

Abstract
The second step in the biosynthesis of the cellular antioxidant glutathione (GSH) is catalyzed by human glutathione synthetase (hGS), a negatively cooperative homodimer. Patients with mutations in hGS have been reported to exhibit a range of symptoms from hemolytic anemia and metabolic acidosis to neurological disorders and premature death. Several patient mutations occur in the S-loop of hGS, a series of residues near the negatively cooperative γ-GC substrate binding site. Experimental point mutations and molecular dynamic simulations show the S-loop not only binds γ-GC through a salt bridge and multiple hydrogen bonds, but the residues also modulate allosteric communication in hGS. By elucidating the role of S-loop residues in active site structure, substrate binding, and allostery, the atomic level sequence of events that leads to the detrimental effects of hGS mutations in patients are more fully understood.
Abbreviations: GSH, glutathione; hGS, human glutathione synthetase; γ-GC, γ-glutamylcysteine; WT, wild-type; IPTG, isopropyl-1-thio-β-galactopyranoside; γ-GluABA, L-γ-glutamyl-L-α-aminobutyrate; PK, pyruvate kinase; LDH, lactate dehydrogenase; PEP, phosphoenolpyruvic acid; DSC, differential scanning calorimetry; Td, denaturation temperature midpoint; MD, molecular dynamics; Eint, average energy of interaction.
Graphical Abstract
1. Introduction
The tripeptide glutathione (L-γ-glutamyl-L-cysteinylglycine, GSH) relieves oxidative stress, aids in the excretion of toxins, and serves as a coenzyme for several cellular processes [1]. Deficiencies in GSH are associated with a variety of diseases, including Parkinson's disease, Alzheimer's disease, cystic fibrosis, and HIV [[2], [3], [4]]. Regulation of glutathione synthesis is crucial for maintaining cellular GSH levels to combat oxidative stress and to balance cellular cysteine levels for protein synthesis and sulfur metabolism [5,6]. Mutations in the enzymes responsible for the biosynthesis of GSH have been shown to result in metabolic acidosis, hemolytic anemia, and moderate to severe neurological disorders [7,8]. In the first step of the glutathione biosynthetic pathway, γ-glutamylcysteine synthetase ligates glutamate and cysteine to form γ-glutamylcysteine (γ-GC) [1]. Human glutathione synthetase (hGS) then catalyzes the synthesis of a peptide bond between γ-glutamylcysteine and glycine to form glutathione in an ATP-dependent reaction [1]. The research presented herein focuses on elucidating key aspects of substrate binding in hGS in an effort to better understand both glutathione biosynthesis and regulation.
An obligate homodimer with two 52 kDa subunits, hGS has protein-protein interface maintained by a handful of strong electrostatic interactions [9,10]. Two catalytic loops (A- and G-loop) regulate access to the active sites [[11], [12], [13]], which bind the ATP and associated Mg2+ needed to fuel peptide bond formation [14]. Interestingly, hGS exhibits negative cooperativity towards γ-GC (Hill coefficient of 0.69) [15,16], a type of allosteric regulation wherein the binding of γ-GC to one subunit of the enzyme results in a decreased affinity for γ-GC at the second active site ca. 30 Å away. While allosteric communication at the protein-protein interface of hGS is modulated almost exclusively by weak, hydrophobic interactions, most notably those involving Val44 and Val45, the residues responsible for allostery at the substrate binding sites remain unexplored [9,17]. The relatively simple homodimer hGS thus serves as a model for understanding negatively cooperative enzymes and allosteric regulation, which it is thought to be pivotal in pathway fluxes of key metabolites, like cysteine and glutathione [[18], [19], [20], [21]].
A variety of biological processes exhibit allosteric regulation, including the transport of O2 through the blood by hemoglobin and the activation of cell signaling pathways by G-protein coupled receptors [[22], [23], [24], [25], [26]]. Allosteric binding sites are increasingly pursued as drug targets for cancers and autoimmune diseases [25,27,28]. Although negative cooperativity is exhibited by important biosystems, such as insulin receptors and β-adrenergic receptors [29,30], little is understood about the pathways by which such allosteric communication occurs [18,31]. Exploring the key components of allosteric regulation in hGS can thus provide valuable insight into mechanisms of protein allostery.
The present research investigates the function of the S-loop of hGS. The S-loop comprises a series of eleven residues, which were identified using the first hGS crystal structure10 as forming a wall of the active site of hGS near the negatively cooperative (γ-GC) substrate: (F266-R267-D268-G269-Y270-M271-P272-R273-Q274-Y275-S276), Fig. 1. Previous work from our group has shown that the S-loop does not undergo significant catalytic motion during peptide bond formation as compared to the G- and A-loops but suggested that the S-loop has a role in substrate binding [12].
Fig. 1.
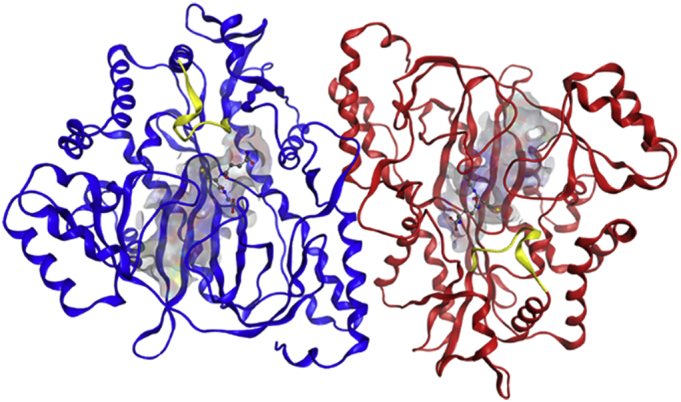
S-loop of hGS in dimeric hGS from crystal structure (2HGS). S-loop shown as yellow ribbon, GSH shown as ball-and-stick model, and active site shown as grey-blue-red surface. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Two S-loop residues have been implicated in patients with mutations in hGS: R267 and Y270. Patients with a single hGS mutation at Y270 to either cysteine or histidine exhibit low GSH levels, severe reductions in hGS activity, hemolytic anemia, and metabolic acidosis [7,8]. A patient with a double mutation (R267W and R283C) had similar symptoms. Most drastically, a patient born with R267W and D469E mutations had the aforementioned symptoms in addition to seizures and psychomotor retardation; the patient died at 5 days of age [7,8]. Rare mutations in hGS thus have acute physiological consequences, which underscore the importance of hGS.
The severity of S-loop patient mutations, coupled with high sequence conservation (results section) and proximity to the bound negatively cooperative γ-GC substrate suggests that the region has a crucial role in the activity of hGS. In the presented work, molecular dynamics simulations and experimental point mutations are used probe the role of S-loop residues in tertiary structure, substrate binding, and allosteric communication. Alterations in bonding architectures, activity, cooperativity, and thermal stability indicate S-loop residues modulate both substrate binding and allostery in hGS.
2. Materials and Methods
2.1. Materials
Oligomer primers (Supplementary Table S1) were purchased from Integrated DNA Technologies, Inc. Sequencing was conducted by GeneWiz, Inc. QuickChange™ Mutagenesis Kits were purchased from Stratagene, while Wizard® Plus Midiprep DNA Purification Systems were from Promega. Novagen, Inc. supplied expression vector pET-15b, E. coli XL1 Blue competent cells and Ni-NTA His-Bind® resin. Isopropyl-1-thio-β-galactopyranoside (IPTG) was purchased from American Bioanalytical, Inc. L-γ-glutamyl-L-α-aminobutyrate (γ-GluABA) was supplied by Bachem, Inc. or synthesized [15]. Other reagents were obtained in the highest purity from Sigma-Aldrich, US Biological, Fisher Scientific or Amresco.
2.2. Preparation and Purification of hGS
Wild-type hGS with an N-terminal 6× histidine tag was inserted in a pET-15b expression vector (hGS-pET-15b) [14]. Site-directed mutagenesis of the vector was conducted through PCR with the QuickChange™ Mutagenesis Kit. Resulting plasmids were transformed into E. coli XL1 Blue competent cells. DNA was purified with Wizard® Plus Midiprep DNA Purification Systems; wild-type and mutant cDNA was sequenced by GeneWiz, Inc. After hGS-pET-15b plasmids were expressed in E. coli BL21(DE3) cells, the protein was purified as previously reported [14].
Purified wild-type or mutant protein was dialyzed overnight in a Tris buffer (20 mM Tris-Cl, 1 mM EDTA, pH 8.6) then stored in sterile cryogenic tubes at 4 °C. Pure protein was confirmed by SDS-PAGE. The Lowry method was used with a bovine albumin standard to ascertain protein concentration [32].
2.3. Enzyme Activity and Kinetic Assays
Activity assays of purified hGS were conducted using a pyruvate kinase (PK)/lactate dehydrogenase (LDH) coupled assay at 37 °C [14]. To avoid complications associated with thiol oxidation, the native substrate (γ-GC) was replaced by γ-GluABA, which exhibits the same activity and kinetics as γ-GC [15]. An assay mixture of 100 mM Tris (pH 8.2, 25 °C), 50 mM KCl, 20 mM MgCl2, 5 mM phosphoenolpyruvic acid (PEP), 10 units/assay LDH (type II rabbit muscle), 10 units/assay PK (type II rabbit muscle), 0.3 mM NADH and 10 mM each of γ-GluABA, ATP and glycine was incubated at 37 °C for 11 min prior to addition of 10 μL of hGS for a total volume of 0.2 mL. The rate was continuously monitored at 340 nm. A unit of activity is defined as the amount of enzyme required to catalyze the formation of 1 μmol of product/min at 37 °C. Assays without γ-GluABA served as controls, confirming specificity within the assay. Kinetics assays were conducted in the same manner, with concentrations of γ-GluABA varied 10-fold above and below the standard. The Sigma Plot software was used to determine kinetic parameters (Km, Vmax, and Hill coefficient) [11,13,14].
2.4. Differential Scanning Calorimetry (DSC)
Purified proteins were dialyzed overnight in sodium phosphate buffer (10 mM, pH 7.5, 4 °C). Enzyme samples were concentrated (1–2 mg/mL) and degassed for 15 min.17 The Calorimetry Sciences Nano Series III differential scanning calorimeter was used to conduct scans at 1.0 atm, from 10 to 90 °C at a rate of 1.0 °C/min. Scans were baseline corrected against the sodium phosphate buffer specified above.
2.5. Computational Methods
A sequence analysis of hGS was conducted with a non-redundant BLAST of the NCBI database (442 sequences) [[33], [34], [35]]. Theoretical and hypothetical sequences were eliminated; the remaining sequences were aligned with the BLOSUM62 matrix [36]. Percent conservations are listed relative to wild-type hGS.
An hGS crystal structure (2HGS, 2.1 Å resolution10) was used as a starting structure for molecular dynamics (MD) simulations of the dimer in GROMACS with an AMBER99sb force field [[37], [38], [39], [40]]. The simple point charge water model was used to solvate the protein in a dodecahedron box with borders at least 10.0 Å from the protein [41]. Charge was neutralized with randomly dispersed Na+ and Cl− ions at a concentration of 0.15 M. Long range electrostatic interactions were modeled with the Particle Mesh Ewald method [42,43]. Structures were optimized in GROMACS until forces converged at ≤10 kJ/mol before beginning unconstrained MD runs. The temperature was ramped from 1 to 300 K over a 1 ps interval. Runs were conducted under constant NPT, using the Parrinello-Rahman barostat with 1.0 ps coupling constant [44]. MD simulations of free wild-type (10 ns) and mutant (5 ns) hGS were conducted in 0.5 fs time steps with data saved every 0.5 ps. As mutant structures began from low energy WT structures, the potential energy and RMSD of mutant structures converged within the 5 ns simulation. The lowest energy structure in the last ns of each simulation was extracted for bond analysis and docking studies. All hydrogen bonds are compared to an ideal bond (3.0 Å, 180°).
The negatively cooperative substrate (γ-GC) was docked into a single active site of each low energy structure with the induced fit method in MOE, using the London and GBVI/WSA scoring functions [[45], [46], [47]]. A pose was selected for further MD simulations based on similarity to the crystal structure. The ligand was added to the protein with the acpype (AnteChamber PYthon Parser interfacE) script, before conducting MD simulations in GROMACS with the procedure outlined above. Utilizing GROMACS, the average interaction energy of each residue within 4.5 Å of the ligand was determined over the course of the MD run.
3. Results
3.1. Sequence Analysis of hGS
The identity of S-loop residues near the γ-GC binding site of hGS is relatively well conserved in both higher eukaryotes and mammals (Table 1). S-loop residues F266, R267, G269, Y270, P272, Y275 and S276 have identify conservations above the 43% average for the entire enzyme in higher eukaryotes. Although D268 is not well conserved in higher eukaryotes (conservation 18%), it has a very high identity conservation in mammals (71%). Several S-loop residues have sequence conservations >70% in mammals: F266, R267, D268, G269, Y270, M271, P272, and Y275. It is difficult to prioritize residues for further study based solely upon sequence conservation. Thus, several S-loop residues were also selected for further study due to their prevalence in patient mutations [7,8] and their position within the crystal structure [10].
Table 1.
Percent identity conservation of hGS S-loop residues in higher eukaryotes and mammals.
| Residue | Higher Eukaryotes | Mammals |
|---|---|---|
| F266 | 61.0 | 71.4 |
| R267 | 64.6 | 71.4 |
| D268 | 18.3 | 71.4 |
| G269 | 64.6 | 71.4 |
| Y270 | 68.3 | 71.4 |
| M271 | 20.7 | 71.4 |
| P272 | 63.4 | 76.1 |
| R273 | 8.5 | 23.8 |
| Q274 | 17.1 | 66.7 |
| Y275 | 59.8 | 76.2 |
| S276 | 48.8 | 47.6 |
| Averagea | 42.6 | 70.5 |
| Std Deva | 18.2 | 17.3 |
Average and standard deviation of all amino acids in sequence relative to hGS.
3.2. Substrate Binding and S-Loop Structure in WT hGS
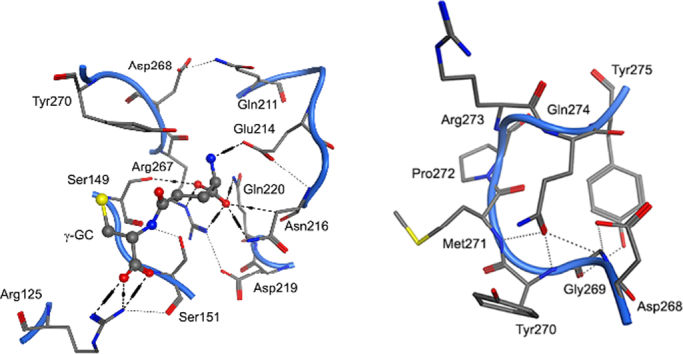
Computational models show two S-loop residues, R267 and Y270, revealed as important by their high conservation identity and from known patient mutations [7,8], bind γ-GC within the active site of hGS. Over a 10 ns MD run, the average interaction energy (Eint) between γ-GC and R267 of −117 kJ/mol corresponds to a salt-bridge and hydrogen bond(s) formed between R267 and γ-GC (Fig. 2 and Supplementary Table S2). In the lowest energy structure from the last ns of the MD run, the R267 guanidyl forms a salt bridge and hydrogen bond with the α-carboxyl of the γ-GC glutamyl, Fig. 3 (next page). At some steps of the MD run, the backbone carbonyl of R267 forms an additional hydrogen bond with the glutamyl α-amino of γ-GC. To a lesser extent, Y270 also binds γ-GC in WT hGS through the formation of a hydrogen bond between the Y270 hydroxyl and the terminal γ-GC amine with an average interaction energy of −27 kJ/mol. In WT hGS, both R267 and Y270 strongly bind γ-GC.
Fig. 2.

Average interaction energy (kJ/mol) between γ-GC and important S-loop residues over the course of 5–10 ns MD runs for WT and S-loop mutant hGS.
Fig. 3.

Important residues in binding γ-GC near the S-loop of hGS in the lowest energy structure in the last ns of MD simulations of (A) WT hGS, (B) R267W, (C) D268A, and (D) Y270F.
Several residues outside of the S-loop also interact with the negatively cooperative substrate (γ-GC), Fig. 2. Most notably, R125 forms a salt bridge and 2 hydrogen bonds with the cysteine carboxyl group of γ-GC in the lowest energy structure from the last ns of the MD simulation, which corresponds to an average interaction energy of −111 kJ/mol. Hydrogen bonds at S151, E214, N216 and Q220 stabilize γ-GC in the active site (Eint = −29, −67, −37 and − 42 kJ/mol, respectively), while S149 and F152 exhibit more moderate substrate binding (Eint = −11 and − 18 kJ/mol, respectively). The comparison of interaction energies and bonds thus shows that the S-loop region plays a critical role in γ-GC binding.
Another S-loop residue, D268, helps maintain the structure of the active site near γ-GC. The carboxyl side chain of D268 forms hydrogen bonds with G269 and Q274, which vary across several low energy MD structures. More interestingly, D268 forms a consistent hydrogen bond with Q211, a residue on a neighboring active site loop. One may posit that this motion may help to bring these two γ-GC binding regions into close proximity (Fig. 3), thereby playing a critical role in the experimentally observed cooperativity. Over the course of a 10 ns MD run, D268 has an average interaction energy with γ-GC (−7 kJ/mol), which is very small compared with the previously discussed substrate binding residues, R267 and Y270. While playing only a minor role in γ-GC binding, the flexible hydrogen bonding network of D268 maintains the active site structure of hGS.
As one of the “walls” of the hGS active site, the S-loop plays a crucial role in maintaining an ideal environment for γ-GC binding. Interactions between three S-loop residues (G269, P272, Y275) serve to bend the flexible loop into an optimal geometry, which is maintained in throughout the catalytic cycle. In low energy frames from both free and γ-GC containing WT hGS MD simulations, G269 forms backbone hydrogen bonds with the hydroxyl of Y275 and the carbonyl of Q274; these interactions on either side of G269 form the tiny loop of the C-terminus of the S-loop (G269-Y257), Fig. 4. As a surface residue, P272 forms the tight curve of the S-loop and has varied weak interactions with F99 and M271. The steady interactions and positions of G269, Y275 and P272 are independent of substrate binding and stabilize the structure of the S-loop nearest the surface of the enzyme.
Fig. 4.

Structure of the S-loop of hGS in the lowest energy structure in the last ns of MD simulations of (A) WT hGS, (B) G269 V, (C) P272A, and (D) Y275A.
Computational models thus suggest that several S-loop residues have an important role in (a) γ-GC binding, (b) active site structure, and (c) the structure of the S-loop. Hydrogen bonding with γ-GC, strong interaction energies, and patient mutations suggest R267 and Y270 have important roles in substrate binding. The variety of hydrogen bonds D268 forms with neighboring regions may be important in maintaining the active site of hGS. Finally, bonds within the S-loop and the position of residues indicates G269, P272, and Y275 conserve the structure of the S-loop throughout the catalytic cycle. Thus, the molecular dynamics simulations of free and γ-GC bound WT hGS guided the selection of residues for experimental analysis.
3.3. Substrate Binding Residues (R267 and Y270) of the S-Loop
Njålsson et al. probed the kinetic properties of three S-loop hGS mutants (Y270C, Y270H and R267W) as part of their studies of missense mutations in hGS deficient patients [48,49]. Three experimental point mutations were chosen to probe the role of R267 in substrate binding of hGS: R267A/K/W. These mutants exhibit severe reduction in activity relative to WT hGS (kcat = 18.2 s−1, Table 2). The activity of R267A is only 2% of WT (kcat = 0.36 s−1), too low to determine kinetic parameters. Substitution to a bulky aromatic group in R267W yields even lower activity (kcat = 0.05 s−1). In contrast, R267K retains ~15% of WT activity (kcat = 2.7 s−1) by preserving the positive charge with a flexible lysine substitution. The γ − GluΑΒΑ Michaelis constant of R267K (Km = 13.34 mM) suggests the mutant hGS does not bind the negatively cooperative substrate as effectively as WT (Km = 1.31 mM). Despite the large shifts in activity, mutations in R267 have only moderate impact on the structural stability of hGS (Td = 60.3, 52.8, 54.3, and 53.7 °C, for WT, R267A/K/W, respectively). Mutations to R267 have a dramatic impact on the activity and kinetics of hGS, without significant alterations in thermal stability.
Table 2.
Impact of S-loop mutations on experimental activity, kinetic parameters and thermal stabilities of hGS.
| Enzymea | Kcat (s−1) | Kmapp γ-gluABA (mM) | keff (s−1 M−1) | Hill Coef. | Td (°C) |
|---|---|---|---|---|---|
| WT | 18.2 ± 2.0 | 1.31 ± 0.13 | 1.4 × 104 | 0.69 ± 0.03 | 59.6 ± 0.7 |
| R267A | 0.36 ± 0.02 | NDb | ND | ND | 52.8 ± 0.01 |
| R267K | 2.7 ± 0.4 | 13.34 ± 1.6 | 2.0 × 102 | 1.03 ± 0.09 | 54.3 ± 0.1 |
| R267W | 0.05 ± 0.02 | ND | ND | ND | 53.7 ± 0.04 |
| D268A | 11.8 ± 0.1 | 1.33 ± 0.17 | 8.9 × 103 | 0.78 ± 0.01 | ND |
| D268E | 11.3 ± 0.7 | 0.19 ± 0.09 | 5.9 × 104 | 0.68 ± 0.06 | 54.4 ± 0.1 |
| G269 V | 0.6 ± 0.3 | ND | ND | ND | 45.5 ± 0.5 |
| Y270A | 0.33 ± 0.06 | ND | ND | ND | 59.6 ± 0.2 |
| Y270F | 8.6 ± 0.8 | 8.73 ± 0.99 | 9.9 × 102 | 0.78 ± 0.03 | 58.5 ± 1.0 |
| P272A | 1.19 ± 0.01 | 0.06 ± 0.02 | 2.0 × 104 | 1.02 ± 0.01 | 60.8 ± 0.3 |
| Y275A | 9.2 ± 1.0 | 0.05 ± 0.03 | 1.8 × 105 | 0.73 ± 0.08 | ND |
| Y275F | 6.6 ± 0.7 | 0.10 ± 0.04 | 6.6 × 104 | 0.70 ± 0.03 | 59.4 ± 0.4 |
Data from 2 to 3 independent purifications, with duplicate assays on each.
ND = activity too low to determine.
In a low energy frame from an MD run of R267A, γ-GC fails to stay deep within the binding pocket, instead forming new bonds with A-loop residues usually responsible for binding glycine (V461 and A462). Interactions between γ-GC and E214, Q220 and R267 weaken by 33–83 kJ/mol, relative to WT; γ-GC interactions with S149 and G370 strengthen by 16 and 10 kJ/mol, Fig. 2, respectively. The R267W mutant displays the largest shift in γ-GC binding, as the substrate binds in a twisted orientation and is unable to fit deep into the binding pocket, Fig. 3. Of the 11 residues that interact with γ- GC in WT, only R125, N146, and Y270 form hydrogen bonds with the substrate in R267W. Average interactions energies with γ-GC at E214, N216, Q220, R267, and Y270 decrease by 40–96%. The hydrogen bond between active site residues D268 and Q211 is also disrupted. In contrast, the positively charged amine of the R267K mutant binds γ-GC and allows the substrate to adopt a similar orientation to WT hGS. Over a 5 ns MD run, average interaction energies with γ-GC at S151 and E214 strengthen by 63 and 37 kJ/mol, while interactions at R125, S149, and A150 increase moderately (13–19 kJ/mol) relative to WT hGS. Binding of γ-GC at F152, N216, and Q220 weakens by 14–25 kJ/mol and repulsive interactions with D219 increase by 7 kJ/mol. According to computational models, mutations of R267 drastically impact γ-GC binding.
Experimental alanine and phenylalanine mutations at Y270 yield large drops in activity. While Y270A shows a dramatic 98% reduction in activity (kcat = 0.33 s−1), the Y270F mutant has a more moderate impact (kcat = 8.6 s−1). Indeed, the presence of the aromatic ring within the active site is enough to maintain 48% of WT activity. The Y270F mutant exhibits lowered substrate affinity (Km = 8.73 mM), similar to that displayed by R267K. With denaturation temperature midpoints of 59.6 and 58.5 °C, respectively, Y270A/F mutant enzymes have structural thermal stabilities comparable to WT hGS (Td = 59.6 °C).
MD simulations of Y270A and Y270F show the mutated hGS enzymes bind γ-GC in a slightly twisted orientation, wherein the glutamyl moiety of γ-GC displays many hydrogen bonds, but its cysteine moiety is rotated further away from the S-loop than in WT, Fig. 3. The negatively cooperative substrate γ-GC maintains bonds with S151, E214, N216, and Q220 in the lowest energy frames from the last ns of MD simulations of Y270A and Y270F (Eint = −46 to −109 kJ/mol). Interestingly, Y270F exhibits stronger bonding of γ-GC than Y270A at R125 (Eint = −135 and − 73 kJ/mol) and R267 (Eint = −174 and − 150 kJ/mol), Fig. 2. The Y270F mutant also forms a long hydrogen bond at the sulfur of γ-GC and the S149 hydroxyl (4.0 Å S-O distance, 137°). The bond between G269 and Q274 is disrupted in the Y270A mutant, allowing for a more flexible S-loop. The aromatic ring maintained in Y270F allows for substrate binding and an S-loop structure more similar to WT than that exhibited by the Y270A mutant.
3.4. Role of D268 in Active Site Structure
Two experimental point mutations were made to explore the role of D268 in active site structure: D268A/E. The D268 mutants demonstrate similar activity of 62–65% of WT (kcat = 11.8 and 11.3 s−1, respectively). Despite these comparable activities, D268E binds γ-GluABA tightly (Km = 0.19 mM), while D268A has a Michaelis constant nearly identical to WT (Km = 1.33 mM). In addition to altered substrate binding, D268E has a lower thermal stability, relative to WT hGS (Td = 54.4 °C). While both D268 mutations result in moderate decreases in activity, the D268E mutation also alters substrate binding and thermal stability in hGS.
Low energy frames from MD simulations provide insight into the structural shifts induced by D268 mutations. In D268A, the loss of a side chain carboxyl at 268 disrupts the hydrogen bond between the residue and the side chain amide of Q211. The neighboring hydrogen bond between Q220 and E214 exhibited by free WT hGS is replaced by a hydrogen bond between the side chain carboxyl of E214 to the backbone α-amine of N216 in D268 mutant hGS. Although the D268E maintains the interaction between the side chain carboxyl at 268 and the Q211 amide, the mutant displays a similar shift in binding at E214. The MD simulations show mutations to D268 alter hydrogen bonding interactions within the active site of hGS.
The binding of γ-GC is distorted in both D268 mutants. In an MD simulation of D268A, the glutamyl side of the substrate twists away from the S-loop; additional hydrogen bonds form at R267 and E214, but the distance from Y270 increases, Fig. 3. Over the course of the MD run, average γ-GC interaction energies strengthen at R125, S151, and E214 by 32, 65, and 41 kJ/mol, relative to WT. Interactions between γ-GC and S149, A150, and R267 also increase by 13–19 kJ/mol, Fig. 2. An MD simulation of D268E reveals a more dramatic shift in γ-GC binding. Within the binding pocket, γ-GC adopts a twisted orientation wherein the glutamyl α-amine of γ-GC forms a hydrogen bond with the thiol of the cysteine moiety of γ-GC. Hydrogen bonding between γ-GC and residues deep in the pocket (E214, Q220, and R267) are disrupted; interaction energies between these residues and γ-GC weaken by 65, 37, and 51 kJ/mol in D268E, relative to WT. As the negatively cooperative substrate does not fit as deeply into the pocket of D268E, interactions between the cysteine moiety of γ-GC and G368 strengthen by 23 kJ/mol over the course of the MD run, relative to WT. Binding of γ-GC is significantly altered upon mutation of D268 in hGS.
3.5. S-Loop Structure Residues (G269, P272, Y275)
Experimental point mutations of G269 and Y275 serve to explore the role of hydrogen bonding within the S-loop of hGS. Insertion of a large hydrophobic residue into the S-loop with the G269 V mutation results in a remarkable 97% drop in activity (kcat = 0.6 s−1). Mutations at Y275 reveal a striking contrast, maintaining 36–50% activity of WT hGS. Both Y275A and Y275F exhibit similar activities (kcat = 9.2 and 6.6 s−1, respectively) and comparable increases in γ-GC binding affinities (Km = 0.05 and 0.10 mM, respectively). Although Y275F has a thermal stability near that of WT (Td = 59.4 °C), the denaturation temperature midpoint of G269 V is much lower at 45.5 °C. Mutations to residues involved in hydrogen bonds within the S-loop can have severe impacts on the activity, kinetics, and thermal stability of hGS.
An analysis of low energy frames from MD simulations of G269 V provide insight into the structural changes behind these activity change. Although an MD simulation of free G269 V shows the enzyme maintains the hydrogen bond between the backbone carbonyl of residue 269 and the amide of Q300, the hydrogen bonds with Q274 and Y275 are disrupted. A compensating hydrogen bond stretches across the S-loop from the side chain amine of Q274 to the backbone amine of Y270. Interestingly, the hydrogen bond between Y275 and R283 is lost in both free and γ-GC bound G269 V. The γ-GC bound form of G269 V exhibits a disruption of backbone hydrogen bonds at V269 with Y275 and Q300. Not only is the hydrogen bond between Q274 and the backbone of residue 269 conserved in G269 V, an additional bond between Q274 and the backbone of Y270 is present, Fig. 4. MD simulations show the G269 V mutation of hGS impacts the structure of the S-loop through disruptions of hydrogen bonds between the backbone of residue 269 and the side chains of Q274, Y275, and Q300.
Although γ-GC binds to G269 V hGS in a similar orientation to WT, interactions with S149, N216 and Q220 are disrupted in a low energy MD frame; the substrate does not nestle as tightly into the pocket. Over the course of a 5 ns MD run of G269 V, residues R267 and D268 have average interaction energies with γ-GC of −172 and − 56 kJ/mol, respectively, much stronger than in WT (−117 and − 7 kJ/mol, respectively), Fig. 2. Interactions with γ-GC at R125 and E214 in G269 V weaken by 39–48 kJ/mol, while interactions at S149, S151, N216, Q220, and Y270 are within 12 kJ/mol of WT. Computational models show the G269 V mutation has a large impact on γ-GC binding in hGS.
Analysis of low energy frames from MD simulations of Y275 mutants show deviations in the bonds between residues of the S-loop. With the loss of the Y275 hydroxyl in the free Y275A and Y275F mutants, the hydrogen bond between residue 275 and G269 is lost. Yet, hydrogen bonds between the side chain carbonyl of Q274 and the backbone amines of G269 and Y270 are maintained in Y275 mutants. While the hydrogen bond from G269 to Q300 is disrupted in Y275A, it remains in Y275F. Larger shifts in S-loop structure are apparent in MD simulations of the γ-GC bound form of Y275A, Fig. 4. Without the hydroxyl group of Y275, the hydrogen bond between G269 and residue 275 is disrupted. Hydrogen bonds between the Q274 side chain amine and the backbones of Y270 and M271 remain, helping stabilize the twist in the S-loop. In contrast, the γ-GC bound Y275F mutant exhibits a loss of the interaction between residue 275 and G269, while retaining three hydrogen bonds from Q274 (backbones of G269, Y270, and M271). In both γ-GC bound Y275 mutants, interactions between the S-loop and R283 are disrupted, while only Y275A exhibits a disruption of the G269-Q300 hydrogen bond. The bonding network within the S-loop is impacted by mutations to Y275, especially in the case of the alanine mutant.
Although MD simulations show Y275 mutants bind γ-GC in orientations similar to WT hGS, the interaction energies between γ-GC and active site residues vary, Fig. 2. Relative to WT, both Y275A and Y275F exhibit strengthened γ-GC interactions at S151 and E214 by 7 to 12 kJ/mol. Interestingly, interactions with γ-GC at R125 and R267 strengthen by 32 and 58 kJ/mol compared to WT in Y275A. The Y275F mutant has more moderate shifts of 3 to 9 kJ/mol at these same residues. While γ-GC interactions with G370 are ca. -3 kJ/mol in Y275A, Y275F exhibits much stronger binding of −25 kJ/mol. Mutations to G269 and Y275 have long range impacts on the interaction of hGS with γ-GC.
Experimental mutation of P272A reveals a marked 94% drop in activity with a kcat of 1.19 s−1. Despite the low activity of P272, a Michaelis constant of 0.06 mM was determined for the mutant, which is much lower than WT (Km = 1.31 mM). Interestingly, the mutant exhibits a denaturation temperature midpoint nearly identical to WT hGS (Td = 60.8). In the lowest energy frames from the last ns of a 5 ns of MD runs of free and γ-GC bound P272A, the bend in the S-loop is reinforced through two new hydrogen bonds from the M271 sulfur to the backbone amines of R273 and Q274, Fig. 4. Although the position of γ-GC in the active site of P272A is similar to that in WT hGS, the interaction energies over the course of a 5 ns MD run vary significantly. The P272 mutant has average γ-GC interaction energies at S151 and R267 (−80 and − 176 kJ/mol) that are 51–58 kJ/mol stronger than in WT. The P272A mutant also exhibits strengthened interactions at R125, S149, N216 (10–13 kJ/mol stronger than WT), and E214 (30 kJ/mol stronger than WT). In contrast, interaction between γ-GC and Y270 is weaken by 7 kJ/mol in P272A. Clearly, the P272A mutation has a large impact on the activity and binding properties of hGS.
3.6. Allosteric Modulation in the S-Loop
WT hGS is negatively cooperative with respect to the γ-GC substrate with a Hill coefficient of 0.69 (Table 2). Due to insufficient activity, kinetic parameters of R267A, R267W, G269 V and Y270A could not be determined. For P272A, a Km of 0.06 ± 0.02 mM and Hill coefficient of 1.02 ± 0.01 were determined from γ-GluABA kinetic assays. While duplicate assays from independent purifications have high reproducibility, the low kcat of P272A (1.19 s−1) could impact the uncertainty of the measured kinetic parameters. Mutations that increased γ-GC binding affinities (D268E, Y275A and Y275F) have little impact on the cooperativity of hGS (Hill coefficients of 0.68, 0.73 and 0.70, respectively). Both D268A and Y270A display a slight decrease in negative cooperativity (Hill coefficients of 0.78). While the Michaelis constant of D268A mirrors that of WT, MD simulations suggest that the mutation significantly increases interactions with R267 and E214, which bind and position γ-GC. In contrast, the decreased negative cooperativity of Y270A is matched by a decrease in γ-GC binding. Most notably, the R267K mutation results in a dramatic decrease in γ-GC binding, and is completely noncooperative (Hill coefficient = 1.03).
4. Discussion
Extensive experimental and computational studies determined the role of S-loop residues responsible for forming the curve of the S-loop (G269, Y275, and P272), maintaining active site structure (D268), and binding the negatively cooperative substrate (R267 and Y270). Those mutations that strongly alter γ-GC binding, either directly or indirectly, also impact allosteric communication in hGS. The crucial role of the S-loop in allosteric communication is directly linked to the importance of S-loop residues in both active site structure and γ-GC binding.
The tight curve at the end of the S-loop is maintained by P272 at the top of the loop and a hydrogen bond between G269 and Y275 stretching across the region. While Y275 mutants exhibit moderate reductions in activity upon the disruption of a hydrogen bond from G269 to Y275, the severe reduction in activity in G269 V likely results from the loss of an additional hydrogen bond between G269 and Q274. Although the thermal stabilities of Y275 mutants suggest the residue does not have a large impact on overall structure, G269 V exhibits a low denaturation temperature midpoint that may indicate a folding problem in the mutant enzyme, which occurs on a larger time scale than can be feasibly modeled with MD. According to MD simulations, mutation at P272A impacts γ-GC binding, most notably at R267, which yields a severe reduction in activity. A large increase in γ-GC binding affinity is coupled with a complete loss of cooperativity in P272A. The interactions and positions of G269, Y275, and P272 form the tight curve in the S-loop hGS, which is needed for the enzyme to maintain tertiary structure and properly bind the negatively cooperative substrate.
Computational models indicate the positively charged guanidyl group of R267 binds and positions γ-GC, while the aromatic ring and hydroxyl group of Y270 bind γ-GC near its peptide bond. The preserved aromatic stabilization in Y270F allows for moderate activity not seen in Y270A; without the aromatic stabilization of Y270, the cysteine moiety of γ-GC is not properly oriented relative to glycine within the active site. Those mutants with sufficient activity to determine kinetic parameters show decreased negative cooperativity, with a complete loss of cooperativity in the case of R267K. With moderate reductions in the thermal stabilities, the changes activity and allostery exhibited by R267 and Y270 mutants are likely the result of disrupted γ-GC binding by these residues, rather than an overall denaturation of the enzyme.
The extensive experimental and computational research on the S-loop of hGS presented here provides insight into patient mutations in hGS: Y270C, Y270H and R267W. The hemolytic anemia and metabolic acidosis exhibited by patients with mutations at Y270 (Y270C and Y270H) may be associated with the dramatic reduction in GSH production expected from the disruption of interactions with γ-GC at the position [7,8]. It is conceivable that γ-GC is not binding in these patient mutations since Y270F has better binding than R267K. The conservation of an aromatic interaction with γ-GC in Y270H and the conservation of the hydrogen bonding capacity in Y270C allows patients with such mutations to maintain a moderate level of hGS activity and thus function without severe neurological disorders. More dramatically, the extremely low activity of R267W at least partially explains the extremely low GSH levels, psychomotor retardation, and premature death of the patient with an R267W/D469E mutation [6,7]. Of all the substrate binding residues in hGS, R267 is perhaps the most important, serving to mediate repulsive interactions and bind γ-GC at both backbone and terminal atoms.
Residues that form or mediate salt bridges and hydrogen bonds with γ-GC, such as R267, initiate allosteric communication in hGS in a highly specific manner. In contrast, allosteric communication at the dimer interface of hGS occurs through hydrophobic interactions, such as those at V44 and V45, as shown in previous work by our groups [17]. Insights gained through the study of negative cooperativity in hGS can be applied to a wider range of allosteric communication. It can be posited that residues with strong substrate interactions serve to activate allosteric communication at binding sites, while more flexible weak interactions modulate allostery at protein-protein interfaces. The types of residues that modulate allosteric communication vary depending on position within the protein in order to permit both a wide variety of structural conformation while ensuring sensitivity to substrate.
Funding
This work was supported in part by the National Institute of Health (NIH) Grant R15GM086833 (M.E.A). This work was supported in part by a Welch Grant to Texas Woman's University Department of Chemistry and Biochemistry. This work was also funded in part by a TWU Faculty Research Grant to M.E.A and H.C.W. This article was published with support from the Texas Woman's University Libraries' Open Access Fund.
Notes
The authors declare no competing financial interests.
Acknowledgments
The authors would like to thank Kerri Slavens, Anna Stopper, Jodi Johnston, Perla Gonzalez, Stacey Griger and Aisha Chughtai for assistance. Much appreciation goes out to Richard Sheardy for assistance with instrumentation. The authors thank the Chemical Computing Group for generously providing the MOE software.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2018.11.008.
Contributor Information
Mary E. Anderson, Email: manderson3@mail.twu.edu.
Thomas R. Cundari, Email: t@unt.edu.
Appendix A. Supplementary data
Supplementary material
References
- 1.Meister A., Anderson M.E. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 2.Ansari M.A., Scheff S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulz J.B., Lindenau J., Seyfried J., Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 4.Townsend D.M., Tew K.D., Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57:145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffith O.W. Mammalian sulfur amino acid metabolism: an overview. Methods Enzymol. 1987;143:366–376. doi: 10.1016/0076-6879(87)43065-6. [DOI] [PubMed] [Google Scholar]
- 6.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 7.Ristoff E., Mayatepek E., Larsson A. Long-term clinical outcome in patients with glutathione synthetase deficiency. J Pediatr. 2001;139:79–84. doi: 10.1067/mpd.2001.114480. [DOI] [PubMed] [Google Scholar]
- 8.Dahl N., Pigg M., Ristoff E., Gali R., Carlsson B., Mannervik B. Missense mutations in the human glutathione synthetase gene results in severe metabolic acidosis, 5-oxoprolinuria, hemolytic anemia and neurological dysfunction. Hum Mol Genet. 1997;6:1147–1152. doi: 10.1093/hmg/6.7.1147. [DOI] [PubMed] [Google Scholar]
- 9.De Jesus M.C., Ingle B.L., Barakat K.A., Shrestha B., Slavens K.D., Cundari T.R. The role of strong electrostatic interactions at the dimer interface of human glutathione synthetase. Protein J. 2014;33:403–409. doi: 10.1007/s10930-014-9573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polekhina G., Board P.G., Gali R.R., Rossjohn J., Parker M.W. Molecular basis of glutathione synthetase deficiency and a rare gene permutation event. EMBO J. 1999;18:3204–3213. doi: 10.1093/emboj/18.12.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dinescu A., Brown T.R., Barelier S., Cundari T.R., Anderson M.E. The role of the glycine triad in human glutathione synthetase. Biochem Biophys Res Commun. 2010;400:511–516. doi: 10.1016/j.bbrc.2010.08.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinescu A., Anderson M.E., Cundari T.R. Catalytic loop motion in human glutathione synthetase: a molecular modeling approach. Biochem Biophys Res Commun. 2007;353:450–456. doi: 10.1016/j.bbrc.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 13.Brown T.R., Drummond M.L., Barelier S., Crutchfield A.S., Dinescu A., Slavens K.D. Aspartate 458 of human glutathione synthetase is important for cooperativity and active site structure. Biochem Biophys Res Commun. 2011;411:536–542. doi: 10.1016/j.bbrc.2011.06.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinescu A., Cundari T.R., Bhansali V.S., Luo J., Anderson M.E. Function of conserved residues of human glutathione synthetase: implications for the ATP-grasp enzymes. J Biol Chem. 2004;279:22412–22421. doi: 10.1074/jbc.M401334200. [DOI] [PubMed] [Google Scholar]
- 15.Oppenheimer L., Wellner V.P., Griffith O.W., Alton M. Glutathione synthetase: purification from rat kidney and mapping of the substrate binding sites. J Biol Chem. 1979;254:5184–5190. [PubMed] [Google Scholar]
- 16.Luo J.L., Huang C.S., Babaoglu K., Anderson M.E. Novel kinetics of mammalian glutathione synthetase: characterization of γ-glutamyl substrate cooperative binding. Biochem Biophys Res Commun. 2000;275:577–581. doi: 10.1006/bbrc.2000.3337. [DOI] [PubMed] [Google Scholar]
- 17.Slavens K.D., Brown T.R., Barakat K.A., Cundari T.R., Anderson M.E. Valine 44 and valine 45 of human glutathione synthetase are key for subunit stability and negative cooperativity. Biochem Biophys Res Commun. 2011;410:597–601. doi: 10.1016/j.bbrc.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornish-Bowden A. Understanding allosteric and cooperative interactions in enzymes. FEBS J. 2014;281:621–632. doi: 10.1111/febs.12469. [DOI] [PubMed] [Google Scholar]
- 19.Bocedi A., Fabrini R., Bello M.L., Caccuri A.M., Federici G., Mannervik B. Evolution of negative Cooperativity in Glutathione Transferase Enabled Preservation of Enzyme Function. J Biol Chem. 2016;291:26739–26749. doi: 10.1074/jbc.M116.749507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cornish-Bowden A. The physiological significance of negative cooperativity revisited. J Theor Biol. 2013;319:144–147. doi: 10.1016/j.jtbi.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Bush E.C., Clark A.E., Deboever C.M., Haynes L.E., Hussain S., Ma S. Modeling the Role of negative Cooperativity in Metabolic Regulation and Homeostasis. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bohr C., Hasselbach K.A., Krogh A. Ubereinen in biologischen Beziehung wiechtigen Einfluss den die kohlensauresparnung de bluter auf dessen sauerstoff binduing üft. Skand Arch Physiol. 1904;16:401–412. [Google Scholar]
- 23.Perutz M.F. Stereochemistry of cooperative effects in haemoglobin. Nature. 1970;228:726–734. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 24.Laine J.M., Amat M., Morgan B.R., Royer W.E., Massi F. Insight into the allosteric mechanism of Scapharca dimeric hemoglobin. Biochemistry. 2014;(46):7199–7210. doi: 10.1021/bi500591s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keov P., Sexton P.M., Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology. 2011;60:24–35. doi: 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 26.May L.T., Leach K., Sexton P.M., Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 27.Geitmann M., Elinder M., Seeger C., Brandt P., de Esch I.J.P., Danielson U.H. Identification of a novel scaffold for allosteric inhibition of wild type and drug resistant HIV-1 reverse transcriptase by fragment library screening. J Med Chem. 2011;54:699–708. doi: 10.1021/jm1010513. [DOI] [PubMed] [Google Scholar]
- 28.Scheuermann T.H., Li Q., Ma H., Key J., Zhang L., Chen R. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. 2013;9:271–278. doi: 10.1038/nchembio.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Meyts P. The insulin receptor: a prototype for dimeric, allosteric membrane receptors? Trends Biochem Sci. 2008;33:376–385. doi: 10.1016/j.tibs.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Limbird L.E., De Meyts P., Lefkowitz R.J. β-Adrenergic receptors: evidence for negative cooperativity. Biochem Biophys Res Commun. 1975;64:1160–1168. doi: 10.1016/0006-291x(75)90815-3. [DOI] [PubMed] [Google Scholar]
- 31.Changeux J., Edelstein S.J. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 32.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 33.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henikoff S., Henikoff J.G. Proc. Natl. Acad. Sci USA. Vol. 89. 1992. Amino acid substitution matrices from protein block; pp. 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hess B., Kutzner C., van der Spoel D., Lindahl E. GROMACS 4: algorithms for highly efficient load-balanced, and scalable molecular simulations. J Chem Theory Comp. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 38.Cornell W.D., Cieplak P., Bayly C.L., Gould I.R., Merz K.M., Jr., Ferguson D.M. A second generation force fields for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 39.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 40.Hornak V., Abel R., Okur A., Strockbine B., Roitber A., Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct, Funct, Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., and Hermans, J. (1981) Interactions models for water in relation to protein hydration, in Intermolecular Forces (Pullman, B.) pp 331–3432, D. Reidel Publishing, Dordrecht.
- 42.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:100089–100092. [Google Scholar]
- 43.Essmann U., Perera L., Berkowitz M.L., Darde T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8592. [Google Scholar]
- 44.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 45.Molecular Operating Environment (MOE), 2013.08 (2014) Chemical Computing Group Inc. Montreal, QC, Canada.
- 46.Labute P. The generalized born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J Comput Chem. 2008;29:1693–1698. doi: 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
- 47.Corbeil C.R., Williams C.I., Labute P. Variability in docking success rates due to dataset preparation. J Comput Aided Mol Des. 2012;26:775–786. doi: 10.1007/s10822-012-9570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Njålsson R., Carlsson K., Bhansali V., Luo J.L., Nilsson L., Ladenstein R. Human hereditary glutathione synthetase deficiency: kinetic properties of mutant enzymes. Biochem J. 2004;381:489–494. doi: 10.1042/BJ20040114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Njålsson R., Carlsson K., Olin B., Carlsson B., Whitbread L., Polekhina G. Kinetic properties of missense mutations in patients with glutathione synthetase deficiency. Biochem J. 2000;349:275–279. doi: 10.1042/0264-6021:3490275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material