

Abstract
For many years, our research group worked to develop gene transfer approaches for salivary gland disorders that lacked effective conventional therapy. The purpose of this chapter is to describe and update key methods used in this process. As described in our original chapter from the 2010 volume, we focus on one clinical condition, irradiation-induced salivary hypofunction, and address the choice of transgene and vector to be used, the construction of recombinant viral vectors, how vector delivery is accomplished, and methods for assessing vector function in vitro and in an appropriate animal model.
Keywords: Gene therapy, Salivary glands, Adenovirus, Adeno-associated virus, Radiation damage, Salivary hypofunction
1. Introduction
There are two major disorders that lead to the irreversible loss of salivary gland function: (1) irradiation damage that occurs during the course of treatment for a head and neck cancer and (2) the autoimmune exocrinopathy Sjögren’s syndrome. Both disorders are fairly common. For 2015, the estimated number of new cases of oro-pharyngeal cancers diagnosed in the US was 45,780, accounting for 2.76 % of all estimated new malignancies [1]. The treatment for most such patients, in developed societies, includes surgery and irradiation±chemotherapy. Sjögren’s syndrome has a prevalence of ~0.5–1 %, making it the second most common rheumatic disease after rheumatoid arthritis [2]. Although the etiologies of these two disorders are dramatically different, both conditions result in the loss of salivary acinar cells, the only cell type that normally secretes the fluid component of saliva. With both conditions, the predominant remaining epithelial cells are of duct origin, and incapable of fluid secretion. Patients lacking saliva suffer considerable morbidity, including dysphagia, oral infections, delayed mucosal healing, and considerable pain and discomfort.
If patients in either disease group have a reasonable mass of acinar cells remaining, treatment with sialogogues (salivary stimulants, e.g., pilocarpine, civemaline) can be beneficial. For patients who lack most or all salivary acinar cells there currently is no suitable treatment available, a situation that provided the impetus for our beginning to explore the use of in vivo gene transfer (gene therapy). It also is important to recognize a key difference in these two conditions: irradiation-induced salivary hypofunction is a localized gland problem. While this condition certainly leads to some systemic concerns (e.g., dysphagia, infections), a primary treatment for it needs only to be targeted to the damaged gland. Furthermore, the pathologic etiology, i.e., the radiation treatment, is time-limited. A patient presenting with irradiation-induced salivary hypofunction was treated with radiotherapy in the past and is without any ongoing active disease process. Conversely, a patient with Sjögren’s syndrome experiences a systemic autoimmune disease, albeit one commonly having salivary glands as a major target organ. Localized salivary gland gene therapy for a patient with Sjögren’s syndrome can address their salivary hypofunction [3], but in all likelihood, at least for the present, will have no beneficial effects on the systemic disease process. Additionally, it is important to recognize that Sjögren’s syndrome patients show continuous disease activity, e.g., the presence of serum autoimmune markers. Based on its more localized nature, and the absence of an active disease process, irradiation-induced salivary hypofunction is a disorder more readily treatable by salivary gland gene therapy. It also lends itself to a more useful presentation for a chapter such as this. Hence, the focus of this chapter is only on the “repair” of irradiation-induced salivary hypofunction.
As with the development of a therapy for any disease condition, an essential initial element is to have a good understanding of the physiology of the normal target tissue, in addition to a good understanding of the pathophysiological situation. Fortunately, our laboratory has such a background, as a result of working many years in the salivary gland field [4]. Based on that understanding, we decided that surviving duct cells in the irradiated gland were capable of generating an osmotic gradient into the gland lumen, but needed water permeability pathways to allow fluid to follow this gradient [5]. Accordingly, the gene of choice for our approach was aquaporin-1 (AQP1), the first described water channel [6]. Given that salivary epithelial cells are slowly dividing post-mitotic cells, it was appropriate to employ nonintegrating vectors for the actual gene transfer. Accordingly, we have used recombinant serotype 5 adenoviral (rAd5) and serotype 2 adeno-associated viral (rAAV2) vectors [7]. Each vector type has distinct advantages. For example, rAd5 vectors are relatively easy to produce, lead to high transgene expression, but induce a potent immune response that in animal models renders the expression transient (<2–4 weeks). rAAV2 vectors elicit a modest immune response and generally lead to low, but stable, transgene expression; however, they are more difficult to produce. We initially used rAd5 vectors for proof of concept studies in both animal models and humans, and now are working on developing a long-lived therapeutic correction of irradiation-induced salivary hypofunction by using a rAAV2 vector. Herein, we will describe the production of both vector types.
1.1. Recombinant Serotype 5 Adenoviral Vector Production
Adenoviruses are non-enveloped, double stranded DNA viruses. There are more than 50 human adenoviral serotypes. A common adenoviral gene transfer vector is based on adenovirus serotype 5 and we typically use an E1 gene-deleted, replication-deficient rAd5 vector in our studies. Importantly, these vectors can efficiently transduce salivary acinar and duct cells [8]. To construct, propagate and purify an E1 deleted rAd5 vector, various products and services from different companies can be used, including ABL, Inc (Rockville, MD), Agilent Technologies (Santa Clara, CA), Cell Biolabs, Inc (San Diego, CA), Clontech (Mountain View, CA), Creative Biogene (Shirley, NY), Life Technologies (Grand Island, NY), Microbix Biosystems (Toronto, Canada), SignaGen Laboratories (Rockville, MD), ViGene Biosciences (Rockville, MD), Vector Biolabs (Malvern, PA), and Viral Vector Core Facility (Iowa City, IA). In the first generation (simplest) rAd5 vectors, the E1 gene is deleted. This gene encodes the E1a and E1b proteins, which are required for the replication of a wild-type Ad5. This gene deletion creates space in the vector genome to insert a foreign gene or cDNA (the transgene).
Currently, there are three principal ways to generate E1 deleted rAd5 vectors; using a two-plasmid co-transfection method with eukaryotic (293) cells, a one-plasmid transfection method (Viral Power Adenoviral Expression System from Life Technologies) and an E. coli-based system (from Agilent Technologies). Herein, we will describe the two-plasmid co-transfection method to generate a rAd5 vector, as this is the method we routinely use. In the two-plasmid system, one plasmid is termed a shuttle vector (the one used in our laboratory is pACCMV-pLpA) and can express in both prokaryotic and eukaryotic cells. This plasmid has a human cytomegalovirus (CMV) promoter, a multiple cloning site, and a SV40 polyadenylation signal that can be placed in the E1 region between map units 1.3 and 9.1 of the adenoviral genome. This plasmid also contains 455 base pairs of Ad5 sequence (0.0–1.3 map units) upstream of the CMV promoter and 3039 base pairs of Ad5 sequence (9.3–17.0 map units) downstream of the SV40 polyadenylation signal. The second plasmid is pJM17, and it provides most of the Ad5 sequence except for the E1 gene and a small part of the E3 gene region. The pJM17 plasmid (Fig. 1) contains overlapping sequences with the Ad5 sequences found in pACCMV-pLpA, which permit homologous exchange when they are co-transfected into 293 cells, a human embryonic kidney cell line [9]. The 293 cells stably express the E1 proteins necessary to allow replication of the constructed E1-deleted rAd5 vector [10].
Fig. 1.

(a) Map of pJM17 plasmid showing HindIII restriction sites. (b) Gel showing HindIII digestion results. There are 10 HindIII restriction endonuclease sites in the pJM17 plasmid. After digestion, it yields 10 fragments (8010, 5446, 5322, 4597, 4370, 3795, 3437, 2937, 2081, and 75 bp). Eight of these fragments from the HindIII-digested pJM17 are readily visualized as bands on 1 % agarose gels after electrophoresis
1.2. Recombinant AAV Serotype 2 Vector Production
The production of rAAV2 can be generally accomplished with either of two methods: the use of stable packaging cell lines, or transient transfection of production cell lines. Both methods require three genetic elements: (1) the “ recombinant AAV2 genome,” consisting of the wild-type AAV2 inverted terminal repeats (ITRs) framing the expression cassette. The ITRs contain the cis sequence information needed for replication and encapsidation. (2) the genes coding for the replication and structural proteins (rep and cap, respectively), expressed in trans. (3) the required helper functions naturally encoded by a helper virus such as adenovirus, also expressed in trans.
To generate rAAV2 using stable packaging cell lines, the expression cassette is stably transfected together with rep and cap genes into a cell line (e.g., HeLa cells). These cells can be stored and expanded for rAAV2 production, which then only requires a wild-type Ad5 infection. This method is easily scalable and thus very suitable for large-scale production in vector core facilities. However, this process is quite laborious if used for only a few preparations. Also, stable transfection of the rep and cap genes, encoding toxic proteins, can be technically challenging. Contamination with wild-type Ad5 can be a problem due to the infection of stably transfected cell lines with Ad5 to start the production process, but new purification methods to overcome this are being developed.
In contrast, the relatively undemanding transient transfection method is ideally suited for mid-scale production, and is what we use in our laboratory. This classical rAAV2 production method is based on a trans-complementary co-transfection of plasmids containing the required genetic elements into an appropriate target cell (we use 293T cells). It allows flexible and quick rAAV2 production using the straightforward protocol described below. The expression cassette is present in the pAAV plasmid flanked by ITRs [11]. For optimal yield and convenience, we use a packaging/helper plasmid, pmmtvRC, which contains the rep and cap sequences, and an Ad5 helper plasmid, pRS449B. With this method, the typical yield, per 108 cells, after CsCl centrifugation and subsequent dialysis is between 1 × 1012 and 6 × 1012 total particles, as determined by QPCR.
The significant interest in the use of AAV vectors clinically has led to recent advances in scalable production and purification methods. Transfection of cells with a set of plasmids, such as described above, has been used for many years to produce a cell lysate containing large amounts of AAV vector particles that are free of contaminating helper virus. The AAV particles are then purified from this crude lysate by a combination of methods such density gradient centrifugation (with CsCl or iodixanol), affinity or ion exchange chromatography, and gel filtration.
Although effective, these methods lack easy scalability, which is required for clinical applications. The use of suspension cells [12], roller bottles [13], or cell factories in combination with cell lines [14], which stably maintain the rAAV vector and some of the AAV helper functions required to produce rAAV, has addressed some of the scalability issues. These cells can then be “induced” to produce rAAV vector by infection with replication defective lytic viruses such as derived from Ad5 or herpes simplex virus [15, 16]. Similarly, the use of Baculovirus-based systems [17], in which the AAV vector and AAV helper functions are delivered to suspensions of insect cells by infection, has increased both the scale and the yield of particles per cell to the levels sufficient for large-scale manufacturing as required for either preclinical studies in large animal models or clinical studies for localized gene therapy applications, such as in the salivary glands.
1.3. Cannulation of Salivary Glands
Among the most common target organs for in vivo gene therapy are the liver, lung, and muscle. The liver and lung have considerable metabolic activity and bulk protein production, but are not easy to access. Muscle, while easy to access for vector delivery, is not a tissue known for protein production and secretion. Salivary glands present an attractive target with some particular advantages for in vivo gene transfer. First, they are easy to access, as the ductal orifices of the glands open into the mouth and typically can be visualized and cannulated without significant complications. Indeed, contrast imaging of cannulated glands (sialography) is a routine clinical diagnostic procedure. Secondly, although salivary glands are densely packed, almost all acinar and ductal cells have their apical membranes directly in contact with the oral cavity along the lumen. Third, the majority of salivary parenchymal cells (acinar) are capable of producing high amounts of protein for both exocrine and endocrine secretion. Therefore, a transgene can be targeted to a large number of protein-producing cells with little difficulty.
1.4. Assessing Functional Response (Saliva Collection)
After delivery of the transgene of interest into salivary glands using a viral vector, it is essential to assess the function of the transgene. For the purpose outlined in this chapter, the key biological function to measure is salivary flow rate. We typically collect whole saliva from the floor of the mouth in rodent experiments (Fig. 2), although saliva can be collected from individual glands as well. Whole saliva is much easier and is better for animal survival, which is particularly important if you are conducting long-term studies with multiple sample collections.
Fig. 2.
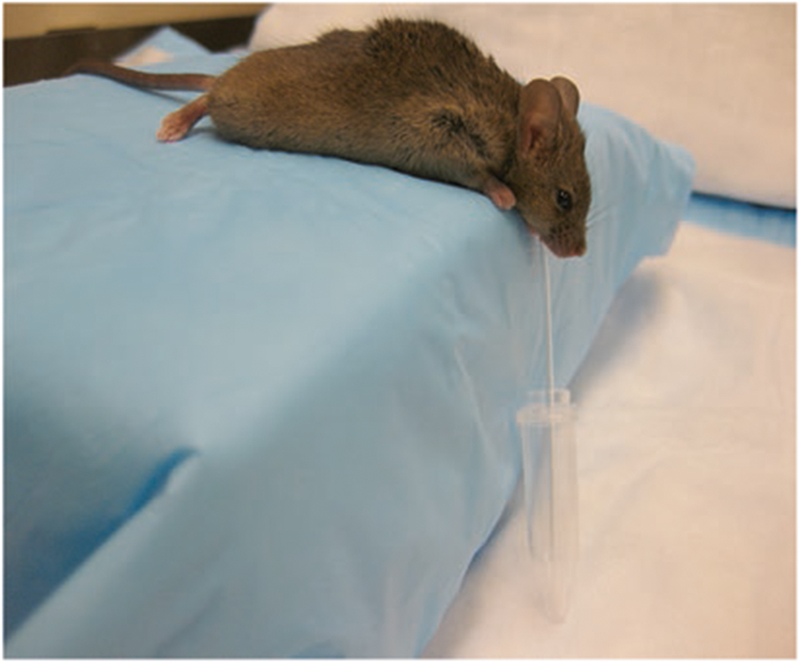
Figure of mouse during the whole saliva collection procedure. Note the positioning of the capillary tube draining the saliva from the floor of the mouth into a pre-weighed Eppendorf tube
1.5. Animal Models
An animal model provides an in vivo, nonhuman experimental tool that mimics a disease or injury that is similar to a human condition. The use of animal models allows the investigation of pathology and pathogenesis in ways that are impossible in a human patient. Thus, animal models provide scientists with a rich experimental resource, but also require high ethical behavior and great care when used. All animal studies should be conducted using a detailed, written experimental protocol that is reviewed by an appropriate and independent oversight body to ensure that minimal animal pain or discomfort occurs, and that the study is scientifically valid.
1.5.1. Irradiation of Salivary Glands
Irradiation in animal models has long been used to mimic salivary hypofunction in patients who receive irradiation to treat head and neck cancer. Many different animals can be used and our group has employed rhesus monkeys, miniature pigs, rats, and mice at various times and for various purposes as preclinical models of salivary gland irradiation damage. Mice are advantageous, as they are small, easy to handle, and inexpensive. Thus, for the purpose of this chapter, we will provide details on irradiating mice and the particular procedures we use. Anyone who is contemplating studying irradiated animals, of whatever species, is urged to consult radiation biology experts at your institution.
For our studies, the mice are 7–10 weeks old (young adults), and typically weigh 20–30 g. We irradiate salivary glands by placing each animal into a specially built Lucite jig in such a way that the animal can be immobilized without the use of anesthesia. The use of anesthesia can change the sensitivity to irradiation, so if anesthesia must be used, it must be used in all experimental and control groups for proper comparison. Additionally, the jig is fitted with a Lucite cone that surrounds the animal’s head and prevents head movement during the radiation exposure (see Fig. 3). For single dose irradiation experiments in mice, we use a 15Gy dose [18]. Single dose studies are very convenient, but they do not fully mimic clinical irradiation protocols. Clinically, patients receive fractionated doses, commonly ~1.5–2.5 Gy/day 5 days/week, for up to 6 weeks. For our studies in mice we utilize an adapted fractionated dose scheme, five daily fractions of 6 Gy. This is more convenient than the human regimen, yet one that leads to significant salivary hypofunction [19]. Radiation is delivered to the animal’s head with a Therapax DXT300 X-ray irradiator (Precision X-ray Inc., North Branford, CT) using 2.0 mm Al filtration (300 KVp) at a dose rate of 1.9 Gy/min. The single dose and fractionated dose schemes described achieve comparable salivary hypofunction, resulting in ~50–60 % reduction when measured 8 weeks after irradiation was completed [18, 19]. Immediately after radiation, animals are removed from the Lucite jig and housed (4 animals/cage) in a climate and light controlled environment, and allowed free access to food and water.
Fig. 3.

Figure of three mice in the specially made Lucite jig prior to irradiation. The animals are immobilized without the use of anesthesia. Note that the jig is fitted with a Lucite cone that surrounds the animal’s head and prevents head movement during the radiation exposure
2. Materials
2.1. Generating a rAd5
293 Cell line (Microbix Biosystems, Toronto, Canada).
IMEM medium supplemented with 10 % fetal bovine serum, 100 U/mL penicillin G, and 100 μg/mL streptomycin for 293 cell line.
pJM17 plasmid (Microbix Biosystems).
Calcium Phosphate Transfection Kit.
Ultra-clear centrifuge tubes (Beckman Coulter).
Cesium Chloride (CsCl) gradients (1.25 mg/mL, 1.33 mg/mL, and 1.4 mg/mL) for rAd5 purification. CsCl is dissolved in TD buffer: 140 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 25 mM Tris–HCl, pH 7.4.
SW41 rotor (Beckman Coulter) or equivalent.
PIERCE Slide-A-Lyzer dialysis cassette (ThermoFisher Scientific, Rockford, IL).
Virus dialysis buffer: 100 mM Tris–HCl, pH 7.4, 10 mM MgCl2, and 10 % v/v glycerol.
Virus dilution buffer: 5 mM MgCl2, 20 % v/v glycerol, 10 mM Tris–HCl, pH 7.4.
SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA).
ABI StepOne Plus Sequence Detector (Applied Biosystems).
2.2. Generating a rAAV2 Vector
293T cells (American Type Culture Collection, Manassas, VA).
150 mm plates.
DMEM low glucose (4.51 g/L D-glucose, Gibco) media: 100 U/mL penicillin G, 100 μg/mL streptomycin, 2 mM glutamine, 10 % fetal bovine serum.
pAAV2 plasmid containing expression cassette with the transgene and ITRs (6 μg/plate).
pmmtv2RC plasmid ([20]; termed p2RepCap in the citation; 6 μg/plate).
pRS449B Ad5 helper plasmid ([21]; 12 μg/plate).
Solution A (stock for ten 150 mm plates): 2× Hepes buffered Saline pH 7.03±02, 10 mL.
Solution B (for ten 150 mm plates): 9.0 mL double distilled water, followed by adding plasmid DNA at 60 μg pmmtv2RC, 120 μg p449B, 60 μg pAAV2-transgene, followed by adding 1000 μL of 2 M CaCl2 and mixing by pipetting up and down.
TD buffer: As described in Subheading 2.1, item 6.
Benzonase (also called endonuclease from Sigma, 25,000 units).
50 mL conical tubes.
250 mL conical tubes.
CsCl (as above for rAd5 production).
Beckman Ultra Clear Tube.
Ultra centrifuge and Beckman SW41 Ti rotor and buckets.
Refractometer.
“Butterfly needle” (21G ¾ Vacutainer; Becton Dickinson).
Several Eppendorf micro-centrifuge tubes.
Dialysis cassette (Pierce, Slide-A-Lyzer 10 K).
Saline.
2.3. Delivery of Vector to Murine Submandibular Glands
Mice.
Rack made by us for oral cavity display (Fig. 4a).
Spring for spreading cheeks (made from paperclip).
Cotton to hold back tongue.
Microfine IV needle BD insulin syringe 0.3 cc, 28-gauge needle.
Preformed PE10 tubing (Intramedic PE10 polyethylene tubing, Clay Adams Brand, Becton Dickinson), heated and pulled to make cannulae (see Note 2).
Scalpel blade to bevel cannulae.
Binocular dissecting microscope.
Krazy glue (ethyl cyanoacrylate; Elmer’s Products, Inc; Columbus, OH).
Curved forceps with very fine tip.
Ketamine (100 mg/mL).
Xylazine (20 mg/mL).
Atropine (Sigma) made by dissolving powder in water at 0.5 mg/mL (can be stored in aliquots at −20 °C).
Fig. 4.
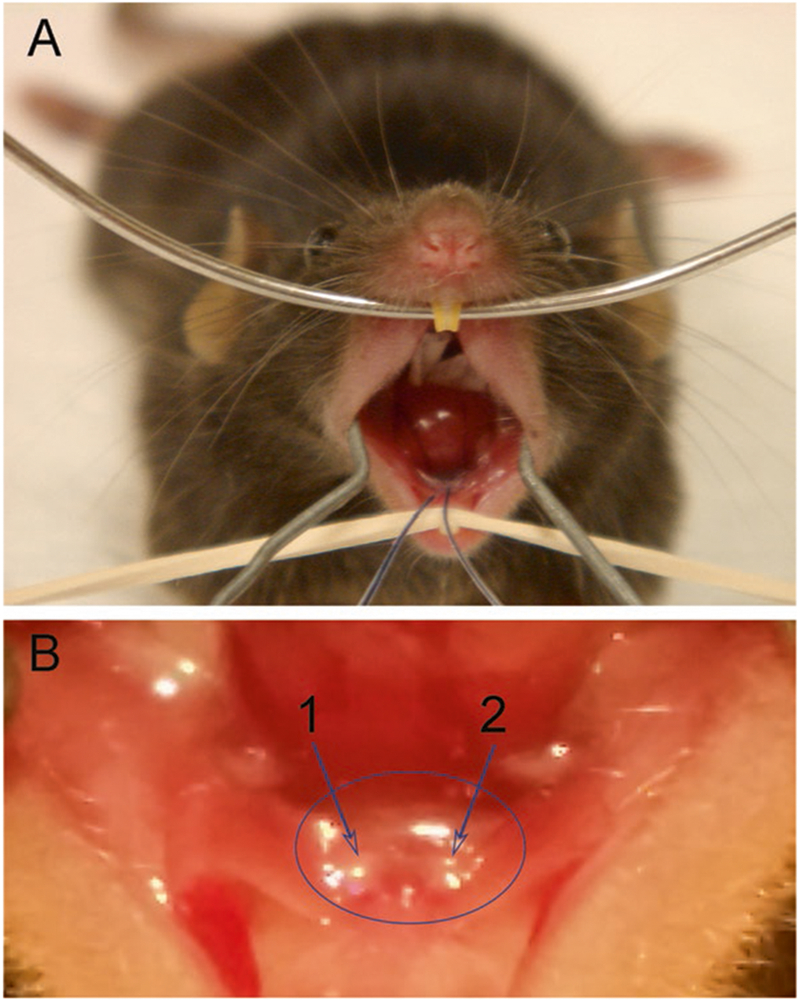
(a) Mouse with cannulas inserted into the orifices of Wharton’s duct (submandibular gland) prior to vector delivery (note needle and syringe attached to each cannula are out of the plane of this picture). Place the upper jaw of the mouse on a wire, i.e., teeth over the wire, and pull the lower jaw down with rubber band onto rack. Finally, expand cheeks with wire spring made from a bent paper clip. (b) Close-up view of right (“1 arrow”) and left (“2 arrow”) Wharton’s ducts in a mouse
2.4. Mouse Whole Saliva Collections
Heparinized Micro-Hematocrit capillary tubes.
Pilocarpine.
Ketamine (100 mg/mL) and xylazine (20 mg/mL).
3. Methods
3.1. Generating a rAd5
The following protocol is what we use routinely to produce an El-deleted rAd5 vector.
Insert transgene of interest into multiple cloning site of pACCMV-pLpA using conventional molecular cloning methods to create the shuttle vector.
Make a high quality preparation of shuttle vector.
Make a high quality preparation of the plasmid pJM17 (see Fig. 1; Note 1).
Grow 293 cells in IMEM.
Co-transfect pJM17 and the shuttle vector into 293 cells by calcium phosphate co-precipitation using the Calcium Phosphate Transfection Kit. Add 15 μg of pJM17, 5 μg of shuttle vector, 36 μL of 2 M CaCl2 and use double-distilled H2O to bring volume to 300 μL in a 14 mL polypropylene tube, and then mix well.
Quickly add 300 μL 2× Hepes buffered saline and bubble with an automatic pipet for 1 min.
Incubate at room temperature for 30 min until a fine precipitate is formed.
Add the entire mixture to a 100 mm plate of 293 cells, and gently rotate the plate to mix well.
Put the plate back into the tissue culture incubator, and replace the growth medium every 2 days. To increase the chances of successful vector formation, usually 4–5 plates of cells are co-transfected.
After ~10–12 days, plaques (holes in the 293 monolayer) can be observed, which indicate the replication of the recombinant adenovirus. Continually, and carefully, replace the medium until ~50 % of 293 cells are lysed.
Harvest the entire plate (293 cells and growth medium) by pipetting the growth medium present to dislodge the cell monolayer, i.e., do not use trypsin, and collect into a 50 mL conical test tube.
Completely lyse the 293 cells by freezing (dry ice) and thawing (37 °C water bath) five times, vortexing for 1 min after each time, to release rAd5 vector from the cells. Centrifuge at ~700 × g for 5 min.
Transfer supernatant [crude viral lysate (CVL)] to a new 50 mL tube. Use 50 μL of the CVL to infect one well of 293 cells (105 cells/well in a 12-well plate). Measure the recombinant protein production by an appropriate method (e.g., by Western blot, ELISA, enzyme assay) in the lysate or medium of these cells after 24 h to confirm that the CVL contained functional virus (See Note 3).
To make a large-scale preparation of the recombinant rAd5 vector, use the above CVL to transduce three fresh 100 mm plates of 293 cells.
Harvest the plates of cells at day 3. Freeze and thaw five times as above, combine and again centrifuge to collect supernatant.
Use this CVL to transduce 20 150 mm plates of 293 cells. Harvest the cells at day 3, centrifuge the harvested cells and aspirate all but ~6 mL of the media. Use the remaining 6 mL to resuspend the cell pellet. Freeze and thaw the cell suspension five times, then centrifuge at ~1600 × g for 10 min.
Make the first CsCl gradient by placing 2.5 mL of density 1.25 CsCl in sterile ultra-clear centrifuge tubes, then slowly underlay with 2.5 mL of density 1.40 CsCl. Transfer the CVL supernatant to the top of the CsCl gradient.
Spin the tubes in a SW41 rotor at ~151,000 × g, 22 °C for 1 h (balance carefully with culture medium or phosphate-buffered saline).
Following centrifugation, clean the outside of the tube with 70 % alcohol, then use a 21-gauge needle and syringe to poke through the side of the tube. Collect the lower opalescent band, which contains the rAd5 vector.
Place 8 mL of density 1.33 CsCl into sterile ultra-clear centrifuge tubes and transfer the vector band to this second CsCl gradient.
Spin the tubes in a SW41 rotor at ~151,000 × g, 22 °C for 18 h (again balancing carefully). As above, clean the outside of the tube with 70 % alcohol, and again use a 21-gauge needle and syringe to collect the opalescent band and add glycerol to 10 %.
Transfer the vector into a Slide-A-Lyzer dialysis cassette and dialyze two times in 500 mL dialysis buffer for 30 min, and then in 1-L fresh dialysis buffer for 1 h, three times.
Remove the vector suspension from the dialysis cassette and aliquot in sterile Eppendorf tubes at ~100 μL/tube and store at −80 °C.
Use real-time PCR (QPCR) to titer the rAd5 vector with primers from the E2 region of adenovius; E2q1 (5′-GCAGAACCACCAGCACAGTGT-3′) and E2q2 (5′-TCCACGCATTTCCTTCTAAGCTA-3′). Titers are expressed as particles/mL. The plasmid pACCMV-pLpA can be used as a standard for QPCR, with 1 μg of the plasmid (~8570 base pairs) being equivalent to 9.0 × 1010 molecules. Standard curves are established from 101 molecules to 108 molecules of shuttle vector, and rAd5 vectors are tested at three dilutions over a 100-fold range. These QPCR assays are typically carried out with the SYBR Green PCR Master Mix using a StepOne Plus Sequence Detector, with the following conditions: stage 1, 95 °C for 2 min; stage 2, 95 °C for 10 min; stage 3, 95 °C for 15 s, 60 °C for 1 min, repeated 40 times.
3.2. Generating a rAAV2 Vector
Culture ten 150 mm plates of 293 T cells in DMEM media at ~1.5 × 106 cells/plate.
Incubate cells in DMEM with 10 % FBS overnight.
For transfection of plasmids, allow 10 mL 2× Hepes (Solution A) to come to room temperature in 50 mL tube.
Place a 2 mL pipette in solution A and continuously bubble air while adding solution B drop by drop from a pipet containing 10 mL of solution B.
Incubate until a white precipitate is seen (at least 30 min), mix up and down with pipet.
Add 2.0 mL of this mixture per plate, drop by drop while gently swirling the plate.
24 h after transfection, remove medium and add 20 mL fresh DMEM with 10 % FBS.
48 h after transfection, harvest the cells by scraping them in their own media and transferring to 250 m > conical tube.
Centrifuge cells at ~500 × g for 15 min.
Discard media and while washing the cell pellet with 50 mL 1× PBS transfer to 50 mL conical tube and centrifuge again.
Discard 1× PBS and resuspend cell pellet with 10 mL 1× TD buffer (this can be safely stored at −20 °C).
Freeze cell pellets on dry ice, then thaw pellets (37 °C), and vortex cell lysate for 2 min. Repeat twice.
Add Benzonase to a final concentration of 20 U/mL of cell lysate.
Sodium deoxycholate is then added to a final concentration of 0.5 %, and incubate for 60 min at 37 °C.
Centrifuge lysate at 340 × g for 15 min.
Collect supernatant and add 0.55 g of CsCl per mL of supernatant and mix until dissolved, about 10 min at room temperature.
Adjust Refractive Index (RI) of supernatant to 1.372 with refractometer. If the RI is too high, add small amounts of TD buffer. If RI is too low, add small amounts of CsCl.
Add supernatant to one Ultra Clear Tube and fill tube to the top (approximately 11 mL) with TD buffer adjusted with CsCl to an RI of 1.372.
Centrifuge tubes for 72 h at ~182,300 × g using low acceleration and low deceleration settings.
As above with rAd5 vector preparation, clean the outside of the tube with 70 % alcohol.
With a butterfly needle, puncture tube 1-cm above the bottom of the tube. Be sure to twist needle while pushing so that the plastic plug from the tube wall does not get stuck in the needle preventing the fractions from going through.
Using a clamp to control the flow, collect ~700 μL fractions of purified virus in several 1.5 mL microcentrifuge tubes. Before collecting the next fraction, measure the RI of the previous fraction, keeping only those fractions with an RI between 1.371 and 1.372. These fractions typically contain the highest titers of the rAAV2 vector. The fraction in which the vector is found depends on the size of the transgene (i.e., larger transgene would mean fraction with a higher RI).
Perform QPCR to measure vector titer from the different fractions, on 1 μL aliquots from 1:1000 μL dilution in double distilled water, as described in Subheading 3.1, step 24. The sequences for the forward primer, reverse primer are based on the promoter of the expression cassette. The sequences used are as follows for CMV promoter: Forward—CATCTACGTATTAGTCATCGCTATTACCAT; Reverse – TGGAAATCCCCGTGAGTCA. The reaction mixture is incubated at 95 °C for 2 min (stage 1), 95 °C for 8 min (stage 2), then denaturation at 95 °C for 15 s (step 3), and annealing and extension at 60 °C for 1 min, repeated 40 times (stage 4). After completion, store selected fractions at 4 °C.
This stock virus can be stored in aliquots as desired and kept at −80 °C. Once an aliquot is thawed on ice, it can be stored at 4 °C for up to 2 weeks. Note that repeat thawing and refreezing will result in loss of vector activity.
Before using in vivo dialyze fractions against 0.9 % Sodium Chloride, putting fraction in dialysis cassette using a 21-gauge needle with a 1.5 mL syringe (see Pierce manual) and dialyzing by floating the cassette in 400 mL of saline for 30 min, twice, while stirring at room temperature.
Change saline and repeat twice (total of 1.2 L of saline).
3.3. Methods for Delivery of Vector to Submandibular Glands
Weigh animal.
Inject 1 μL/g ketamine: xylazine (3:2) intramuscularly into animal’s hind leg—the animal is usually anesthetized within 5 min.
Place animal on a specially constructed rack (Fig. 4a) with the upper jaw on wire—teeth over wire, the lower jaw pulled down with rubber band on the rack, the cheeks expanded with a wire spring, and a ball of cotton (~0.8 cm) positioning the tongue back toward the throat.
Adjust binocular dissecting microscope to focus on both submandibular gland duct orifices, which are located slightly lateral to the midline of the floor of the mouth, about 4–5 mm posterior from the lower incisors. You should see two whitish papillae. The duct orifice is located on the ventral aspect, about half way down the papillae (Fig. 4b).
Pick up preformed cannula made from PE10 tubing for insertion. Using the scalpel blade, cut a bevel (~45°) on the narrow end in the middle of the thinnest part of the tubing. Note that younger animals tend to require thinner tubing, while older animals require thicker tubing.
Using very delicate forceps, pick up the beveled (narrow) end of the tubing (cannula), approximately 2 mm from the end of the tubing, and push it gently into the duct orifice. The angle of the cannula should be 45° to the floor of the mouth. Push the cannula approximately 3–4 mm into the duct. Be sure that the cannula is well sealed with the duct orifice by visualizing the fitness of the orifice rim to the cannula.
After successful cannula insertion, place a drop of Krazy glue at the site of insertion to secure the cannula.
Inject 0.5 mg/kg atropine intramuscularly, wait 10 min. While waiting, fill a syringe with sample or control solution for delivery. For mice the optimal volume to infuse is 50 μL. Dilute vector in saline to the desired concentration.
Place distal end of cannula around the needle of a 0.3 cc syringe and inject sample slowly through the cannula into the gland.
Wait 10 min before gently removing the syringe to prevent backflow.
Gently remove cannulae and Krazy glue by pulling on tubing.
3.4. Methods for Murine Saliva Collections
Pre-weigh 1.5 mL Eppendorf tube (one/mouse) and record.
Anesthetize mouse with ketamine (60 mg/kg) + xylazine (8 mg/kg).
Inject pilocarpine (0.25 μg/g; this dose may need to be adjusted depending on the mouse strain used) subcutaneously in back of neck. Pilocarpine will stimulate the salivary glands to secrete saliva. Normally, the saliva will start to flow about 3–5 min after pilocarpine is injected.
Place the mouse on a box about 8 cm high, with the head of the mouse over the edge of the box (see Fig. 2).
Put one end of a Micro-Hematocrit capillary tube under the mouse’s tongue and the other end into the pre-weighed Eppendorf tube. Check capillary tube position and saliva flow constantly.
Monitor mouse carefully for signs of difficulty breathing, and supply with oxygen if necessary.
Stop saliva collection after 20 min, making sure that all of the saliva drains from mouse’s mouth and capillary tube into the Eppendorf tube.
Weigh the tube, then subtract the pre-weighed value of the tube. The amount of saliva will be based on a specific gravity of 1 (i.e., 1 g/mL) and typically will be expressed as μL/20 min. You can also convert the saliva output to μL/g-body weight or milligram-gland weight.
The saliva can be directly used as output (end point measure) or be used to assess transgenic protein production or activity, if appropriate. Additionally, salivary composition can be determined, e.g., calcium, sodium, amylase, total protein, etc. Saliva should be stored at −80 °C until assayed.
Acknowledgment
The authors’ research is supported by the intramural research program of the National Institute of Dental and Craniofacial Research.
4 Notes
Difficulty with the preparation of the pJM17 plasmid (see gel picture of high quality pJM17 preparation in Fig. 1). The pJM17 plasmid is a large plasmid, 40.07 kb. When preparing this plasmid it is necessary to be careful about two things. First, remember when making this plasmid it results in a low yield. It is best to use 100 μg/mL of ampicillin for cultures of bacteria containing pJM17, and to culture the bacteria for less than 16 h. Second, during the plasmid extraction procedure, because of its size, this plasmid breaks easily. One needs to be very careful at each step to prevent artificial shearing of the plasmid. A high quality preparation of pJM17 will show eight bands following 1 % agarose gel electrophoresis after HindIII digestion (Fig. 1).
Making a cannula.
-
(a)Cut a segment of tubing about 5 cm long.
-
(b)Hold both ends of tubing between index finger and thumb (keep tubing a little bit loose).
-
(c)Hold tubing about 15 cm above a delicate flame for a very short time—when the tubing begins to SOFTEN—remove from the flame and pull gently at both ends (not too much!).
-
(d)Wait approximately 20 s for tubing to cool and solidify, store at room temperature.
Test for viral activity of produced rAd5 and rAAV2.
Seed a 12-well dish of 293 or 293T cells. At 90 % confluency, transduce individual wells with equal amounts of viral particles (as determined by QPCR) from fractions being tested from the CsCl gradients. For rAd5 vectors use 10–100 viral particles/cell and for rAAV2 vectors use 1000–100,000 viral particles/cell. Perform an ELISA or Western immune blot on media or cell extracts later, as appropriate, from each transduced well to determine the viral fractions that best express transgenic protein and use those fractions for in vivo experiments.
References
- 1.Siegel RL, Miller KD, Jemal A (2015) Cancer statistics 2015 CA. Cancer J Clin 65:5–29 [DOI] [PubMed] [Google Scholar]
- 2.Mitsias DI, Kapsogeorgou EK, Moutsopoulos HM (2006) Sjögren’s syndrome: why autoimmune epithelitis. Oral Dis 12:523–532 [DOI] [PubMed] [Google Scholar]
- 3.Kok MR, Yamano S, Lodde BM, Wang J, Couwenhoven RI, Yakar S, Voutetakis A, Leroith D, Schmidt M, Afione S, Pillemer SR, Tsutsui MT, Tak PP, Chiorini JA, Baum BJ (2003) Local adeno-associated virus-mediated interleukin 10 gene transfer has disease-modifying effects in a murine model of Sjögren’s syndrome. Hum Gene Ther 14:1605–1618 [DOI] [PubMed] [Google Scholar]
- 4.Baum BJ (1993) Principles of saliva secretion. Ann NY Acad Sci 694:17–23 [DOI] [PubMed] [Google Scholar]
- 5.Vitolo JM, Baum BJ (2002) The use of gene transfer for the protection and repair of salivary glands. Oral Dis 8:183–191 [DOI] [PubMed] [Google Scholar]
- 6.Preston GM, Agre P (1991) Isolation of the cDNA for erythrocyte integral membrane protein of 28 kilodaltons: member of an ancient channel family. Proc Natl Acad Sci US A 88:11110–11114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baum BJ, Wellner RB, Zheng C (2002) Gene transfer to salivary glands. Int Rev Cytol 213:93–146 [DOI] [PubMed] [Google Scholar]
- 8.Mastrangeli A, O’Connell B, Aladib W, Fox PC, Baum BJ, Crystal RG (1994) Direct in vivo adenovirus-mediated gene transfer to salivary glands. Am J Physiol 266:G1146–G1155 [DOI] [PubMed] [Google Scholar]
- 9.McGrory WJ, Bautista DS, Graham FL (1988) A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology 163:614–617 [DOI] [PubMed] [Google Scholar]
- 10.Graham FL, Smiley J, Russell WC, Nairn R (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–74 [DOI] [PubMed] [Google Scholar]
- 11.Chiorini JA, Wendtner CM, Urcelay E, Safer B, Hallek M, Kotin RM (1995) High-efficiency transfer of the T cell co-stimulatory molecule B7–2 to lymphoid cells using high-titer recombinant adeno-associated virus vectors. Hum Gene Ther 6:1531–1541 [DOI] [PubMed] [Google Scholar]
- 12.Park JY, Lim BP, Lee K, Kim YG, Jo EC (2006) Scalable production of adeno-associated virus type 2 vectors via suspension transfection. Biotechnol Bioeng 94:416–430 [DOI] [PubMed] [Google Scholar]
- 13.Qu G, McClelland A, Wright JF (2000) Scaling-up production of recombinant AAV vectors for clinical applications. Curr Opin Drug Discov Devel 3:750–755 [PubMed] [Google Scholar]
- 14.Clark KR, Voulgaropoulou F, Johnson PR (1996) A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity titration of adeno-associated virus vectors. Gene Ther 3:1124–1132 [PubMed] [Google Scholar]
- 15.Conway JE, Rhys CM, Zolotukhin I, Zolotukhin S, Muzyczka N, Hayward GS, Byrne BJ (1999) High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene Ther 6:986–993 [DOI] [PubMed] [Google Scholar]
- 16.Gao GP, Lu F, Sanmiguel JC, Tran PT, Abbas Z, Lynd KS, Marsh J, Spinner NB, Wilson JM (2002) Rep/Cap gene amplification and high-yield production of AAV in an A549 cell line expressing Rep/Cap. Mol Ther 5:644–649 [DOI] [PubMed] [Google Scholar]
- 17.Urabe M, Ding C, Kotin RM (2002) Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Sene Ther 13:1935–1943 [DOI] [PubMed] [Google Scholar]
- 18.Vitolo JM, Cotrim AP, Sowers AL, Russo A, Wellner RB, Pillemer SR, Mitchell JB, Baum BJ (2004) The stable nitroxide Tempol facilitates salivary gland protection during head and neck irradiation in a mouse model. Clin Cancer Res 10:1807–1812 [DOI] [PubMed] [Google Scholar]
- 19.Cotrim AP, Hyodo F, Matsumoto K, Sowers AL, Cook JA, Baum BJ, Krishna MC, Mitchell JB (2007) Differential radiation protection of salivary glands versus tumor by Tempol with accompanying tissue assessment of Tempol by magnetic resonance imaging. Clin Cancer Res 13:4928–4933 [DOI] [PubMed] [Google Scholar]
- 20.Walters RW, Yi SM, Keshavjee S, Brown KE, Welsh MJ, Chiorini JA, Zabner J (2001) Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J Biol Chem 273:20610–20616 [DOI] [PubMed] [Google Scholar]
- 21.Smith RH, Afione S, Kotin RM (2002) Transposase-mediated construction of an integrated adeno associated virus type 5 helper plasmid. Biotechniques 208:210–211 [DOI] [PubMed] [Google Scholar]