CsF, 2 MgCl2, 10 EGTA, and 10 HEPES adjusted to pH 7.4 with CsOH. For cell-attached single channel recordings, the pipette contained (mM) 280 NaCl, 5.4 KCI, 1 MgC12, 2 CaC12, 10 glucose, and 10 HEPES (pH 7 0.3). To adjust the resting membrane potential to 0 mV, a bathing solution containing 150 mM KCl and 10 mM HEPES (pH 7.4) was used.
Electrophysiological recordings.
Patch-clamp recordings were carried out in the whole cell configuration at room temperature (∼22°C) as previously described (2). Ionic currents were recorded with an amplifier (Axopatch 200B, Axon Instruments). Patch pipettes (Corning Kovar Sealing code 7052, WPI) had resistances of 1.9–2.5 MΩ. Currents were filtered at 5 kHz (−3 dB, 8-pole low-pass Bessel filter) and digitized at 30 kHz (NI PCI-6251, National Instruments, Austin, TX). Data were acquired and analyzed with Clampfit software (Molecular Device).
To measure peak INa amplitude and determine current-voltage relationships (current-voltage curves), currents were elicited by 50-ms test potentials from −100 to +60 mV by increments of 5 mV from a holding potential of −120 mV. Single channel recordings were obtained from a protocol of 100 sweeps at 0.2 Hz, depolarizing at −20 mV from a holding potential of −100 mV.
Single channel events were recorded from cells with a minimum of channels under the patch to ensure single channel recordings. The single channel conductance was determined by recording over a thousand sweeps in each condition at 1-kHz frequency; no additional filtration was applied. Clampfit analysis software was used to determine single (level 1)- and double (level 2)-level events after subtraction of the pipette capacitance. This allowed us to determine open level 1 and open level 2. After establishing these two open levels, we segmented the recorded traces into 0.2-ms segments, which allowed us to capture totally closed, open level 1, or open level 2 events for each 0.2-ms segment. Gating was considered simultaneous or “coupled” as opposed to “stacked” events when it reached the second level within 0.4 ms (two segments).
Experiments performed at physiological temperature were monitored by the ThermoClamp II Temperature Control System from Automate Scientific. While cells were maintained at 37°C, a succession of pulse at −20 mV from a holding potential of −120 mV allowed us to record peak INa density. Once steady state was reached, the temperature control system was set up to 42°C. The time necessary to reach 42°C was 1 min ± 5 s, thus ensuring that the effect on INa from temperature would not be caused by trafficking.
Cell surface biotinylation assays.
Surface biotinylation of HEK-293 cells was completed as previously described (6). Briefly, cells were incubated with cleavable EZ-Link Sulfo-NHS-SS-Biotin (0.5 mg/ml, Pierce) in ice-cold PBS (pH 7.4) for 30 min at 4°C. Free biotin was quenched with Tris-saline solution (10 mM Tris and 120 mM NaCl, pH 7.4), and detergent-soluble cell lysates were prepared. Biotinylated cell surface proteins were affinity purified using NeutrAvidin-conjugated agarose beads (Pierce), and purified cell surface proteins were analyzed by Western blot analysis. Nav1.5 channel protein was detected using mouse monoclonal anti-NavPAN antibody (S0819, Sigma), and the transferrin receptor (TransR) was used as an endogenous control protein (mouse monoclonal anti-TransR, no. 13-6890, Invitrogen). Bands corresponding to Nav1.5 were normalized to bands corresponding to TransR from the same sample. Nav1.5 protein expression (total or cell surface) was expressed relative to Nav1.5 protein expression (total or cell surface) in cells transfected with Nav1.5-WT alone.
Coimmunoprecipitation.
After protein extraction, cell pellets were pipetted up and down 20 times, rocked 15 min at 4°C, and finally centrifuged for 10 min at 6,000 g. Magnetic Dynabeads (Dynal, Oslo, Norway) were washed twice with 25 mM citric acid and 50 mM Na2HPO4 (pH 5), incubated with rat anti-hemagglutinin (HA; Roche) from Santa Cruz Biotechnology for 2 h at room temperature, washed three times again with 25 mM citric acid, 50 mM Na2HPO4, and 0.1% Tween, and incubated with precleared lysate samples (total 400 mg protein) while being rotated overnight at 4°C. After beads had been washed three times with lysis buffer, proteins were eluted with Laemmli sample buffer at 37°C for 30 min with agitation and analyzed by Western blot analysis.
Cross-linking experiments.
Protein cross-linking experiments were performed using either 50 or 100 μM disuccinimidyl suberate (DSS; ThermoFisher Scientific), a noncleavable and membrane-permeable cross-linker according to the manufacturer’s instructions. Briefly, cross-linking of Nav1.5, Nav1.5-L325R, or Nav1.5 + Nav1.5-L325R expressed in HEK-293 cells was performed before cell lysis to allow identification of the interaction between Na+ channel α-subunits. Cross-linking incubation time was 20 min. After protein isolation, Western blots were performed. To assess size, a Hi-Mark Pre-Stained Protein Ladder was used (Thermo Fisher Scientific).
Blue Native.
The Blue Native gel method has been adapted from the extensively described methods in Schagger et al. (13). Briefly, Blue Native gels were run in 3–12% Novex bis-Tris gels from Invitrogen with lysis buffer containing 1% digtonin, 150 mM NaCl, 10 mM Tris (pH 7.4), and protease inhibitors (Complete, Roche Diagnostics). The running buffer contained 50 mM Tricine, 15 mM bis-Tris (pH 7.0), and 0.002% Coomassie G-250. The loading buffer contained 5 mM bis-Tris (pH 7.0), 75 mM aminocaproic acid, 0.5% Coomassie G-250, 2% glycerol. The transfer buffer contained 50 mM Tris, 380 mM glycine, and 20% methanol.
Coupled Markov chain model.
A parameterized Markov chain model was used to demonstrate the distinct coupling behavior between the WT voltage-gated Na+ channel and the Nav1.5-L325R mutant. This Markov chain model was first developed by Chung and Kennedy (1) for coupled channel activity and recently used by Navedo et al. (8) to describe the coupling of voltage-gated Ca2+ channels. The single channel currents recorded by patch clamping were used by a hidden Markov model algorithm (Matlab) to estimate a transition matrix. From the transition matrix, a parameter set including the coupling coefficient (κ) and lumped parameters for channel open-to-open probability (ρ) and close-to-close probability (ζ) were estimated using the gradient descent algorithm. Parameters of a lumped model, ρ and ζ, are not equivalent to rate constants and therefore should not be used to describe the gating kinetics of the Na+ channel.
Statistical analysis.
Data are presented as means ± SE; n values are included in the respective figures. Statistical significance was estimated with SigmaPlot software by Student’s t-test. P < 0.05 was considered significant.
RESULTS
Nav1.5-L325R mutant traffics to the cell surface and exerts a DN effect.
We (3) have previously demonstrated a mechanism by which the DN effect was due to the retention of WT/mutant interacting channels within the ER. Our most recent work also showed that Na+ channel α-subunits dimerize and that the dimers have coupled gating properties (2). We therefore explored here whether the DN effect was exerted only through trafficking or whether coupled gating could also be implicated. For these experiments, we used the previously identified DN mutant Nav1.5-L325R (5). We first recorded whole cell Na+ currents from HEK-293 cells expressing WT, L325R, or WT + L325R channels. When Nav1.5-L325R was expressed alone, very little current was observed (~3% of the WT channels expressed alone; Fig. 1), suggesting that the channel at the cell surface was partially functional. When WT and mutant channels were expressed together, mimicking the heterozygous state of the patient, we observed only 25% of INa compared with cells expressing WT channels alone, thus resulting in a DN effect. Importantly, we were able to reproduce Keller et al.'s (5) results with this mutant (Fig. 1). To determine whether the DN effect was due to retention of WT + L325R dimers in the ER, we performed cell surface biotinylation assays. Surprisingly, these experiments showed that Nav1.5-L325R was able to traffic to the cell surface but with reduced expression compared with WT channels alone (40% compared with WT channels; Fig. 2). However, when the L325R mutant was coexpressed with WT channels, cell surface expression of total Nav channels was not significantly different than WT channels expressed alone. Since no reduction in total cell surface expression of Nav channels was observed when the mutant was coexpressed with WT channels, this suggests that a trafficking deficiency cannot explain the 75% decrease in INa shown in Fig. 1. Altogether, these results suggest that another mechanism must contribute to the DN effect in cells expressing both channels.
Fig. 1.

Nav1.5-L325R channels exert a dominant negative effect. A: representative family curves recorded from human embryonic kidney-293 cells expressing wild-type (WT), L325R, or WT + L325R channels B: current-voltage (I–V) relationships from WT, L325R, or WT+L325R channels. INa, inward Na+ current.
Fig. 2.

Nav1.5-L325R channels can reach the cell surface A: Western blot analysis and NaV1.5 protein quantification of total lysates from cells expressing wild-type (WT), L325R, or WT + L325R channels. B: cell surface expression levels from cells expressing WT, L325R, or WT + L325R channels. Despite the large (75%) decrease in current density when WT and mutant channels were expressed together, the surface expression of WT + L325R channels was not significantly altered. TransR, transferrin receptor.
Gating probability of cells expressing WT + L325R channels is decreased compared with cells expressing WT channels alone.
If trafficking deficiency was the main cause of the DN effect, we would expect a decrease in the number of channels at the cell surface, since the different compositions of dimers (L325R:L325R, WT:L325R, and L325R:WT) should be retained mainly within the ER as opposed to WT:WT dimers mainly expressed at the cell surface. If so, the coupled gating of the dimer would remain unaffected, and the gating probability and coupled gating under the patch should be identical. To assess whether the gating probability of single channels under the patch was altered between cells expressing WT or WT + L325R channels, we performed single channel recordings. First, our single channel recordings indicated that, in cells expressing WT and L325R channels, the density of channels under the patch was not significantly different from those in cells expressing WT channels alone, since all patches displayed openings. However, our results showed that the gating probability of the channels under each patch was drastically decreased in presence of L325R channels compared with WT channels expressed alone (Fig. 3, A–C). Indeed, although all patches displayed some single channel openings, the gating probability was drastically reduced for cells coexpressing WT + L325R channels, since the number of sweeps presenting openings was decreased by 54% compared with cells expressing WT channels alone (Fig. 3, B and C). One example of sweeps with no openings is shown in Fig. 3A, right (the last example). This result is consistent with a loss in INa of ~54% but does not explain entirely the 75% loss of current density observed at the whole cell level (Fig. 1A). Furthermore, the sum of single channels openings from cells expressing WT channels alone compared with cells expressing WT + L325R channels displayed a much more prominent loss of INa, identical to the one observed at the whole cell level (Fig. 3, D and E). It is worth noting the similitude with the currents recorded at the whole cell level shown in Fig. 3E.
Fig. 3.
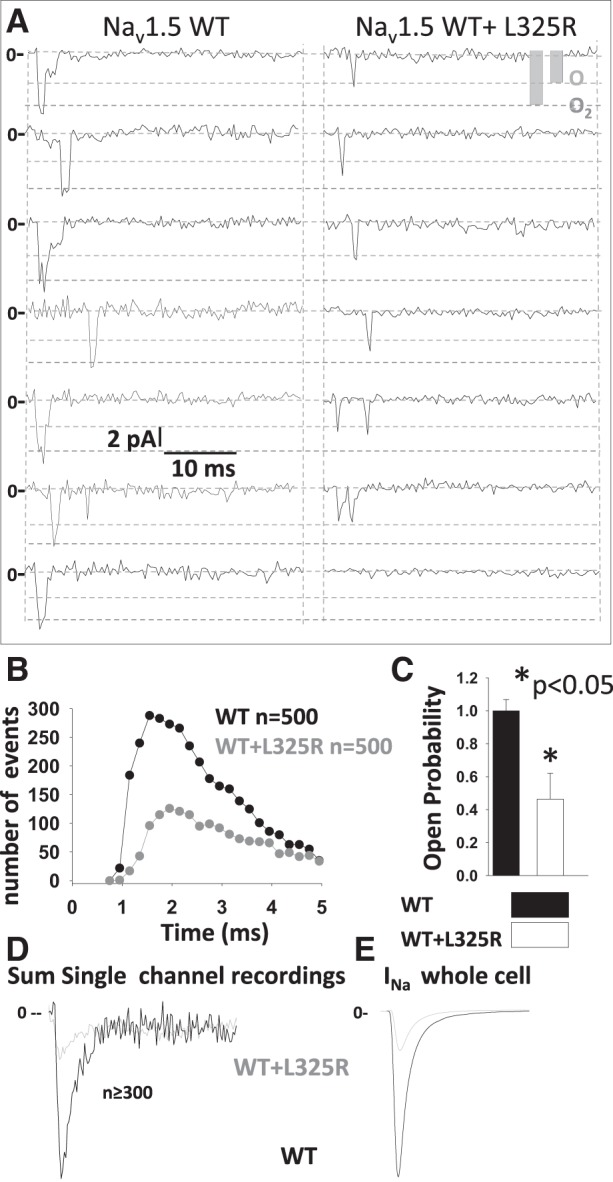
Drastic reduction in single channel gating probability in the presence of Nav1.5-L325R channels. A: representative traces of single channel recordings in human embryonic kidney-293 cells expressing wild-type (WT) or WT + L325R mutant channels. B: number of events as a function of time. C: open probability for WT or WT + L325R channels. D: sum of 500 single channel recordings at −20 mV. E: representative traces at −20 mV in the whole cell configuration for cells expressing Nav1.5-WT or Nav1.5-WT + L325R channels. INa, inward Na+ current.
Coupled gating of Na+ channels for cells expressing WT+L325R is drastically decreased compared with cells expressing WT alone.
Up to this point, the open probability could explain ~50% of the loss of current but not the 75% loss observed at the whole cell level. Therefore, we then investigated the coupled gating of the channel. We assessed whether the mutant leads to a DN effect on the WT channel by impairing WT gating through coupled gating. Thus, we analyzed the pattern of coupled gating by looking at the number of channels displaying double-level (coupled) openings versus single-level (uncoupled) openings. Strikingly, not only was the gating probability affected, as shown in Fig. 3, but also coupled gating (Fig. 4). While overall gating probability was decreased by 54%, we observed that the number of double-level events drastically dropped in cells expressing WT + L325R channels compared with cells expressing WT channels alone. Indeed, we observed a major decrease of double-level openings (by 85%) while also observing a 35% decrease of the single-level openings (Fig. 4A). This suggests that when the WT channel dimerizes with the mutant they almost never gate as dimers. We also assessed the percentage of dimers opening simultaneously or closing simultaneously. Coupled gating or simultaneous opening and closing decreased by 86 and 92%, respectively (Fig. 4C). Examples of coupled gating are shown in Fig. 4D.
Fig. 4.

Drastic reduction in the coupled gating of dimers in the presence of Nav1.5-L325R channels. A: number of double-level and single-level events as a function of time. B: open probability classified by level 2 and level 1. C: percentage of coupled openings and coupled closings. D: representative examples of noncoupled or stacked events as opposed to simultaneous/coupled events. Note that the wild-type (WT) + L325R opening pattern presented a drastic decrease in coupled gating, as illustrated by the drastic loss of coupled openings of level 2. ***P < 0.001. NS, not significant.
Coupled Markov chain model shows reduced coupling when the WT channel is coexpressed with DN mutant L325R.
A Markov chain model developed by Chung and Kennedy (1) was used to estimate the coupling strength between the channels. Coupling coefficients that varied from 0 for independent gating to 1 for tightly coupled gating were obtained from this model. Coupling coefficients for WT (n = 48) and WT + L325R (n = 47) channels are shown in Fig. 5. The high coupling coefficients (close to 1) estimated for many of the single channel traces recorded from WT channels suggest that they are tightly coupled. However, most of the single channel traces obtained from cells coexpressing WT + L325R channels had a lower coupling coefficient compared with WT channels alone. These results demonstrate that the coupling strength for the WT channels alone was much stronger compared with cells that coexpressed WT and L325R channels. Altogether, the data suggest that the dimers formed by WT + L325R are less probable to open as dimers and that the presence of L325R in the dimer impairs WT gating probability as a result of their coupled gating, leading to an overall 75% loss in INa.
Fig. 5.

Histogram of the coupling coefficients obtained from parameterized Markov chain modeling A: coupling coefficients obtained from single channel recordings from human embryonic kidney (HEK)-293 cells expressing wild-type (WT) channels (n = 48). B: coupling coefficients obtained from single channel recordings from HEK-293 cells expressing WT + L325R channels (n = 47).
L325R mutation does not alter dimerization of Na+ channels.
One additional mechanism that could contribute to the DN effect would be the alteration of Na+ channel dimerization in the presence of L325R. To address this, we performed three different sets of experiments, which all showed that WT and L325R channels are dimerizing. First, we performed coimmunoprecipitation between WT and L325R channels. The coimmunoprecipitation results showed that there was still an interaction between the two channels, since the channel tagged with HA could pull down the other channel tagged with cyan fluorescent protein (Fig. 6A). Second, as we did in our previous work (2), we performed cross-linking experiments to see if WT and L325R channels still dimerized. Indeed, we observed dimerization for WT + L325R channels just as we observed for WT channels alone or L325R channels alone, as illustrated by a band at double the molecular weight of a Na+ channel monomer in the presence of the cross-linker DSS (Fig. 6B). Finally, we performed protein chemistry experiments using nondenaturing Blue Native gels from cells that coexpressed WT and L325R mutant channels (Fig. 6C). When run under nondenaturing conditions (no SDS), a band at twice the weight of a Na+ channel monomer was observed (lane 3, no SDS as opposed to lane 1 in the presence of SDS for denaturing conditions). Interestingly, when WT and DN mutant L325R channels were coexpressed, we still observed a band twice the size as the monomer, strongly supporting that WT and L325R mutant channels were also forming dimers (Fig. 6C).
Fig. 6.

L325R mutant dimerizes with the wild-type (WT) channel. A: coimmunoprecipitation (IP) between α-subunits. Hemagglutinin (HA) antibody was used for IP, and Western blot analysis results were revealed with cyan fluorescent protein (CFP) antibody. Lane 1, cells cotransfected with WT-CFP and WT-HA; lane 2, cells cotransfected with WT-CFP and L325R-HA. B: cross-linking experiments performed in human embryonic kidney-293 cells expressing WT, L325R, or WT + L325R channels. Cross-linking was performed using disuccinimidyl suberate (DSS) (DSS) at 50 or 100 μM for 20 min. A band corresponding to a dimer of the Nav1.5 α-subunit was observed in the presence of the cross-linker compared with cells where no cross-linking was performed (0 DSS). C: Blue Native gel (3–15%) from lysates from cells expressing WT or WT + L325R channels. Lanes 1 and 2 included SDS in the loading buffer. All images display a representative example of at least 3 separate experiments. GFP AB, green fluorescent protein antibody.
High temperature does not affect coupled gating.
Since the Brugada phenotype with this mutation appears during fever, it suggests an exacerbated loss of INa at higher temperatures. In the previous work performed by Keller et al. (5), cells were incubated at 28°C before electrophysiological recording. Indeed, lowering the temperature restored currents, as it is known to partially restore the function of misfolded proteins and channels, thus rescuing their function and trafficking. In our present work, to investigate whether fever could exacerbate the loss of INa through the coupled gating mechanism, we measured INa at physiological temperature, 37°C, and then increased the bath temperature to 42°C (temperature reached within 1 min). This quick change in temperature allowed us to specifically assess the effect of temperature on gating while excluding changes due to trafficking. To investigate whether fever could increase the loss of INa mediated through L325R mutant channels, we measured peak current density at 37 and 42°C of HEK-293 cells expressing WT or WT + L325R channels and measured peak INa at −20 mV. No changes in current density between 37 and 42°C was observed, suggesting that gating was not altered at 42°C compared with 37°C (Fig. 7).
Fig. 7.

Effect of temperature on inward Na+ current (INa). A: representative family traces recorded from human embryonic kidney-293 cells expressing wild-type (WT) or WT + L325R channels. B: normalized peak current density at −20 mV subsequently recorded at 37 and 42°C within the same cells at 1-min interval.
DISCUSSION
Voltage-gated Na+ channels display coupled gating properties (2), a mechanism that is crucial for proper propagation of the action potential through the excitable tissues where they are expressed. For years, the Na+ channel α-subunit, encoded by SCNXA genes, was thought to be a monomer or an entity functional on its own. However, we demonstrated that Nav1.5 α-subunits were capable of a DN effect or transcomplementation by a trafficking mechanism (3). Furthermore, we demonstrated that voltage-gated Na+ channels interact as dimers and also possess cooperated gating (2). In the present study, we explored whether mutant Na+ channels could cause a DN effect not only through trafficking deficiency of the dimers but also due to their coupled gating. Our results show that the DN effect was not only due to a trafficking deficiency but also due to their gating cooperation. This discovery has significant implications in understanding the mechanism of channelopathies associated with voltage-gated Na+ channel mutations. Indeed, considering the conservation of voltage-gated Na+ channels through evolution and that we demonstrated coupled gating activity for two neuronal channels, Nav1.1 and Nav1.2, it is highly possible that this mechanism also applies to other channelopathies such as epilepsy, ataxia, or myotonia.
Nav1.5-L325R mutant traffics to the cell surface and exerts a DN effect.
Unlike voltage-gated K+ channel genes, which encode for one polypeptide assembling in a tetrameric complex, voltage-gated Na+ channel genes (SCNXA) encode for an entire functional channel that has traditionally been thought to be a monomer. However, a growing body of literature has started to suggest otherwise. Indeed, different studies have shown that the common polymorphism Nav1.5-H558R could partially restore INa impaired by mutations when expressed on different constructs (12, 14). Additionally, several studies have also demonstrated the presence of DN mutations found in BrS (3, 5, 7, 10, 11). In fact, our previous report described a mechanism by which the DN effect occurred through α-subunit interactions (3). In the present study, we investigated the DN effect of the Nav1.5-L325R mutant channel. To our surprise, we saw through cell surface biotinylation that the mutant channel can traffic to the cell surface, albeit to a lesser degree than the WT channel (Fig. 2). However, if the DN effect was caused by a trafficking deficiency of the dimers containing L325R (L325R:L325R, L325R:WT, and WT:L325R), then one would expect a decrease in total voltage-gated Na+ channel at the plasma membrane to be decreased to a level that could explain the 75% reduction in current density observed (Fig. 1). However, when doing surface biotinylation of the cells expressing both WT and L325R channels, the total amount of Nav channel proteins expressed at the cell surface was similar to that of WT channels expressed alone. This therefore suggests the presence of an additional mechanism to explain the DN effect observed.
Nav1.5-L325R impairs Nav1.5-WT gating.
Interestingly, at the single channel level, we were able to show that, in cells expressing WT and Nav1.5-L325R channels, the density of channels under the patch was not significantly different than in cells expressing WT channels alone, given that all patches displayed single channel openings. This is consistent with the surface biotinylation assays in which we did not see a reduction in total voltage-gated Na+ channel proteins at the cell surface. However, while all patches displayed openings, the overall probability of those channels to open was reduced by 54%, due to an increase in the number of sweeps per patches displaying no openings (Fig. 3). This reduction in open probability could explain ~50% of the loss of current but not the 75% loss observed at the whole cell level. We then turned to the coupled gating properties of voltage-gated Na+ channels (2) as an additional mechanism explaining the DN effect. Strikingly, we observed a major decrease in the number of channels displaying coupled gating and double-level events in presence of the L325R mutant (Fig. 4). This reduction in double-level events resulted in the main loss of INa leading to the DN effect. This change in coupled gating behavior was further assessed using a parameterized Markov chain model. This Markov chain model was first developed by Chung and Kennedy (1) for coupled channel activity and recently used by Navedo et al. (8) to describe coupling of voltage-gated Ca2+ channels. Our results demonstrate that the coupling strength for the WT channel alone is much stronger than cells that coexpressed WT and L325R channels. Altogether, the data suggest that the dimers formed by WT + L325R channels are less likely to open as dimers and that the presence of L325R in the dimer impairs WT gating probability as a result of their coupled gating leading to an overall 75% loss in INa. Thus, our results strongly support that the DN effect is exerted through a direct physical dimerization between WT and mutant channels (Fig. 6) and coupled gating properties between WT and mutant channels.
Potential mechanism of gating modulation.
It remains unclear how the L325R mutant can impair gating of the WT channel. We have previously demonstrated that 14-3-3 was involved in the dimerization and biophysical coupling between Na+ channels. We (2) speculated that 14-3-3 dimers could link the two channels by their long predicted α-helix coming from segment S6 of the first domain. Such conformation would allow 14-3-3 dimers to act as a lever arm allowing the channels to have coupled gating. Hence, we posit that if L325R channels possess a very low gating probability, it could impair WT gating due to the physical coupling through 14-3-3, and the dimer would remain closed. Considering that patients harboring this mutation are affected with BrS, it would be interesting if our findings could be translated into potential therapeutics to prevent this coupling. We have previously demonstrated that in the presence of difopein, a 14-3-3 inhibitor, we could uncouple the α-subunits and, by doing so, abolish the DN effect exerted by all mutants tested including the L325R mutant used in the present study (2). Even though this target is very promising, inhibiting 14-3-3 protein, which is a ubiquitous protein, may have a too important and deleterious impact to be used as a possible treatment. However, considering the potential therapeutic implication, similar avenues to uncouple the channels should be explored in the future.
In conclusion, our results provide evidence that the Nav1.5-L325R channel is able to traffic to the cell surface on its own. In addition, when coexpressed with WT channels, the cell surface expression was not significantly different than when the WT channel was expressed alone. Therefore, trafficking does not seem to be the main cause of the DN suppression exerted by the mutant. Single channel recordings showed that although the cell surface expression was not significantly altered, the biophysical coupling between WT and L325R mutant channels drastically impaired their coupled gating and WT gating probability. Our findings have unveiled a new mechanism (coupled gating) that can contribute to a DN effect for Na+ channel mutations. These results provide new biophysical insights and therapeutic strategies for cardiac arrhythmias or other channelopathies associated with SCNXA mutations.
GRANTS
This work was supported by American Heart Association Scientist Development Grant 0635295N (to I. Deschênes), National Heart, Lung, and Blood Institute Grant R01-HL-094450 (to I. Deschênes), The Kenneth M. Rosen Fellowship in Cardiac Pacing and Electrophysiology from the Heart Rhythm Society (to J. Clatot), and Agence Nationale de la Recherche Grant ANR-15-CE14-0006-01 (to C. Marionneau).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.C., K.R.L., and I.D. conceived and designed research; J.C., Y.Z., A.G., and H.L. performed experiments; J.C., Y.Z., A.G., H.L., K.R.L., and C.M. analyzed data; J.C., Y.Z., K.R.L., C.M., and I.D. interpreted results of experiments; J.C., Y.Z., H.L., C.M., and I.D. prepared figures; J.C., Y.Z., and I.D. drafted manuscript; J.C., Y.Z., K.R.L., C.M., and I.D. edited and revised manuscript; J.C., Y.Z., A.G., H.L., K.R.L., C.M., and I.D. approved final version of manuscript.
REFERENCES
- 1.Chung SH, Kennedy RA. Coupled Markov chain model: characterization of membrane channel currents with multiple conductance sublevels as partially coupled elementary pores. Math Biosci 133: 111–137, 1996. doi: 10.1016/0025-5564(95)00084-4. [DOI] [PubMed] [Google Scholar]
- 2.Clatot J, Hoshi M, Wan X, Liu H, Jain A, Shinlapawittayatorn K, Marionneau C, Ficker E, Ha T, Deschênes I. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun 8: 2077, 2017. doi: 10.1038/s41467-017-02262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clatot J, Ziyadeh-Isleem A, Maugenre S, Denjoy I, Liu H, Dilanian G, Hatem SN, Deschênes I, Coulombe A, Guicheney P, Neyroud N. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 α-subunits. Cardiovasc Res 96: 53–63, 2012. doi: 10.1093/cvr/cvs211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoshi M, Du XX, Shinlapawittayatorn K, Liu H, Chai S, Wan X, Ficker E, Deschênes I. Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ Cardiovasc Genet 7: 123–131, 2014. doi: 10.1161/CIRCGENETICS.113.000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller DI, Rougier JS, Kucera JP, Benammar N, Fressart V, Guicheney P, Madle A, Fromer M, Schläpfer J, Abriel H. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res 67: 510–519, 2005. doi: 10.1016/j.cardiores.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 6.Marionneau C, Carrasquillo Y, Norris AJ, Townsend RR, Isom LL, Link AJ, Nerbonne JM. The sodium channel accessory subunit Navβ1 regulates neuronal excitability through modulation of repolarizing voltage-gated K+ channels. J Neurosci 32: 5716–5727, 2012. doi: 10.1523/JNEUROSCI.6450-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercier A, Clément R, Harnois T, Bourmeyster N, Faivre JF, Findlay I, Chahine M, Bois P, Chatelier A. The β1-subunit of Nav1.5 cardiac sodium channel is required for a dominant negative effect through α-α interaction. PLoS One 7: e48690, 2012. doi: 10.1371/journal.pone.0048690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res 106: 748–756, 2010. doi: 10.1161/CIRCRESAHA.109.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, Kangawa K, Matsuo H, Raftery MA, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312: 121–127, 1984. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 10.Núñez L, Barana A, Amorós I, de la Fuente MG, Dolz-Gaitón P, Gómez R, Rodríguez-García I, Mosquera I, Monserrat L, Delpón E, Caballero R, Castro-Beiras A, Tamargo J. p.D1690N Nav1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm 10: 264–272, 2013. doi: 10.1016/j.hrthm.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 11.Park DS, Cerrone M, Morley G, Vasquez C, Fowler S, Liu N, Bernstein SA, Liu FY, Zhang J, Rogers CS, Priori SG, Chinitz LA, Fishman GI. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest 125: 403–412, 2015. doi: 10.1172/JCI76919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poelzing S, Forleo C, Samodell M, Dudash L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R, Guida P, Chahine M, Pitzalis M, Deschênes I. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 114: 368–376, 2006. doi: 10.1161/CIRCULATIONAHA.105.601294. [DOI] [PubMed] [Google Scholar]
- 13.Schägger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 217: 220–230, 1994. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- 14.Shinlapawittayatorn K, Du XX, Liu H, Ficker E, Kaufman ES, Deschênes I. A common SCN5A polymorphism modulates the biophysical defects of SCN5A mutations. Heart Rhythm 8: 455–462, 2011. doi: 10.1016/j.hrthm.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]