

Abstract
Polycystic ovary syndrome is a complex and common disorder in women, and those affected experience an increased burden of cardiovascular disease. It is an intergenerational syndrome, as affected women with high androgen levels during pregnancy “program” fetal development, leading to a similar phenotype in their female offspring. The effect of excess maternal testosterone exposure on fetal cardiomyocyte growth and maturation is unknown. Pregnant ewes received biweekly injections of vehicle (control) or 100 mg testosterone propionate between 30 and 59 days of gestation (early T) or between 60 and 90 days of gestation (late T). Fetuses were delivered at ~135 days of gestation, and their hearts were enzymatically dissociated to measure cardiomyocyte growth (dimensional measurements), maturation (proportion binucleate), and proliferation (nuclear Ki-67 protein). Early T depressed serum insulin-like growth factor 1 and caused intrauterine growth restriction (IUGR; P < 0.0005). Hearts were smaller with early T (P < 0.001) due to reduced cardiac myocyte maturation (P < 0.0005) and proliferation (P = 0.017). Maturation was also lower in male than female fetuses (P = 0.004) independent of treatment. Late T did not affect cardiac growth. Early excess maternal testosterone exposure depresses circulating insulin-like growth factor 1 near term and causes IUGR in both female and male offspring. These fetuses have small, immature hearts with reduced proliferation, which may reduce cardiac myocyte endowment and predispose to adverse cardiac growth in postnatal life. While excess maternal testosterone exposure leads to polycystic ovary syndrome and cardiovascular disease in female offspring, it may also predispose to complications of IUGR and cardiovascular disease in male offspring.
NEW & NOTEWORTHY Using measurements of cardiac myocyte growth and maturation in an ovine model of polycystic ovary syndrome, this study demonstrates that early gestation excess maternal testosterone exposure reduces near-term cardiomyocyte proliferation and maturation in intrauterine growth-restricted female and male fetuses. The effect of testosterone is restricted to exposure during a specific period early in pregnancy, and the effects appear mediated through reduced insulin-like growth factor 1 signaling. Furthermore, male fetuses, regardless of treatment, had fewer mature cardiomyocytes than female fetuses.
Keywords: androgens, cardiac growth and development, intrauterine growth restriction, ovine, sex differences
INTRODUCTION
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder of reproductive-aged women, affecting up to 1 in 10 women (36, 61). Women affected by PCOS have subfertility and once pregnant have a much greater risk of both maternal and fetal complications (24). Hyperandrogenism is a common characteristic of PCOS, including during pregnancy (81), and women with PCOS affected by high androgen levels are at even greater risk of fetal complications, such as intrauterine growth retardation (IUGR) and induced preterm delivery (24). Experimental models have investigated the fetal effects of excess maternal testosterone exposure and found a host of sex-specific effects on growth, metabolic regulation, and reproductive function that persist into adulthood, essentially recapitulating a PCOS phenotype in female offspring (60) and making PCOS a generationally transmitted syndrome (27). Although excess maternal testosterone exposure has been shown to cause fetal growth restriction (always in female offspring and sometimes in male offspring) (23, 51, 83, 87) and hypertension in adult women (44), the effects on the fetal heart have not been studied.
Cardiovascular disease is the leading cause of death in women (33), and women with PCOS are at an increased risk for cardiovascular disease (3, 26). Some of this increased risk is understood to be the result of excess androgen exposure, insulin resistance, obesity, and hypertension (26). However, there is growing understanding that disruptions to the normal fetal environment cause cardiac adaptations that have consequences for lifelong cardiovascular health (2, 86). Among the many developmental perturbations that can lead to adult cardiovascular outcomes are maternal obesity (70), preterm birth (7), and assisted reproductive technology (77), all factors in PCOS pregnancies. More fundamentally, women affected by PCOS have high androgen levels during pregnancy (81). Timing of exposure to excess maternal androgens differentially masculinizes the female genitalia and brain (71), and alterations in placental morphology are time dependent as well (5). However, the role of excess maternal testosterone exposure in fetal cardiac programming and the critical window of exposure have not been established.
Although sex steroids can have direct effects on the heart (6), excess maternal testosterone exposure strongly affects the fetal insulin-like growth factor (IGF) system (23), and IGF-1 is the main endocrine driver of fetal growth (38, 57). Although variable earlier in gestation, IGF-1 signaling is reduced near term in female fetuses affected by excess maternal testosterone exposure (male fetuses were not tested because differential perinatal growth effects were exclusive to female fetuses in that study) (51). In the fetal heart, cardiac myocytes respond to IGF-1 stimulation by proliferation without induction of terminal differentiation (84), although stimulation of the IGF-2 receptor leads to cellular enlargement (90, 91). Fetal cardiac myocytes expand their numbers through proliferation, a process that is attenuated toward term by terminal differentiation (which transforms mononucleated cells to binucleated cells), after which cardiac myocyte enlargement becomes more prominent (42). Changes in fetal cardiac myocyte growth can affect cardiac myocyte endowment (41), the maximum number of cells that the heart has for life (11), and can contribute to pathology in the adult heart (68).
We hypothesized that excess maternal testosterone exposure early in gestation that led to fetal growth restriction would reduce cardiac myocyte size and proliferation near term, but not terminal differentiation (maturation), and that this effect would be more pronounced in female than male fetuses. We tested this hypothesis by injecting pregnant ewes with testosterone propionate or vehicle between 30 and 59 days of gestational age (dGA) or between 60 and 90 dGA and then studied fetal hearts at 135 dGA (term: 147 dGA). We found that early (30–59 dGA) excess maternal testosterone exposure reduced cardiac myocyte cell cycle activity in both female and male fetuses and that cardiac myocyte size was unaffected by treatment. Contrary to our hypothesis, terminal differentiation was also reduced by early excess maternal testosterone exposure. Additionally, independent of treatment, fewer male cardiac myocytes were terminally differentiated.
MATERIALS AND METHODS
Animal model.
These experiments were conducted in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Oregon State University.
To accurately date pregnancy in Polypay ewes in their second breeding season, estrous cycle synchronization with intravaginal progesterone pessaries and PGF2α injections was carried out as previously described (69). Pregnant ewes (n = 22, singleton or twin pregnancies) were assigned randomly to receive either biweekly intramuscular injections of either corn oil vehicle (control; n = 8 female fetuses and 4 male fetuses) or 100 mg testosterone propionate (Steraloids, Newport, RI) in 2 ml of corn oil from 30 to 59 dGA (early T; n = 4 female fetuses and 5 male fetuses) or from 60 to 90 dGA (late T; n = 5 female fetuses and 5 male fetuses). This dose of testosterone propionate has been previously shown to produce concentrations of testosterone in fetal blood equal to twice that of age-matched control male fetuses (12, 72). Fetal deliveries at 135 ± 1 dGA (mean ± SD, range: 133–137 dGA) were performed surgically under general anesthesia as previously described (69). A blood sample was taken from the umbilical vein, the umbilical cord was ligated, and the fetus was removed. The fetus was rapidly decapitated and weighed, and the heart was removed, trimmed following anatomic landmarks, and then enzymatically digested into individual cardiac myocytes as previously described (40). Cardiac myocytes were fixed with fresh 2% formaldehyde.
IGF-1 measurement.
IGF-1 was measured from frozen fetal plasma by ELISA according to the manufacturer’s protocol (Alpco, Salem, NH). Assay parameters were as follows: sensitivity, 0.091 ng/ml; intra-assay coefficient of variation (CV), 5.1%; and interassay CV, 3.4%. Testosterone was extracted with ether from serum, fractionated on LH20 Sephadex columns (Sigma-Aldrich, St. Louis, MO), and measured by radioimmunoassay as previously described (71). Assay parameters were as follows: mean percentage of recovery, 62.3%; sensitivity, 4.5 pg/ml; and interassay CV 10.5%.
Myocyte measurements.
Calibrated morphometric measurements were taken from no fewer than 50 mononucleated and 50 binucleated cells from each ventricular free wall by light microscopy as previously described (43). At least 300 myocytes from each ventricle of each animal were used to determine the fractions of mono-, bi-, and quadrinucleated cells. Cell cycle activation, as determined from anti-Ki-67 antibody binding (MIB-1, Dako, Carpinteria, CA) in ~500 cells per ventricle per animal was measured as previously described (43).
Statistical analysis.
Statistical analysis was performed using SPSS (version 24.0, IBM SPSS Statistics for Macintosh, Armonk, NY) and Prism (version 6.0h, GraphPad Software, San Diego, CA). Fetal variables, such as body weight, were analyzed by two-way ANOVA with sex (male, female) and treatment (early T, late T, vehicle control) as main effects. Three-way mixed-model ANOVA was used to analyze myocyte parameter with sex, treatment, and ventricle [left ventricle (LV), right ventricle (RV)] as main effects. If indicated, multiple comparisons were tested by Tukey’s honestly significant difference test. Outliers were defined as values of >1.5 times the interquartile range from the boundary of the interquartile range. Elimination of outliers did not meaningfully affect results, and therefore all data are included in the final analysis. Data were screened for maternal effect by inclusions of averaged values of (within treatment) same-sex twin pairs; as the determination of significance was not changed in any analysis, all data are included in the final analysis. The relationship between IGF-1 and fetal weights was tested by Pearson’s moment-product correlation. P < 0.05 was taken as significant. Results are presented as means ± SE unless otherwise noted.
RESULTS
Fetal physiological characteristics.
For circulating IGF-1 levels (Fig. 1A), the interaction term was not statistically significant (P = 0.099). Main effect was significant for treatment (P < 0.01) but not sex. Early T was associated across both sexes with mean IGF-1 of 62 ng/ml (39%) less than control (P = 0.018) and 51 ng/ml (34%) less than late T (P = 0.038). For log-transformed circulating testosterone levels (Fig. 1B), the interaction term was not significant, but main effect was significant for sex. Male fetuses had testosterone levels 0.2 log units higher than female fetuses (P = 0.044).
Fig. 1.
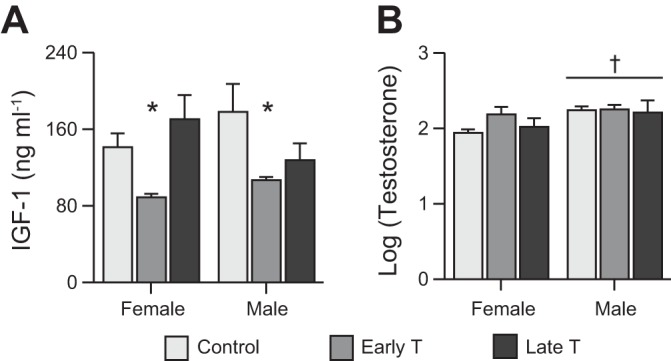
Circulating insulin-like growth factor (IGF-1) and testosterone levels. A and B: circulating IGF-1 levels (A) and testosterone levels (B) (log transformed) in ~135 days of gestation (dGA) fetal sheep after maternal exposure to vehicle or to testosterone propionate from 30 to 59 dGA (early T) or from 60 to 90 dGA (late T). Values are means ± SE. Control: n = 8 (A) or 6 (B) female fetuses and n = 4 male fetuses; early T: n = 4 female fetuses and n = 4 (A) or 3 (B) male fetuses; and late T: n = 5 female fetuses and n = 5 male fetuses. Comparisons were performed by two-way ANOVA. *Different from control and late T; †different from female fetuses (P < 0.05, exact values in text).
For body weight (Fig. 2A), the interaction term was not significant, but main effect was significant for treatment (P < 0.0005). Early T was associated across both sexes with a mean body weight of 1.0 kg (25%) less than control (P < 0.0005) and 1.2 kg (29%) less than late T (P < 0.0005). The Gaussian distribution of control body weights was determined to find the lower 10% confidence interval (3.777 kg), which was used as the threshold for defining IUGR. Female and male early T body weights (individual and group mean) fell below the threshold, with the exception of one male fetus. All female and male late T body weights were above the threshold. There was a strong correlation between circulating IGF-1 and body weight (r = 0.6078, P = 0.0004; Fig. 2B).
Fig. 2.
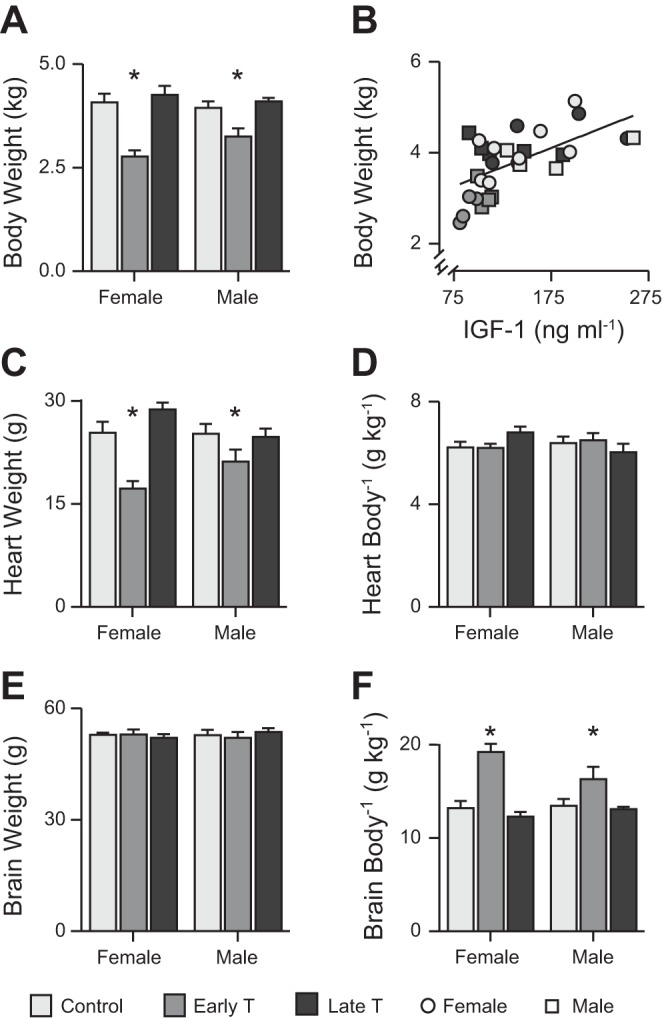
Body, heart, and brain weight. A: body weights differed by treatment and were lower with early testosterone propionate (early T) treatment versus vehicle (control) or late testosterone propionate (late T) treatment. B: fetal body weight was correlated with circulating insulin-like growth factor (IGF-1) levels across treatments (r = 0.6078, P = 0.0004). C: heart weights differed by treatment and were lower in early T than control or late T. D: relationship between heart and body weight was similar across treatments and sexes. E: brain weights were similar across treatments and sexes. F: brain-to-body-weight ratio was greater in early T versus control or late T fetuses. Values are means ± SE. Control: n = 8 female fetuses and n = 4 male fetuses; early T: n = 4 female fetuses and 5 male fetuses; and late T: n = 5 female fetuses and n = 5 male fetuses. Comparisons were performed by two-way ANOVA. Pearson’s product-moment correlation was conducted to test the relationship between IGF-1 and body weight. *Different from control and late T (P < 0.05, exact values in text).
For heart weight (Fig. 2C), the interaction term was not significant (P = 0.056). Main effect was significant for treatment (P < 0.0005). Early T was associated across both sexes with a mean heart weight of 6.1 g (24%) less than control (P < 0.002) and 7.6 g (28%) less than late T (P < 0.0005). There was a strong correlation between circulating IGF-1 and heart weight (r = 0.5214, P = 0.0031; data not shown). For the heart-to-body weight ratio (Fig. 2D), no interaction or main effects were significant, meaning that the relationship between heart and body weight was constant across all comparisons.
Brain weight was measured, as brain sparing is a hallmark of IUGR. For brain weight (Fig. 2E), no interaction term or main effect was significant. For the brain-to-body weight ratio (Fig. 2F), the interaction term was not significant (P = 0.0976), but main effects were significant for treatment (P < 0.0005). Early T was associated across both sexes with a mean normalized brain weight of 4.3 g (32%) greater than control (P < 0.0005) and 4.9 g (39%) greater than late T (P < 0.0005).
Cardiac myocyte growth and maturation.
For proportion binucleate (Fig. 3A), no interaction effect was significant. However, main effects were significant for all factors. Across both sexes and ventricles, early T was associated with a mean proportion binucleate of 15 points (32%) lower than control (P < 0.0005) and 14 points (30%) lower than late T (P < 0.001). Male sex was associated across all treatments and both ventricles with a mean proportion binucleate of 9 points (19%) lower than female sex (P = 0.004). The LV across all treatments and both ventricles was associated with a mean proportion binucleate of 7 points (14%) lower than the RV (P = 0.004).
Fig. 3.
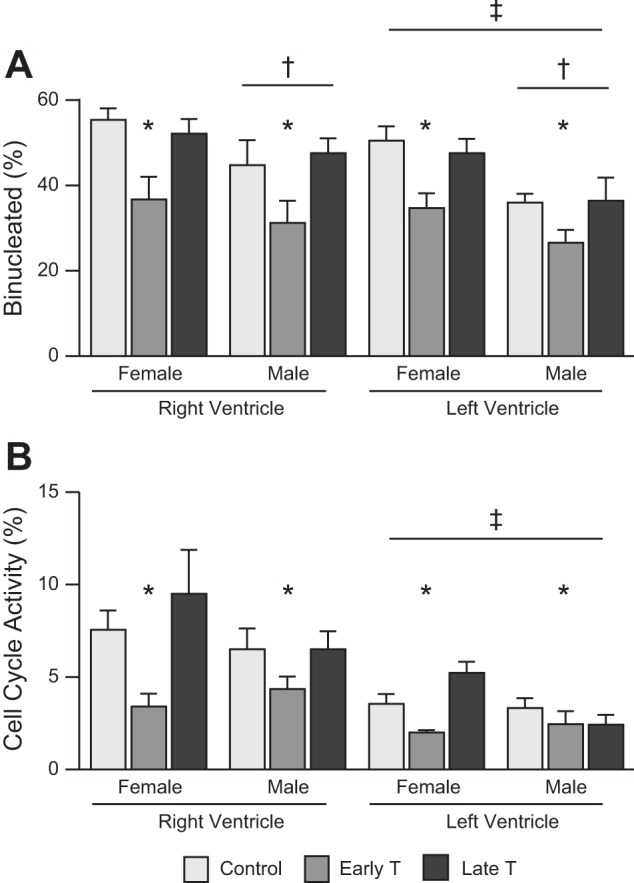
Cardiac myocyte terminal differentiation and cell cycle activity. A: proportionally fewer myocytes were binucleate with early testosterone propionate (early T) treatment compared with vehicle (control) or late testosterone propionate (late T) treatment, fewer myocytes were binucleate in male than female fetuses, and fewer myocytes were binucleate in the left ventricle (LV) than in the right ventricle (RV) in fetal sheep at ~135 days of gestation after maternal exposure to control (n = 8 female fetuses and 4 male fetuses) or early T (n = 4 female fetuses and 5 male fetuses) or late T (n = 5 female fetuses and 5 male fetuses). B: cell cycle activity was less in early T than control or late T and less in the LV than in the RV. Values are means ± SE. Comparisons were performed by three-way mixed-model ANOVA. *Different from control and late T; †different from female fetuses; ‡different from the RV (P < 0.05, exact values in text).
For cell cycle activity (Fig. 3B), no interaction effect was significant. However, main effects were significant for treatment (P = 0.009) and ventricle (P < 0.0005). Early T across both sexes and ventricles was associated with a mean cell cycle activity of 2.2 percentage points (45%) lower than control (P = 0.032) and 2.9 percentage points (49%) lower than late T (P = 0.009). The LV was associated across all treatments and both sexes with a mean cell cycle activity of 3.2 percentage points (51%) lower than the RV (P < 0.0005).
Myocyte dimensions were unaffected by treatment or sex and were larger in the RV than in the LV (Fig. 4). There was no significant interaction between the factors for any myocyte dimension. For mononucleated length (Fig. 4A), main effects were significant for ventricle (P < 0.0005). The LV was associated across all treatments and both sexes with a mean mononucleated length of 6.3 μm (9%) shorter than the RV (P < 0.0005). For binucleated length (Fig. 4C), there was a tendency for a three-way interaction (P = 0.091), and main effects were significant for ventricle (P < 0.0005). The LV was associated across all treatments and both sexes with a mean binucleated length of 10.3 μm (11%) shorter than the RV (P < 0.0005). For mononucleated width (Fig. 4B), main effects were significant for ventricle (P < 0.0005). The LV was associated across all treatments and both sexes with a mean mononucleated width of 2.0 μm (17%) narrower than the RV (P < 0.0005). For binucleated width (Fig. 4D), main effects were significant for ventricle (P < 0.0005). The LV was associated across all treatments and both sexes with a mean binucleated width of 2.9 μm (25%) narrower than the RV (P < 0.0005).
Fig. 4.

Cardiac myocyte size. Right ventricular (RV) dimensions were greater than left ventricular (LV) dimensions for mononucleate length (A), mononucleate width (B), binucleate length (C), and binucleate width (D) in fetal sheep at ~135 days of gestation (dGA) after maternal exposure to vehicle (control: n = 8 female fetuses and 4 male fetuses) or testosterone propionate from 30 to 59 dGA (early T; n = 4 female fetuses and 5 male fetuses) or from 60 to 90 dGA (late T; n = female fetuses and 5 male fetuses). Values are means ± SE. Comparisons were performed by three-way mixed-model ANOVA. ‡Different from the RV (P < 0.05, exact values in text).
Given the relationship between circulating IGF-1 and heart and body weights, the relationship between circulating IGF-1 and myocyte growth parameters was also investigated. Cell cycle activity was significantly correlated with IGF-1 in the RV (r = 0.5403, P = 0.0021) and LV (r = 0.3912, P = 0.0325). Proportion binucleate was correlated with IGF-1 in the RV (r = 0.4248, P = 0.0193) but not the LV (P = 0.2266). There was no correlation between IGF-1 and LV or RV binucleated myocyte length or width.
DISCUSSION
Given the differential effects of timing of excess maternal testosterone exposure on masculinization of female fetuses (71), we investigated whether timing of excess maternal testosterone exposure differentially causes IUGR and affects cardiac growth. In the present study, we show, for the first time, that excess maternal testosterone exposure in early gestation (30–59 dGA but not 60–90 dGA) affects cardiac growth in IUGR female and male fetuses near term. The reduction in cardiac weight, which was proportional to somatic growth restriction, was the result of reduced cardiac myocyte terminal differentiation (because mononucleated cells are smaller than binucleated cells) and reduced proliferation (because myocyte sizes were not different) (42). These changes may result in a less mature heart with a small cardiac myocyte endowment at birth (41, 68, 74).
At every stage in development, there is a typical relationship between body and heart size (42); excess maternal testosterone exposure did not disrupt this relationship in near-term fetuses. However, it has been shown in many large epidemiological as well as experimental studies that being born small profoundly raises risk for cardiovascular disease later in life (2, 86). Mediators include increased obesity, reduced lean body mass, and insulin resistance (18, 29, 65), but important changes occur within the heart itself, such as reduced cardiomyocyte endowment (14, 67). Therefore, while the small hearts in the early T group are appropriate for body size, they also reflect the aberrant myocardium formed during development that later in life may contribute to the increased cardiac disease in women affected by PCOS (3, 26), as has been found following IUGR from other causes (7, 8, 10, 14, 63, 67). Animal models of PCOS that have explored the mechanisms contributing to such cardiac dysfunction find progressive cellular hypertrophy and fiber disarray, interstitial fibrosis, and myocardial apoptosis as well as increased myocardial molecular markers of insulin signaling, hypertrophy, and stress (32, 88). Cardiac changes associated with PCOS have developmental origins. Increased LV mass is noted early in life (76) and is sustained into young adulthood together with left atrial dilation (89). Our findings of reduced prenatal cardiac myocyte proliferation and potential underendowment in the context of cardiac hypertrophy later in life may help explain the etiology of cardiac pathophysiology of PCOS.
Sex effects.
Padmanabhan and colleagues (23, 44, 51, 60, 61, 83, 87) have extensively studied the sheep excess maternal testosterone model with exposure between 30 and 90 dGA. Those investigations have inconsistently found differences between female and male offspring, sometimes finding that male offspring are not growth-restricted near term or at birth (5, 51, 87). We did not find any interaction of sex with treatment on body weight, suggesting that early T leads to growth restriction at term similarly in both sexes (Fig. 1B), in concordance with the finding of IUGR in both female and male offspring of women affected by PCOS (80). It is possible that testosterone supplementation between 60 and 90 dGA partially ameliorates the growth-retarding effects of excess testosterone earlier in gestation only in male fetuses. Early gestation estrogen and progesterone exposure has been suggested to increase the risk of cardiovascular disease (58, 66), but the results of this study suggests that in addition to predisposing female offspring to PCOS, excess maternal testosterone exposure may predispose male offspring to IUGR and cardiovascular disease. Further studies are necessary to understand how fetal sex changes cardiac growth and development responses to in utero challenges.
Independent of treatment, we found that male sex reduced terminal differentiation by ~9 points compared with female fetuses (Fig. 3A). Few studies in fetal sheep have been powered to separately analyze male and female fetuses. However, as less mature cardiac myocytes in the male fetus have been previously described (50), a careful analysis of sex differences in cardiomyocyte maturation at term may be valuable, especially given sex differences in fetal growth strategies and risk of myocardial damage at birth (28, 47).
Timing of testosterone exposure and the role of IGF-1.
We have previously found that exposure to excess maternal testosterone in the first half of this period masculinizes the female genitalia, but excess testosterone in the second half of that period masculinizes the sexually dimorphic nucleus of the preoptic area (71). We found low circulating IGF-1 and reduced body weight (Fig. 1) in fetuses that were exposed to high maternal testosterone between 30 and 59 dGA (early T) but not between 60 and 90 dGA (late T). The somatic and cardiac growth effects in early T fetuses may result from alterations in the fetal IGF system. Changes in circulating IGF and IGF binding proteins are present after 30- to 90-dGA excess maternal testosterone exposure as early in gestation as has been previously investigated (23), secondary to altered placental development (5). After excess maternal testosterone exposure from 30 dGA, by 65 dGA, placenta weight is reduced, whereas efficiency is increased and fetal weight is not yet impacted (5). In sheep, implantation occurs by day 16 (82), and the placenta grows very rapidly, attaining close to the maximum size and weight by day ~60 (53), which may explain the critical window of 30–60 dGA for the impact of excess maternal testosterone exposure.
Body weight across all treatments was strongly correlated with IGF-1 levels (Fig. 1C), supporting a role for low IGF-1 in IUGR after early excess maternal testosterone exposure, as has been previously described in clinical IUGR and other experimental IUGR models (34, 38, 57, 73). Although a 1-wk infusion of an IGF-1 analog with a low affinity for IGF-binding proteins into the late-term fetus leads to increased heart growth disproportionate to body growth (84), in this model with low circulating IGF-1, heart growth was regulated proportional to body weight. Heart growth occurs proportional to body growth, resulting in a characteristic weight ratio that is formulaic according to developmental age (42), but the mechanisms linking normal body and heart growth have not been determined. IGF-1 stimulates fetal cardiomyocyte growth (84, 91), and thus the low levels of IGF-1 may play a role in the proportional slowing of heart growth in early T fetuses. This possibility is supported by the correlation between cardiac myocyte cell cycle activity and circulating fetal IGF-1 levels, suggesting that IGF-1 regulation of cardiac myocyte proliferation might contribute to the coordination of fetal heart and body growth. Conclusive evidence of a mechanistic link awaits further study.
In contrast to reduced proliferation and maturation, it is notable that cardiac myocyte dimensions were not changed in this study (Fig. 4). Fetal cardiac myocytes are sensitive to mechanical load (4, 39, 59, 75, 78), suggesting that venous and arterial pressures may not be different in fetuses of ewes exposed to early excess testosterone during pregnancy. It further supports the cardiac role of IGF-1 in utero as being primarily proproliferative (84), although one study has shown in utero myocyte hypertrophy in male fetuses (50), potentially through the IGF-2 receptor (90). However, unchanged myocyte size is consistent with most models of IUGR (13, 15, 49, 52, 54, 56, 85).
A direct effect of testosterone on the heart is possible, given that fetal cardiomyocytes express androgen receptors (64). It is notable that testosterone induces cardiomyocyte differentiation from embryonic stem cells (1, 35, 37), an effect that may be relevant to differential timing effects of excess maternal testosterone exposure. However, given substantially reduced heart weight in early T fetuses but normal cellular dimensions, it is likely that these fetuses have fewer rather than more cardiomyocytes. Testosterone is also known to regulate cardiomyocyte apoptosis, with a prosurvival benefit under conditions of stress with low added levels of testosterone (modeling hormone replacement) (92, 93) but proapoptosis with high levels of testosterone (modeling anabolic steroid abuse) (25, 62). However, testosterone levels are normal at 135 dGA in fetuses of ewes transiently exposed to excess testosterone earlier in gestation (72). Therefore, the role for direct action of testosterone on the developing heart remains to be determined.
Retarded maturation.
The cause of the reduced cardiac myocyte maturation that is a hallmark of IUGR has not been demonstrated (15, 49, 52, 54, 56, 85). Here, we showed a relationship between terminal differentiation and circulating IGF-1 in the RV but not in the LV. Given this inconsistency, it is possible that that IGF-1 is only incidentally correlated with cardiac myocyte terminal differentiation or does not have much influence. This is difficult to distinguish because proportion binucleate is the cumulative result of several processes (proliferation, terminal differentiation, and apoptosis), and thus it may be an insensitive indicator of the role of IGF-1 on terminal differentiation. It is notable, however, that 1 wk of IGF-1 analog infusion in the near-term fetus did not increase proportion binucleate in near-term fetal sheep (84), supporting a noncausal role of IGF-1 in terminal differentiation. Furthermore, IGF-1 levels were not different between female and male fetuses, although proportion binucleate was different.
Thyroid hormone is a hormone that has been implicated in mammalian cardiac myocyte terminal differentiation (17, 19–21) that may play a role in different terminal differentiation in early T fetuses. Low thyroid hormone levels are associated with fetal growth restriction, and there are low circulating total and free thyroid hormones in growth-restricted fetuses (30, 45), although there was an insufficient sample to measure thyroid hormone levels in this study. However, free thyroxine and triiodothyronine are not different in cord blood between male and female fetuses (31, 48). Whether female and male fetal cardiomyocytes differently express thyroid hormone receptors or critical downstream signaling components remain to be determined.
Finally, with regard to the cause of differences in terminal differentiation, maturation can have a large effect on heart mass, as binucleated myocytes are, on average, twice the volume as mononucleated myocytes (42). A simple calculation suggests that ~10 points of the 24% reduction in heart weight in the early T group could be due to cardiac myocyte immaturity. It is possible that a component of the signal driving cardiac myocyte terminal differentiation is linked to somatic growth, which is retarded in IUGR fetuses. How these potential regulators of cardiac maturation interact in the normally growing and IUGR fetus remains to be dissected in further experiments.
Inhibition of proliferation.
In contrast to maturational retardation, the relationship between IGF-1 and fetal cardiomyocyte proliferation is clear. In the fetal sheep, IGF-1 stimulates fetal cardiomyocyte proliferation both in vivo and in culture (22, 84), supporting the correlation found here between cell cycle activity and circulating IGF-1 levels. Many other factors contribute to cell cycle regulation in cardiomyocytes, however, and the variability shown in Fig. 3 likely reflects the complexity of the intact physiological system being studied. Furthermore, variability in cell cycle activity may also reflect saltatory growth, as growth in infants and children occurs episodically rather than continuously (46).
As heart weights were substantially reduced near term in early T fetuses and as their cardiomyocytes were not significantly different in size, it is likely that they have fewer cardiomyocytes at birth. This is associated with cardiomyocyte hypertrophy and cardiac pathology later in life (7, 8, 10, 14, 63, 67), which may be a factor contributing to cardiac disease in women affected by PCOS (and men exposed in utero to excess maternal testosterone). We speculate that as IGF-1 deficiency contributes to reduced cardiomyocyte proliferation, prenatal IGF-1 treatment may correct it and improve lifelong cardiac outcomes. Furthermore, cardiomyocyte maturation is retarded at birth, but whether this represents quiescence or senescence is unknown. The abrupt reduction in myocyte proliferation after preterm birth (8) reduces myocyte number in the adult heart (55) and suggests senescence, given the failure of postnatal mononucleated myocytes to undergo further proliferation. Further studies are required to clarify the dynamics of maturational state of mononucleated myocytes at term birth.
LV versus RV.
We found systemic differences between myocytes based on whether their origin was the LV or RV. Normal fetal growth and maturation profiles indicate differences between LV and RV cells (16, 41, 43). In consideration of such differences, we and others have often analyzed the LV and the RV separately, although inclusion of a ventricular term in analysis allows a more complete determination of differences in LV and RV growth and maturation. When this has been done in fetal sheep studies, it has been noted that RV myocytes are bigger (15, 54, 79). This is the first study, however, demonstrating that the near-term fetal sheep LV is less mature (Fig. 3A) and less proliferative (Fig. 3B) than at the RV at ~135 dGA. These indexes are variable near term, and that we were able to show this difference may be due to the number of fetuses included in our analysis. They are concordant with RV myocytes demonstrating accelerated maturation, as indicated by RV myocytes in the fetus being of a larger size (Fig. 4) (15, 54, 79).
Conclusions.
Excess maternal testosterone during a specific window in early pregnancy alters fetal IGF-1 and body mass in both female and male fetal sheep near term. Cardiac growth is reduced proportional to body growth due to delayed maturation and reduced proliferation. The changes in cardiac myocyte growth driven by early excess maternal testosterone exposure in this study may underlie some of the predisposition to cardiovascular disease experienced by women with PCOS. Given similar findings of retarded myocyte maturation and proliferation in both sexes, this study suggests that excess maternal testosterone exposure may also increase cardiovascular disease in male offspring. As an immature heart with fewer cardiomyocytes is common to pregnancies affected by excess maternal testosterone, placental insufficiency and IUGR, and premature birth, perinatal interventions to mitigate lifelong risk may be found that target similar cardiomyocyte growth and maturational mechanisms. Thus, the findings of this study have important implications for both women and men exposed to excess maternal testosterone during early pregnancy.
GRANTS
C. E. Roselli was supported by National Institutes of Health Grant R01-OD-011047. S. S. Jonker was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant R01-HD-071068.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S.J. and C.E.R. conceived and designed research; S.S.J. performed experiments; S.S.J. analyzed data; S.S.J. interpreted results of experiments; S.S.J. prepared figures; S.S.J. drafted manuscript; S.S.J., S.L., and C.E.R. edited and revised manuscript; S.L. and C.E.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Fred Stormshak, Mary Smallman, and the Oregon State University Sheep Center for supplying the healthy ewes bred for these experiments; our work study students for helping with the care of the animals; and Dr. Charles Estill and Dr. Hernan Montilla as well as Oregon State University veterinary students for helping with the surgical delivery of fetuses.
REFERENCES
- 1.Al Madhoun AS, Voronova A, Ryan T, Zakariyah A, McIntire C, Gibson L, Shelton M, Ruel M, Skerjanc IS. Testosterone enhances cardiomyogenesis in stem cells and recruits the androgen receptor to the MEF2C and HCN4 genes. J Mol Cell Cardiol 60: 164–171, 2013. doi: 10.1016/j.yjmcc.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Alexander BT, Dasinger JH, Intapad S. Fetal programming and cardiovascular pathology. Compr Physiol 5: 997–1025, 2015. doi: 10.1002/cphy.c140036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appelman Y, van Rijn BB, Ten Haaf ME, Boersma E, Peters SA. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis 241: 211–218, 2015. doi: 10.1016/j.atherosclerosis.2015.01.027. [DOI] [PubMed] [Google Scholar]
- 4.Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol 279: R1157–R1164, 2000. doi: 10.1152/ajpregu.2000.279.4.R1157. [DOI] [PubMed] [Google Scholar]
- 5.Beckett EM, Astapova O, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programing: impact of testosterone on placental differentiation. Reproduction 148: 199–209, 2014. doi: 10.1530/REP-14-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell JR, Bernasochi GB, Varma U, Raaijmakers AJ, Delbridge LM. Sex and sex hormones in cardiac stress−mechanistic insights. J Steroid Biochem Mol Biol 137: 124–135, 2013. doi: 10.1016/j.jsbmb.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 7.Bensley JG, De Matteo R, Harding R, Black MJ. The effects of preterm birth and its antecedents on the cardiovascular system. Acta Obstet Gynecol Scand 95: 652–663, 2016. doi: 10.1111/aogs.12880. [DOI] [PubMed] [Google Scholar]
- 8.Bensley JG, Moore L, De Matteo R, Harding R, Black MJ. Impact of preterm birth on the developing myocardium of the neonate. Pediatr Res 83: 880–888, 2018. doi: 10.1038/pr.2017.324. [DOI] [PubMed] [Google Scholar]
- 10.Bensley JG, Stacy VK, De Matteo R, Harding R, Black MJ. Cardiac remodelling as a result of pre-term birth: implications for future cardiovascular disease. Eur Heart J 31: 2058–2066, 2010. doi: 10.1093/eurheartj/ehq104. [DOI] [PubMed] [Google Scholar]
- 11.Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andrä M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H, Frisén J. Dynamics of cell generation and turnover in the human heart. Cell 161: 1566–1575, 2015. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 12.Birch RA, Padmanabhan V, Foster DL, Unsworth WP, Robinson JE. Prenatal programming of reproductive neuroendocrine function: fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology 144: 1426–1434, 2003. doi: 10.1210/en.2002-220965. [DOI] [PubMed] [Google Scholar]
- 13.Botting KJ, McMillen IC, Forbes H, Nyengaard JR, Morrison JL. Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia-responsive genes. J Am Heart Assoc 3: 3, 2014. doi: 10.1161/JAHA.113.000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Botting KJ, Wang KC, Padhee M, McMillen IC, Summers-Pearce B, Rattanatray L, Cutri N, Posterino GS, Brooks DA, Morrison JL. Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clin Exp Pharmacol Physiol 39: 814–823, 2012. doi: 10.1111/j.1440-1681.2011.05649.x. [DOI] [PubMed] [Google Scholar]
- 15.Bubb KJ, Cock ML, Black MJ, Dodic M, Boon WM, Parkington HC, Harding R, Tare M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J Physiol 578: 871–881, 2007. doi: 10.1113/jphysiol.2006.121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burrell JH, Boyn AM, Kumarasamy V, Hsieh A, Head SI, Lumbers ER. Growth and maturation of cardiac myocytes in fetal sheep in the second half of gestation. Anat Rec A Discov Mol Cell Evol Biol 274A: 952–961, 2003. doi: 10.1002/ar.a.10110. [DOI] [PubMed] [Google Scholar]
- 17.Burton PB, Raff MC, Kerr P, Yacoub MH, Barton PJ. An intrinsic timer that controls cell-cycle withdrawal in cultured cardiac myocytes. Dev Biol 216: 659–670, 1999. doi: 10.1006/dbio.1999.9524. [DOI] [PubMed] [Google Scholar]
- 18.Camm EJ, Brain KL, Niu Y, Allison BJ, Botting KJ, Itani N, Skeffington KL, Beck C, Giussani DA. Chronic fetal hypoxia in ovine pregnancy triggers a fetal origin of heart disease that amplifies with ageing: prevention with maternal antioxidant treatment (Abstract). Reprod Sci 22: 1, 2015. [Google Scholar]
- 19.Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J 26: 397–408, 2012. doi: 10.1096/fj.10-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chattergoon NN, Giraud GD, Thornburg KL. Thyroid hormone inhibits proliferation of fetal cardiac myocytes in vitro. J Endocrinol 192: R1–R8, 2007. doi: 10.1677/JOE-06-0114. [DOI] [PubMed] [Google Scholar]
- 21.Chattergoon NN, Louey S, Stork P, Giraud GD, Thornburg KL. Mid-gestation ovine cardiomyocytes are vulnerable to mitotic suppression by thyroid hormone. Reprod Sci 19: 642–649, 2012. doi: 10.1177/1933719111432860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chattergoon NN, Louey S, Stork PJ, Giraud GD, Thornburg KL. Unexpected maturation of PI3K and MAPK-ERK signaling in fetal ovine cardiomyocytes. Am J Physiol Heart Circ Physiol 307: H1216–H1225, 2014. doi: 10.1152/ajpheart.00833.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crespi EJ, Steckler TL, Mohankumar PS, Padmanabhan V. Prenatal exposure to excess testosterone modifies the developmental trajectory of the insulin-like growth factor system in female sheep. J Physiol 572: 119–130, 2006. doi: 10.1113/jphysiol.2005.103929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Wilde MA, Lamain-de Ruiter M, Veltman-Verhulst SM, Kwee A, Laven JS, Lambalk CB, Eijkemans MJC, Franx A, Fauser BCJM, Koster MPH. Increased rates of complications in singleton pregnancies of women previously diagnosed with polycystic ovary syndrome predominantly in the hyperandrogenic phenotype. Fertil Steril 108: 333–340, 2017. doi: 10.1016/j.fertnstert.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 25.do Nascimento AM, de Lima EM, Boëchat GA, Meyrelles SD, Bissoli NS, Lenz D, Endringer DC, de Andrade TU. Testosterone induces apoptosis in cardiomyocytes by increasing proapoptotic signaling involving tumor necrosis factor-α and renin angiotensin system. Hum Exp Toxicol 34: 1139–1147, 2015. doi: 10.1177/0960327115571766. [DOI] [PubMed] [Google Scholar]
- 26.Dokras A. Cardiovascular disease risk in women with PCOS. Steroids 78: 773–776, 2013. doi: 10.1016/j.steroids.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med 32: 159–165, 2014. doi: 10.1055/s-0034-1371087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eriksson JG, Kajantie E, Osmond C, Thornburg K, Barker DJ. Boys live dangerously in the womb. Am J Hum Biol 22: 330–335, 2010. doi: 10.1002/ajhb.20995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fall CH. Evidence for the intra-uterine programming of adiposity in later life. Ann Hum Biol 38: 410–428, 2011. doi: 10.3109/03014460.2011.592513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franco B, Laura F, Sara N, Salvatore G. Thyroid function in small for gestational age newborns: a review. J Clin Res Pediatr Endocrinol 5, Suppl 1: 2–7, 2013. doi: 10.4274/jcrpe.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuse Y, Wakae E, Nemoto Y, Uga N, Tanaka M, Maeda M, Tada H, Miyachi Y, Irie M. Influence of perinatal factors and sampling methods on TSH and thyroid hormone levels in cord blood. Endocrinol Jpn 38: 297–302, 1991. doi: 10.1507/endocrj1954.38.297. [DOI] [PubMed] [Google Scholar]
- 32.Gao L, Cao JT, Liang Y, Zhao YC, Lin XH, Li XC, Tan YJ, Li JY, Zhou CL, Xu HY, Sheng JZ, Huang HF. Calcitriol attenuates cardiac remodeling and dysfunction in a murine model of polycystic ovary syndrome. Endocrine 52: 363–373, 2016. doi: 10.1007/s12020-015-0797-1. [DOI] [PubMed] [Google Scholar]
- 33.Garcia M, Mulvagh SL, Merz CN, Buring JE, Manson JE. Cardiovascular disease in women: clinical perspectives. Circ Res 118: 1273–1293, 2016. doi: 10.1161/CIRCRESAHA.116.307547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gentili S, Morrison JL, McMillen IC. Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod 80: 1121–1127, 2009. doi: 10.1095/biolreprod.108.073569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldman-Johnson DR, de Kretser DM, Morrison JR. Evidence that androgens regulate early developmental events, prior to sexual differentiation. Endocrinology 149: 5–14, 2008. doi: 10.1210/en.2007-1123. [DOI] [PubMed] [Google Scholar]
- 36.Goodman NF, Cobin RH, Futterweit W, Glueck JS, Legro RS, Carmina E; American Association of Clinical Endocrinologists (AACE); American College of Endocrinology (ACE); Androgen Excess and PCOS Society . American Association of Clinical Endocrinologists, American College of Endocrinology, and Androgen Excess and PCOS Society Disease State clinical review: guide to the best practices in the evaluation and treatment of polycystic ovary syndrome - part 2. Endocr Pract 21: 1415–1426, 2015. doi: 10.4158/EP15748.DSCPT2. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto H, Yuasa S. Testosterone induces cardiomyocyte differentiation from embryonic stem cells. J Mol Cell Cardiol 62: 69–71, 2013. doi: 10.1016/j.yjmcc.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Hellström A, Ley D, Hansen-Pupp I, Hallberg B, Löfqvist C, van Marter L, van Weissenbruch M, Ramenghi LA, Beardsall K, Dunger D, Hård AL, Smith LE. Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Acta Paediatr 105: 576–586, 2016. doi: 10.1111/apa.13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD. Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol 292: R913–R919, 2007. doi: 10.1152/ajpregu.00484.2006. [DOI] [PubMed] [Google Scholar]
- 40.Jonker SS, Giraud MK, Giraud GD, Chattergoon NN, Louey S, Davis LE, Faber JJ, Thornburg KL. Cardiomyocyte enlargement, proliferation and maturation during chronic fetal anaemia in sheep. Exp Physiol 95: 131–139, 2010. doi: 10.1113/expphysiol.2009.049379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonker SS, Louey S. Endocrine and other physiologic modulators of perinatal cardiomyocyte endowment. J Endocrinol 228: R1–R18, 2016. doi: 10.1530/JOE-15-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jonker SS, Louey S, Giraud GD, Thornburg KL, Faber JJ. Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB J 29: 4346–4357, 2015. doi: 10.1096/fj.15-272013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol 102: 1130–1142, 2007. doi: 10.1152/japplphysiol.00937.2006. [DOI] [PubMed] [Google Scholar]
- 44.King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab 292: E1837–E1841, 2007. doi: 10.1152/ajpendo.00668.2006. [DOI] [PubMed] [Google Scholar]
- 45.Klein RZ, Carlton EL, Faix JD, Frank JE, Hermos RJ, Mullaney D, Nelson JC, Rojas DA, Mitchell ML. Thyroid function in very low birth weight infants. Clin Endocrinol (Oxf) 47: 411–417, 1997. doi: 10.1046/j.1365-2265.1997.2511064.x. [DOI] [PubMed] [Google Scholar]
- 46.Lampl M, Thompson AL. Growth chart curves do not describe individual growth biology. Am J Hum Biol 19: 643–653, 2007. doi: 10.1002/ajhb.20707. [DOI] [PubMed] [Google Scholar]
- 47.Lipshultz SE, Simbre VC II, Hart S, Rifai N, Lipsitz SR, Reubens L, Sinkin RA. Frequency of elevations in markers of cardiomyocyte damage in otherwise healthy newborns. Am J Cardiol 102: 761–766, 2008. doi: 10.1016/j.amjcard.2008.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu R, Xu X, Zhang Y, Zheng X, Kim SS, Dietrich KN, Ho SM, Reponen T, Chen A, Huo X. Thyroid hormone status in umbilical cord serum is positively associated with male anogenital distance. J Clin Endocrinol Metab 101: 3378–3385, 2016. doi: 10.1210/jc.2015-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol 580: 639–648, 2007. doi: 10.1113/jphysiol.2006.122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lumbers ER, Kim MY, Burrell JH, Kumarasamy V, Boyce AC, Gibson KJ, Gatford KL, Owens JA. Effects of intrafetal IGF-I on growth of cardiac myocytes in late-gestation fetal sheep. Am J Physiol Endocrinol Metab 296: E513–E519, 2009. doi: 10.1152/ajpendo.90497.2008. [DOI] [PubMed] [Google Scholar]
- 51.Manikkam M, Crespi EJ, Doop DD, Herkimer C, Lee JS, Yu S, Brown MB, Foster DL, Padmanabhan V. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology 145: 790–798, 2004. doi: 10.1210/en.2003-0478. [DOI] [PubMed] [Google Scholar]
- 52.Mayhew TM, Gregson C, Fagan DG. Ventricular myocardium in control and growth-retarded human fetuses: growth in different tissue compartments and variation with fetal weight, gestational age, and ventricle size. Hum Pathol 30: 655–660, 1999. doi: 10.1016/S0046-8177(99)90090-4. [DOI] [PubMed] [Google Scholar]
- 53.Metcalfe J, Meschia G, Hellegers A, Prystowsky H, Huckabee W, Barron DH. Observations on the growth rates and organ weights of fetal sheep at altitude and sea level. Q J Exp Physiol Cogn Med Sci 47: 305–313, 1962. [DOI] [PubMed] [Google Scholar]
- 54.Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL, McMillen IC. Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol 293: R306–R313, 2007. doi: 10.1152/ajpregu.00798.2006. [DOI] [PubMed] [Google Scholar]
- 55.Mrocki MM, Nguyen VB, Lombardo P, Sutherland MR, Bensley JG, Nitsos I, Allison BJ, Harding R, De Matteo R, Schneider M, Polglase GR, Black MJ. Moderate preterm birth affects right ventricular structure and function and pulmonary artery blood flow in adult sheep. J Physiol. In press. doi: 10.1113/JP275654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murotsuki J, Challis JR, Han VK, Fraher LJ, Gagnon R. Chronic fetal placental embolization and hypoxemia cause hypertension and myocardial hypertrophy in fetal sheep. Am J Physiol Regul Integr Comp Physiol 272: R201–R207, 1997. doi: 10.1152/ajpregu.1997.272.1.R201. [DOI] [PubMed] [Google Scholar]
- 57.Murray PG, Clayton PE. Endocrine control of growth. Am J Med Genet C Semin Med Genet 163: 76–85, 2013. doi: 10.1002/ajmg.c.31357. [DOI] [PubMed] [Google Scholar]
- 58.Nora JJ, Nora AH, Blu J, Ingram J, Fountain A, Peterson M, Lortscher RH, Kimberling WJ. Exogenous progestogen and estrogen implicated in birth defects. JAMA 240: 837–843, 1978. doi: 10.1001/jama.1978.03290090031012. [DOI] [PubMed] [Google Scholar]
- 59.O’Tierney PF, Anderson DF, Faber JJ, Louey S, Thornburg KL, Giraud GD. Reduced systolic pressure load decreases cell-cycle activity in the fetal sheep heart. Am J Physiol Regul Integr Comp Physiol 299: R573–R578, 2010. doi: 10.1152/ajpregu.00754.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Padmanabhan V, Veiga-Lopez A. Reproduction symposium: developmental programming of reproductive and metabolic health. J Anim Sci 92: 3199–3210, 2014. doi: 10.2527/jas.2014-7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol 373: 8–20, 2013. doi: 10.1016/j.mce.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papamitsou T, Barlagiannis D, Papaliagkas V, Kotanidou E, Dermentzopoulou-Theodoridou M. Testosterone-induced hypertrophy, fibrosis and apoptosis of cardiac cells−an ultrastructural and immunohistochemical study. Med Sci Monit 17: BR266–BR273, 2011. doi: 10.12659/MSM.881930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paradis AN, Gay MS, Zhang L. Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state. Drug Discov Today 19: 602–609, 2014. doi: 10.1016/j.drudis.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pedernera E, Gómora MJ, Meneses I, De Ita M, Méndez C. Androgen receptor is expressed in mouse cardiomyocytes at prenatal and early postnatal developmental stages. BMC Physiol 17: 7, 2017. doi: 10.1186/s12899-017-0033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Phillips DI, Barker DJ, Hales CN, Hirst S, Osmond C. Thinness at birth and insulin resistance in adult life. Diabetologia 37: 150–154, 1994. doi: 10.1007/s001250050086. [DOI] [PubMed] [Google Scholar]
- 66.Phillips GB, Pinkernell BH, Jing TY. The association of hypotestosteronemia with coronary artery disease in men. Arterioscler Thromb 14: 701–706, 1994. [DOI] [PubMed] [Google Scholar]
- 67.Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, Ritchie RH, Lynch GS, Harrap SB, Thomas WG, Delbridge LM. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am J Physiol Regul Integr Comp Physiol 296: R672–R680, 2009. doi: 10.1152/ajpregu.90919.2008. [DOI] [PubMed] [Google Scholar]
- 68.Porrello ER, Widdop RE, Delbridge LM. Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological ‘catch-up’ growth of the heart? Clin Exp Pharmacol Physiol 35: 1358–1364, 2008. doi: 10.1111/j.1440-1681.2008.05036.x. [DOI] [PubMed] [Google Scholar]
- 69.Reddy RC, Estill CT, Meaker M, Stormshak F, Roselli CE. Sex differences in expression of oestrogen receptor α but not androgen receptor mRNAs in the foetal lamb brain. J Neuroendocrinol 26: 321–328, 2014. doi: 10.1111/jne.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roberts VH, Frias AE, Grove KL. Impact of maternal obesity on fetal programming of cardiovascular disease. Physiology (Bethesda) 30: 224–231, 2015. doi: 10.1152/physiol.00021.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roselli CE, Estill CT, Stadelman HL, Meaker M, Stormshak F. Separate critical periods exist for testosterone-induced differentiation of the brain and genitals in sheep. Endocrinology 152: 2409–2415, 2011. doi: 10.1210/en.2010-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roselli CE, Stadelman H, Reeve R, Bishop CV, Stormshak F. The ovine sexually dimorphic nucleus of the medial preoptic area is organized prenatally by testosterone. Endocrinology 148: 4450–4457, 2007. doi: 10.1210/en.2007-0454. [DOI] [PubMed] [Google Scholar]
- 73.Rozance PJ, Zastoupil L, Wesolowski SR, Goldstrohm DA, Strahan B, Cree-Green M, Sheffield-Moore M, Meschia G, Hay WW Jr, Wilkening RB, Brown LD. Skeletal muscle protein accretion rates and hindlimb growth are reduced in late gestation intrauterine growth-restricted fetal sheep. J Physiol 596: 67–82, 2018. doi: 10.1113/JP275230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rudolph AM. Myocardial growth before and after birth: clinical implications. Acta Paediatr 89: 129–133, 2000. doi: 10.1080/080352500750028681. [DOI] [PubMed] [Google Scholar]
- 75.Samson F, Bonnet N, Heimburger M, Rücker-Martin C, Levitsky DO, Mazmanian GM, Mercadier JJ, Serraf A. Left ventricular alterations in a model of fetal left ventricular overload. Pediatr Res 48: 43–49, 2000. doi: 10.1203/00006450-200007000-00010. [DOI] [PubMed] [Google Scholar]
- 76.Satler F, Vieira RA, Firpo C, Spritzer PM. Association between left ventricular mass, androgens, adiposity and insulin resistance in girls with precocious pubarche: a case-control study. Clin Endocrinol (Oxf) 84: 394–401, 2016. doi: 10.1111/cen.12957. [DOI] [PubMed] [Google Scholar]
- 77.Scherrer U, Rexhaj E, Allemann Y, Sartori C, Rimoldi SF. Cardiovascular dysfunction in children conceived by assisted reproductive technologies. Eur Heart J 36: 1583–1589, 2015. doi: 10.1093/eurheartj/ehv145. [DOI] [PubMed] [Google Scholar]
- 78.Segar JL, Scholz TD, Bedell KA, Smith OM, Huss DJ, Guillery EN. Angiotensin AT1 receptor blockade fails to attenuate pressure-overload cardiac hypertrophy in fetal sheep. Am J Physiol Regul Integr Comp Physiol 273: R1501–R1508, 1997. [DOI] [PubMed] [Google Scholar]
- 79.Segar JL, Volk KA, Lipman MH, Scholz TD. Thyroid hormone is required for growth adaptation to pressure load in the ovine fetal heart. Exp Physiol 98: 722–733, 2013. doi: 10.1113/expphysiol.2012.069435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sir-Petermann T, Hitchsfeld C, Maliqueo M, Codner E, Echiburú B, Gazitúa R, Recabarren S, Cassorla F. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum Reprod 20: 2122–2126, 2005. doi: 10.1093/humrep/dei009. [DOI] [PubMed] [Google Scholar]
- 81.Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Pérez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod 17: 2573–2579, 2002. doi: 10.1093/humrep/17.10.2573. [DOI] [PubMed] [Google Scholar]
- 82.Spencer TE, Johnson GA, Bazer FW, Burghardt RC. Implantation mechanisms: insights from the sheep. Reproduction 128: 657–668, 2004. doi: 10.1530/rep.1.00398. [DOI] [PubMed] [Google Scholar]
- 83.Steckler T, Wang J, Bartol FF, Roy SK, Padmanabhan V. Fetal programming: prenatal testosterone treatment causes intrauterine growth retardation, reduces ovarian reserve and increases ovarian follicular recruitment. Endocrinology 146: 3185–3193, 2005. doi: 10.1210/en.2004-1444. [DOI] [PubMed] [Google Scholar]
- 84.Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJ, Thornburg KL. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol 285: R1481–R1489, 2003. doi: 10.1152/ajpregu.00232.2003. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi N, Nishida H, Arai T, Kaneda Y. Abnormal cardiac histology in severe intrauterine growth retardation infants. Acta Paediatr Jpn 37: 341–346, 1995. doi: 10.1111/j.1442-200X.1995.tb03326.x. [DOI] [PubMed] [Google Scholar]
- 86.Thornburg KL. The programming of cardiovascular disease. J Dev Orig Health Dis 6: 366–376, 2015. doi: 10.1017/S2040174415001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Veiga-Lopez A, Steckler TL, Abbott DH, Welch KB, MohanKumar PS, Phillips DJ, Refsal K, Padmanabhan V. Developmental programming: impact of excess prenatal testosterone on intrauterine fetal endocrine milieu and growth in sheep. Biol Reprod 84: 87–96, 2011. doi: 10.1095/biolreprod.110.086686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vyas AK, Hoang V, Padmanabhan V, Gilbreath E, Mietelka KA. Prenatal programming: adverse cardiac programming by gestational testosterone excess. Sci Rep 6: 28335, 2016. doi: 10.1038/srep28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang ET, Ku IA, Shah SJ, Daviglus ML, Schreiner PJ, Konety SH, Williams OD, Siscovick D, Bibbins-Domingo K. Polycystic ovary syndrome is associated with higher left ventricular mass index: the CARDIA women’s study. J Clin Endocrinol Metab 97: 4656–4662, 2012. doi: 10.1210/jc.2012-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang KC, Brooks DA, Botting KJ, Morrison JL. IGF-2R-mediated signaling results in hypertrophy of cultured cardiomyocytes from fetal sheep. Biol Reprod 86: 183, 2012. doi: 10.1095/biolreprod.112.100388. [DOI] [PubMed] [Google Scholar]
- 91.Wang KC, Brooks DA, Thornburg KL, Morrison JL. Activation of IGF-2R stimulates cardiomyocyte hypertrophy in the late gestation sheep fetus. J Physiol 590: 5425–5437, 2012. doi: 10.1113/jphysiol.2012.238410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang XF, Qu XQ, Zhang TT, Zhang JF. Testosterone suppresses ventricular remodeling and improves left ventricular function in rats following myocardial infarction. Exp Ther Med 9: 1283–1291, 2015. doi: 10.3892/etm.2015.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiao FY, Nheu L, Komesaroff P, Ling S. Testosterone protects cardiac myocytes from superoxide injury via NF-κB signalling pathways. Life Sci 133: 45–52, 2015. doi: 10.1016/j.lfs.2015.05.009. [DOI] [PubMed] [Google Scholar]