

Summary
Irisin is secreted by muscle, increased with exercise and mediates certain favorable effects of physical activity. In particular, irisin has been shown to have beneficial effects in adipose tissues, brain and bone. However, the skeletal response to exercise is less clear and the receptor for irisin has not been identified. Here we show that irisin binds to proteins of the αV class of integrins and biophysical studies identify interacting surfaces between irisin and αV/β5 integrin. Chemical inhibition of the αV integrins blocks signaling and function by irisin in osteocytes and fat cells. Irisin increases both osteocytic survival and production of sclerostin, a local modulator of bone remodeling. Genetic ablation of FNDC5/irisin completely blocks osteocytic osteolysis induced by ovariectomy, preventing bone loss and supporting an important role for irisin in skeletal remodeling. The identification of the irisin receptor should greatly facilitate our understanding of irisin’s function in exercise and human health.
GRAPHICAL ABSTRACT
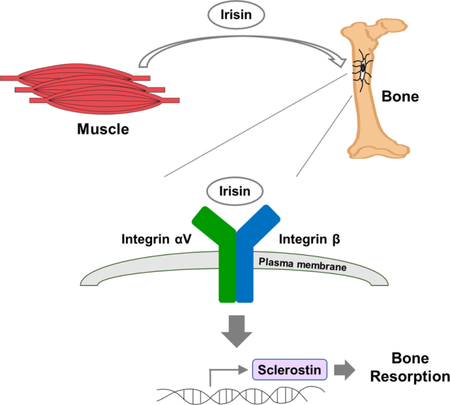
In Brief
Irisin, thorugh its integrin receptor, promotes skeletal remodeling with implications for stemming bone loss
INTRODUCTION
Physical activity has been shown to benefit several metabolic disorders, including obesity, diabetes and fatty liver disease (Kirwan et al., 2017). Older cross-sectional studies suggested exercise might prevent age-related bone loss (Krolner et al., 1983; Prince et al., 1991). Loss of bone mass with age has significant socio-economic and medical implications due to the heightened susceptibility to fractures. Osteoporosis impairs mobility, increases co-morbidities, reduces quality of life and can shorten lifespan, particularly in the elderly (Li et al., 2017).
The evidence that an exercise program can prevent bone loss is somewhat conflicted in part because different types of physical activity impact the skeleton at distinct sites in different ways. For example, several studies have shown that resistance training is associated with relative preservation of femoral but not lumbar bone mass in adults (Eatemadololama et al., 2017; Spindler et al., 1997; Vincent and Braith, 2002). On the other hand, fracture risk reduction has not been established in randomized trials with long term physical activity. Importantly, results from endurance exercise trials, particularly in the elderly, are even less convincing, with some studies showing preservation of bone mass and others showing no effect or even bone loss (Braam et al., 2003; Duckham et al., 2013; Scofield and Hecht, 2012). Consistent with the latter effect, brief bouts of endurance training have been shown to increase bone resorption and stimulate sclerostin, an endogenous inhibitor of bone formation (Baron and Kneissel, 2013; Kohrt et al., 2018; Pickering et al., 2017). Sclerostin is produced almost exclusively by osteocytes, the ‘command and control’ cells of the bone remodeling unit (Bonewald, 2011; van Bezooijen et al., 2004). Osteocytes arise from mature osteoblasts, are imbedded in the cortical matrix, and comprise nearly 90% of the cellular composition of bone (Bonewald, 2011). As such, they are thought to be the transducers of mechanical signals arising from physical activity and loading (Bonewald, 2011). In turn, these cells, through an elaborate network of canaliculi, communicate with both osteoblasts and osteoclasts, tightly regulating remodeling (Bonewald, 2011). Emerging evidence suggests that osteocytes can also directly resorb bone during periods of excessive calcium demand (Qing and Bonewald, 2009) or after ovariectomy (Almeida et al., 2017) and as such these cells have become a prime target for anabolic osteoporotic therapies such as parathyroid hormone and monoclonal anti-sclerostin antibodies (Bellido et al., 2005; Keller and Kneissel, 2005; Li et al., 2009; Ominsky et al., 2010). Anti-sclerostin antibodies increase bone mass dramatically in humans but also may have cardiovascular side-effects that could limit their use in practice (McClung, 2017).
Physical activity doesn’t only target osteocytes but also stimulates the production of several hormone-like molecules from skeletal muscle termed “myokines” (Pedersen and Febbraio, 2012). These include IL-6, irisin and meteorin-like (Bostrom et al., 2012; Keller et al., 2001; Rao et al., 2014). Irisin has been shown to be induced in many (but not all) studies of endurance exercise in both mice and humans (Bostrom et al., 2012; Jedrychowski et al., 2015; Lee et al., 2014; Pekkala et al., 2013). It is a cleaved product from a type I membrane protein, fibronectin type III domain-containing protein 5 (FNDC5), and is shed into the extracellular milieu and circulation (Bostrom et al., 2012). The crystal structure of irisin has been determined and contains an FNIII domain (Schumacher et al., 2013) that is also contained in fibronectin and many other proteins (Bork and Doolittle, 1992; Hynes, 1973; Potts and Campbell, 1994). FNIII domains in polypeptides are quite common, with over 200 polypeptides having these motifs (Bork and Doolittle, 1992; Potts and Campbell, 1994). Importantly, they bind to a wide range of different receptors, including fibroblast growth factor receptor and hemojuvelin (Kiselyov et al., 2003; Yang et al., 2008). Irisin is found in human blood at concentrations of 3–5 ng/ml (Jedrychowski et al., 2015); it has been shown to induce adipose tissue browning when FNDC5 is expressed in the liver through adenoviral vectors, resulting in elevated irisin serum levels (Bostrom et al., 2012). However, the full range of irisin’s effects are just beginning to be explored and, critically, the functioning receptor for irisin has not yet been identified.
Several recent papers have shown that irisin injections can impact skeletal remodeling. For example, very low dose irisin injections, given intermittently, were shown to improve cortical bone mineral density and strength in mice (Colaianni et al., 2015; Colaianni et al., 2017). These effects were consistent with in vitro studies showing that irisin could enhance osteoblast differentiation (Qiao et al., 2016). However, no studies have examined the effects of irisin on the osteocyte, a major regulator of bone structure and function and a cell type critical in the mediation of both mechanical and chemical signals. In addition, the effects of genetic manipulation of FNDC5/irisin on bone have not been reported. In this paper we demonstrate that irisin functions through a subset of αV integrin receptors to promote osteocyte survival and sclerostin secretion. Moreover, genetic deletion of FNDC5/irisin in C57BL/6J mice results in complete resistance at the trabecular and cortical compartments to ovariectomy-induced bone loss. These data identify the functioning receptor for irisin and should facilitate studies of irisin therapeutics for bone and other tissues responsive to physical activity.
Results
Irisin treatment induces the expression of sclerostin in osteocytes for bone remodeling.
To study the functional roles of irisin in osteocytes, we used the MLO-Y4 (osteocyte-like) cell line (Kato et al., 1997). Osteocytes are lost with aging and their death is thought to be an important component in the pathogenesis of age-related osteoporosis. Treatment with hydrogen peroxide has been previously used in these osteocyte-like cells as an assay for apoptotic death (Kitase et al., 2018). Therefore, MLO-Y4 cells were treated with irisin in the presence of hydrogen peroxide at amounts sufficient to induce apoptosis (Figure 1A). Irisin treatment reduced hydrogen peroxide-induced apoptosis at concentrations of 1–500ng/ml. Importantly, these effects were seen within the physiological concentration found in human plasma (3– 5ng/ml) (Jedrychowski et al., 2015) (Figure 1A). Since exercise also raises the levels of plasma sclerostin, a specific product of osteocytes that causes bone resorption and initiates bone remodeling, we also examined expression of this hormone with irisin treatments. Irisin raised the mRNA level of sclerostin in the osteocyte cultures in a dose-dependent manner (Figure 1B). To examine the regulation by irisin in vivo, we injected recombinant irisin protein daily into mice for 6 days (see methods). As shown in Fig. 1C and D, these injections raised the sclerostin mRNA level in osteocyte-enriched bones, as well as the protein level in plasma even though a half-life of recombinant irisin in vivo is less than an hour (Figure S1). These results demonstrate that irisin can protect osteocytes against apoptosis in culture and induce the expression of sclerostin, a key regulator of bone remodeling, in vivo.
Figure 1. Irisin blocks osteocyte cell death and stimulates sclerostin expression at the mRNA and protein levels.
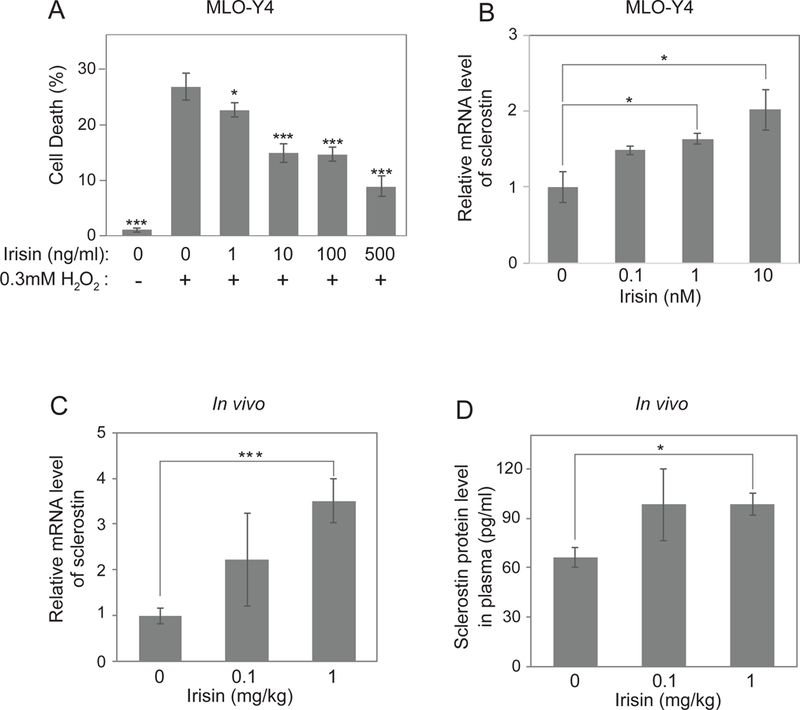
(A) MLO-Y4 cells were treated with indicated concentrations of irisin and hydrogen peroxide followed by analysis of the percentage of cell death. (B) MLO-Y4 cells were seeded and incubated until 60% cell density. The cells were incubated with Freestyle293 medium for 4 hours and were treated with indicated concentrations of irisin for 16 hours. Sclerostin mRNA level was analyzed by qRT-PCR. Cyclophilin was used as a control house-keeping gene. (C-D) 8-week-old wild-type C57BL/6J mice were daily injected with indicated dose of irisin for 6 days. Sclerostin mRNA level from osteocyte-enriched tibia was analyzed by qRT-PCR (C). Cyclophilin was used as a house-keeping gene. Plasma was collected to analyze the sclerostin protein level by ELISA kit (D). Data are represented as mean ± SEM. For C and D, n = 5 animals/group. *; p<0.05, ***; p<0.001
Deletion of FNDC5 prevents ovariectomy-induced trabecular bone loss by inactivating osteocytic osteolysis and osteoclastic bone resorption.
To investigate if irisin plays a role in the endogenous processes of normal bone resorption and remodeling, we first analyzed the femur in mice null for FNDC5 (the precursor of irisin) and littermate wild type mice at 5 months of age (see methods). FNDC5 null mice had significantly lower level of RANKL mRNA in whole bones both in male and female while OPG was not significantly different (Figure S2B-C). RANKL is a key factor in osteoclast activation, so we also analyzed the microarchitecture of bones. FNDC5 null mice had significantly higher femoral trabecular bone mass and greater connectivity density than wild-type mice (Table S1), which is consistent with lower bone resorption and reduced expression of RANKL; on the other hand, there were no differences in cortical bone indices (Table S1). In male mice, there were no differences in bone mass, either in the cortical or trabecular compartment (Table S1).
To further investigate the role of irisin in bone resorption, particularly in this pathological context, we performed ovariectomy (OVX) (Idris, 2012) in mice null for FNDC5 and their littermate controls. As expected, ovariectomy increased bone resorption and caused bone loss in wild-type mice, compared to the sham operated group (Figure 2A-D, Figure S3). This was apparent by the ratio of bone volume to total bone volume, trabecular number and the separation between trabeculae in the lumbar vertebrae (Figure 2E-G, Table S2). However, FNDC5 null mice were strikingly resistant to OVX-induced trabecular bone loss (Figure 2A-D, Figure S3). The maintenance of bone mass in the absence of estrogen in FNDC5 null mice was principally due to marked reduction in bone resorption. (Figure 2H-J, Table S2). Consistent with the lack of resorption in the OVX’d null mice, whole bone RANKL mRNA remained unchanged (Figure S2E). On the other hand, there were no differences in osteoblast number or bone formation rate for the OVX’d FNDC5 null mice compared to OVX’d wild-type mice (Table S2). To ascertain the mechanism responsible for the absence of bone loss and lack of change in RANKL with estrogen deficiency in the FNDC5 KO mice, we compared cortical bone histologically from both controls and null mice after OVX. In the FNDC5 null mice there was a striking lack of osteocytic osteolysis and lacunae enlargement (Figure 3A-E, Table S3) compared to OVX’d control mice, whose cortical bone was characterized by marked enlargement in osteocytic lacunae due to enhanced osteocytic osteolysis (Figure 3A-E, Table S3). Taken together, these data indicate that FNDC5/irisin is required for ovariectomy-induced osteolysis and strongly suggest that endogenous FNDC5/irisin induces bone resorption, at least partly through its actions on osteocytes.
Figure 2. Irisin/FNDC5 global knockout mice are resistant to OVX-induced trabecular bone loss at 9 months age.
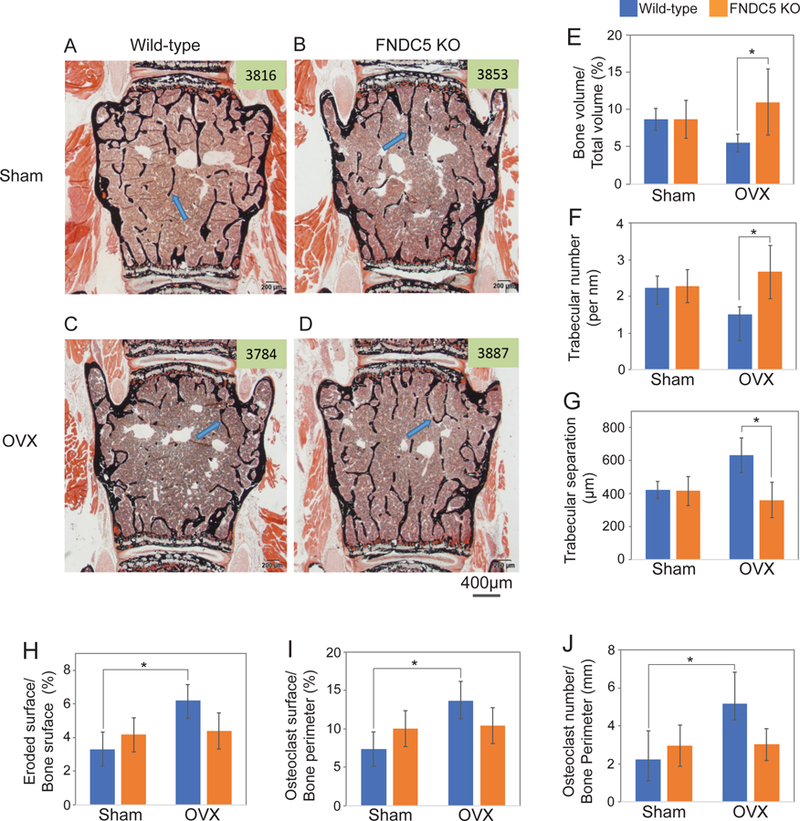
Ovariectomy (OVX) was performed on 9-month-old global FNDC5/irisin knockout mice (FNDC KO) and wild-type littermate mice (WT). (A-D) Representative figures of Von Kossa stained lumbar vertebra from wild-type mice or FNDC5/irisin knockout mice after OVX. Mineralized bone was stained black. Arrow indicates mineralized bone. (E-J) The bone histomorphometric analysis was performed in the lumbar vertebra. Data are represented as mean ± SEM. n = 4–7 animals/group. See also Figure S2–3 and Table S2. *; p<0.05
Figure 3. Deletion of Irisin/FNDC5 prevented OVX-induced osteocytic osteolysis at 9 months of age.
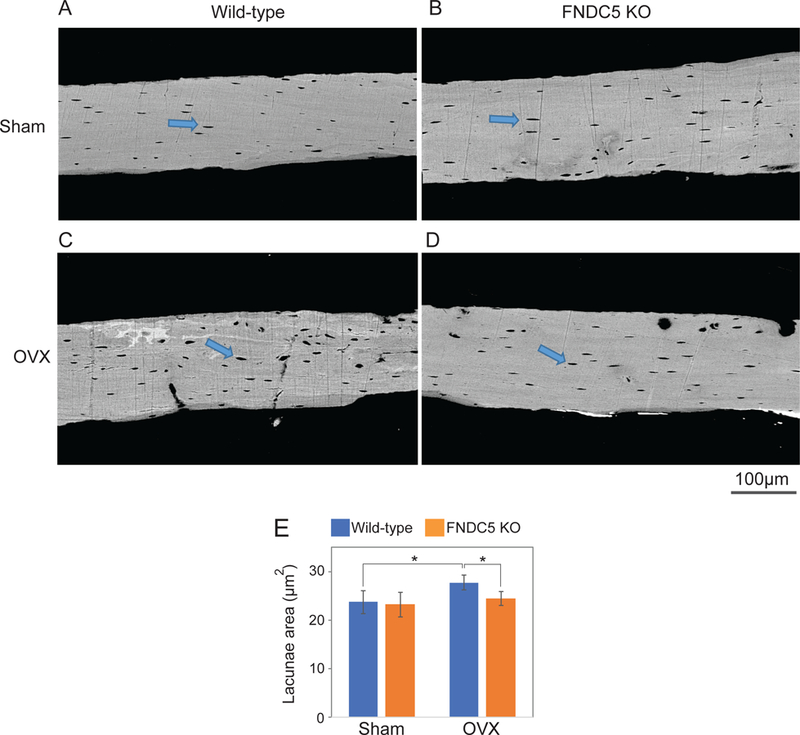
Tibia samples from the OVX experiment in Fig. 2 were analyzed to measure lacunae area using backscatter scanning electron microscopy. (A-D) Representative figures. Arrow indicates lacunae. (E) Analyzed lacunae area. Data are represented as mean ± SEM. n = 4–7 animals/group. See also Table S3. *; p<0.05.
In light of these data, we asked whether ovariectomy changed irisin levels. OVX was performed in 8-week-old wild-type mice; irisin was measured in plasma 2 weeks after OVX using quantitative mass spectrometry by the AQUA method (Jedrychowski et al., 2015). Control (sham operated) mice had 0.3 ng/ml of irisin in plasma, while the OVX mice had 2.4 fold more (Figure S2G). Interestingly, this is 10 fold less than healthy young human males (Jedrychowski et al., 2015).
Quantitative proteomic analysis identified integrin β1 as a candidate for the irisin receptor and irisin treatment triggers integrin-like signaling.
The irisin receptor has not been identified. Since our data showed that MLO-Y4 osteocytes directly respond to low concentration of irisin, we used these cells to identify its receptor. Irisin with a his-tag or an identically tagged control protein (Adipsin) were first incubated with intact cell surfaces at 4°C. A chemical cross-linker was then added and incubated with cells, and the ligands were re-purified with (presumptive) cellular proteins covalently attached. The cross-links were then reversed and the products were subjected to quantitative mass spectrometry (Figure 4A). This quantitative proteomic analysis, using isobaric tagging, revealed five cell surface proteins as potential receptor candidates for irisin (Figure 4B, Table S4). Among them, only integrin β1 is known to bind protein ligands and to trigger downstream signaling. Integrin β1 (like all β-integrins) binds α-integrins to form obligate heterodimers. These heterodimers, upon ligand binding, usually trigger canonical signaling by phosphorylation of focal adhesion kinase (FAK), AKT, and cAMP response element-binding protein (CREB) (D’Amico et al., 2000; Giancotti and Ruoslahti, 1999; Schaller et al., 1994) (Figure 4C). In response to ligand binding to many integrins, FAK is auto-phosphorylated on tyrosine 397 and then downstream signaling follows (Giancotti and Ruoslahti, 1999). MLO-Y4 cells were treated with irisin at 10nM or norepinephrine at the same concentration (as a positive control for phosphorylation of CREB); irisin treatment caused phosphorylation of FAK in 1 minute and the signal decreased after 10 minutes (Figure 4D). AKT was phosphorylated on threonine 308 while phosphorylation of serine at amino acid 473 was not induced. Additionally, CREB was phosphorylated after 5 minutes with irisin and as expected, norepinephrine also did this (Figure 4D). The dose response of these signaling events was then examined. Treatment of these osteocytes with irisin doses as low as 10pM induced the phosphorylation of FAK (Figure 4E). Zyxin, another downstream protein of the integrin signaling pathway (Brancaccio et al., 2006), was phosphorylated potently as well (Figure 4E). These data show that irisin stimulates a very potent pathway of integrin-like signaling.
Figure 4. Irisin stimulates a very potent pathway of “integrin-like” signaling including pFAK, pZyxin and pCREB.
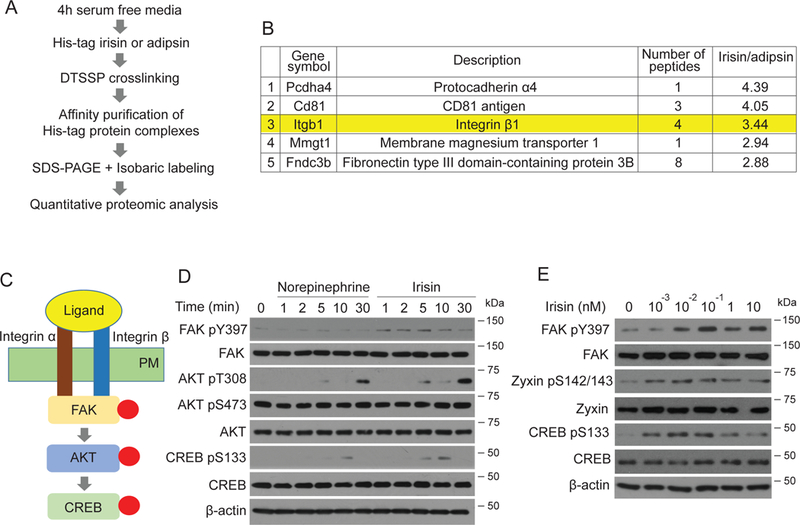
(A) Scheme of crosslinking/co-immunoprecipitation/mass spectrometry experiments to identify irisin receptors. (B) Top 5 enriched proteins with irisin versus adipsin. See also Table S4 for full list. (C) Model of canonical integrin signaling. Integrin heterodimer binds to its ligand. The interaction results in phosphorylation of FAK and Zyxin, followed by phosphorylation of AKT (at T308) and CREB. PM is plasma membrane. (D-E) MLO-Y4 cells were seeded and incubated until 60% cell density. The cells were incubated with FreeStyle293 medium for 4 hours and were treated for indicated time with 10nM norepinephrine or irisin (D) or indicated concentrations of irisin for 10 minutes (E). Cells were lysed to detect the indicated protein level using immunoblot analysis.
Irisin binds directly to integrin complexes through an RGD-analogous motif of irisin and well-known ligand-binding motifs within integrin αV/β5.
To determine whether irisin binds directly to integrins, we performed a binding assay using purified recombinant irisin and many integrin complexes that were commercially available (Figure 5A). Most integrin complexes showed relatively weak binding to irisin (Figure S4A). In particular, several of the β1-containing complexes showed binding to irisin above the background (Figure 5A). However, αV/β5 integrin, both murine and human, showed by far the highest extent of binding.
Figure 5. Irisin directly interacts with integrin complexes and mapping of binding motifs.
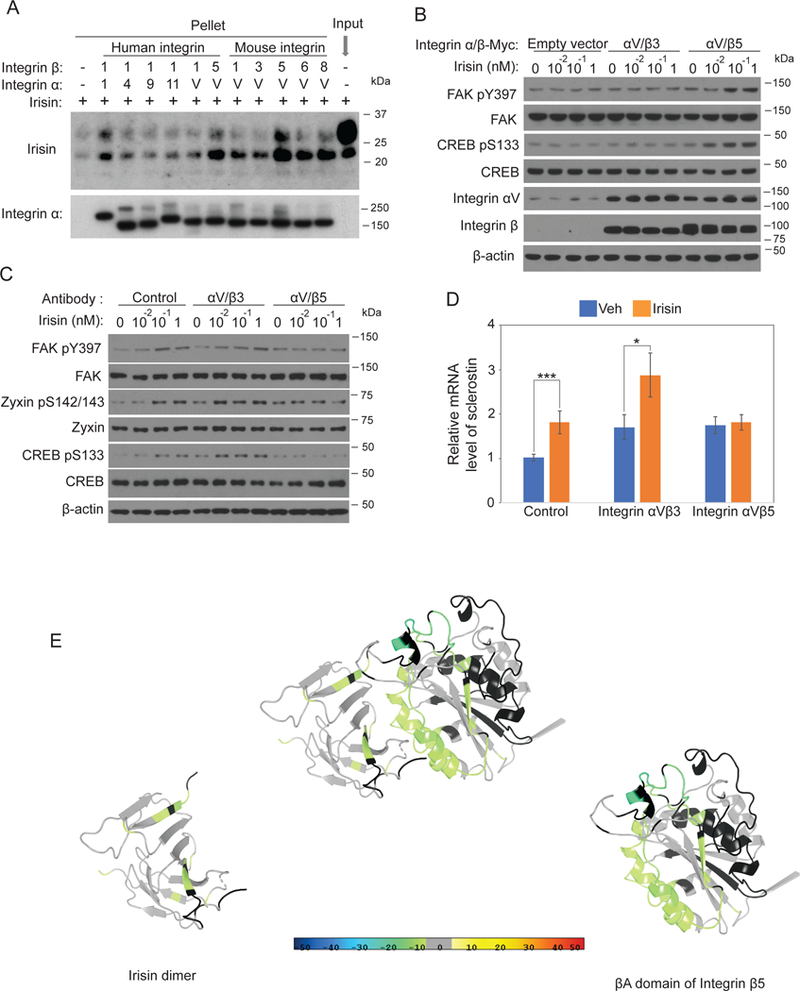
(A) 100nM irisin was incubated with 5nM indicated his-tag integrins followed by immunoprecipitation using Ni-NTA agaroses. Precipitated integrins and co-precipitated irisin were analyzed by immunoblot analysis. (B) HEK293T cells were seeded and incubated until 50% cell density. The cells were transfected with 0.1µ g plasmids of indicated integrins. After 48 hours, the cells were incubated with Freestyle293 medium for 3 hours and were treated with indicated concentration of irisin for 5 minutes. Cells were lysed to detect the indicated protein level using immunoblot analysis. (C) MLO-Y4 cells were treated as described in Fig. 4E with addition of pretreatment of indicated antagonistic antibodies for 10 minutes. Cells were lysed to detect the indicated protein level using immunoblot analysis. (D) MLO-Y4 cells were treated as described in Fig. 1B except with addition of the pretreatment of indicated antagonistic antibodies for 10 minutes. Sclerostin mRNA level was analyzed by qRT-PCR. Cyclophilin was used for house-keeping gene. (E) Docking model of interaction between irisin and integrin αV/β5 (see Methods). The ribbon diagram is colored by HDX stabilization/destabilization. Percentages of deuterium differences are color-coded according to the smooth color gradient key at the bottom of Fig. 5. Crystal structure of irisin dimer is from Protein Data Bank (PDB) (4lsd) and a homology model of integrin β5 was built based on integrin β3 structure from PDB (4MMX).
Using quantitative proteomics with mass spectrometry (spectral counting method), we analyzed expression of multiple integrins in MLO-Y4 that bind to irisin. Integrin αV is the most abundant integrin protein in MLO-Y4 cells, followed by integrin β1, integrin α5, integrin β5 and integrin β3 (Table S5). We also observed minor amounts of integrin β6 and integrin β8. Therefore, we focused mainly on integrin αV/β1, integrin αV/β3, integrin αV/β5 and integrin α5/β1 in cell culture experiments.
Gain of function experiments were next performed, using ectopic expression of integrin subunits in cultured HEK293T cells. These cells showed little basal signaling in response to irisin; cells with forced expression of integrin αV/β5 but not of integrin αV/β3 showed an enhanced level of phosphorylation of FAK upon irisin treatment (Figure 5B). As a positive control, the cells were treated with vitronectin, a ligand for integrin αV family, in the presence of integrin αV/β3 or integrin αV/β5. Vitronectin treatment induced phosphorylation of FAK in both, indicating that the integrins are active forms (Figure S4B). In addition to integrin αV/β5, irisin treatment increased FAK phosphorylation after forced expression of the integrin αV/β1 (Figure S4C). However, cells with forced expression of an empty vector, integrin α5/β1, or integrin α11/β1 showed little phosphorylation of FAK above background upon irisin treatment (Figure S4D).
The response of these cells to irisin was also tested in a loss of function format, namely in the presence of antagonistic antibodies against integrin αV/β3 or integrin αV/β5. MLO-Y4 cells were treated with control mouse monoclonal IgG, or antagonistic antibodies against integrin αV/β3 or integrin αV/β5 before irisin treatment. We observed that anti-integrin αV/β5 completely blocked the irisin-mediated phosphorylation of FAK, Zyxin and CREB, while control Igg or the anti-integrin αV/β3 did not block signaling (Figure 5C). We also observed the same pattern in the irisin-mediated sclerostin gene expression (Figure 5D). These results, taken together, indicate that integrin αV/β5 has both the highest affinity for irisin and is required for the cellular response to irisin; certain other integrins such as αV/β1 also have a significant affinity and response. Importantly, the well-known integrin αV/β3 complex does not trigger a response to irisin in this osteocyte-like cell line.
To confirm a direct interaction between irisin and integrin αV/β5 and to help identify which domains in both irisin and αV/β5 integrin participate in this binding event, we used differential hydrogen-deuterium exchange linked to mass spectrometry (HDX/MS). HDX/MS measures deuterium incorporation of peptides via exchange of backbone amide hydrogens which is sensitive to hydrogen bonding and solvent accessibility. If the protein-protein interaction occurred, we would expect a reduction of solvent exchange in the regions of the protein driving the interaction. We performed the experiment as a differential comparing integrin αV/β5 ± saturating irisin and irisin ± saturating integrin αV/β5. HDX/MS identified putative binding regions in the βA domain of integrin β5 which are stabilized (reduction in solvent exchange) when irisin is bound (Figure S5A). Interestingly, these regions or motifs in integrin β5 have been previously reported to interact with ligands such as fibronectin, osteopontin and vitronectin (Hu et al., 1995; Humphries et al., 2006; Marinelli et al., 2004; Smith et al., 1990; Van Agthoven et al., 2014). HDX/MS also identified a putative integrin-binding region of irisin at amino acids 60– 76 and 101~118 (Figure S5B). Interestingly, this region of irisin is proximal to that which has been suggested as a candidate for receptor binding site based on crystal structural similarity with fibronectin (Schumacher et al., 2013). Moreover, the three-dimensional structure of the proximal motif (amino acid 55–57) is very similar to the well-known “RGD” motif in fibronectin, even though irisin does not have the key amino acid primary sequence(RGD) except for aspartic acid (XXD) (Schumacher et al., 2013). Likely the direct interaction of this loop motif with integrin further stabilizes the proximal region of irisin leading to reduced solvent exchange (Figure S5C). The direct interaction of other identified motifs with integrin also has the same pattern as well (Figure S5D-E). These results demonstrate that irisin directly binds integrin αV/β5 and the regions within each protein that are protected from solvent exchange allow us to make a working model of its three-dimensional interaction (Figure 5E). Further studies will need to be performed to refine this model.
Other integrin inhibitors prevent irisin-induced signaling and sclerostin expression.
Certain peptides with an RGD motif are well-known inhibitors that prevent integrin-ligand binding and function (Plow et al., 2000; Plow et al., 1987).While irisin does not contain an RGD sequence, irisin has a loop that has close structural similarity with certain RGD motifs (Schumacher et al., 2013) and this loop is used by irisin to bind to integrin αV/β5 (Figure 5E).
Therefore, we tested whether RGD inhibitory peptides block the interaction between integrins and irisin. As shown in Fig. 6A, the RGDS peptide, which is a commercially available form of the RGD peptide, dramatically suppressed irisin-induced phosphorylation of FAK, Zyxin, and CREB (Figure 6A). To test whether the αV integrins are major components for FAK signaling in the osteocytes, cells were treated with echistatin, an inhibitor known to affect primarily integrin αV complexes (Kumar et al., 1997). Echistatin also effectively prevented irisin signaling (Figure 6B). In addition, we tested irisin-induced signaling with other specific inhibitors for integrin αV, such as cyclo RGDyK and SB273005 (Chen et al., 2004; Dechantsreiter et al., 1999; Lark et al., 2001; Miller et al., 2000; Yu et al., 2014). These inhibitors all block irisin-induced signaling (Figure S6A).
Figure 6. Integrin inhibitors such as RGD peptide and echistatin block irisin-induced signaling and gene expression.
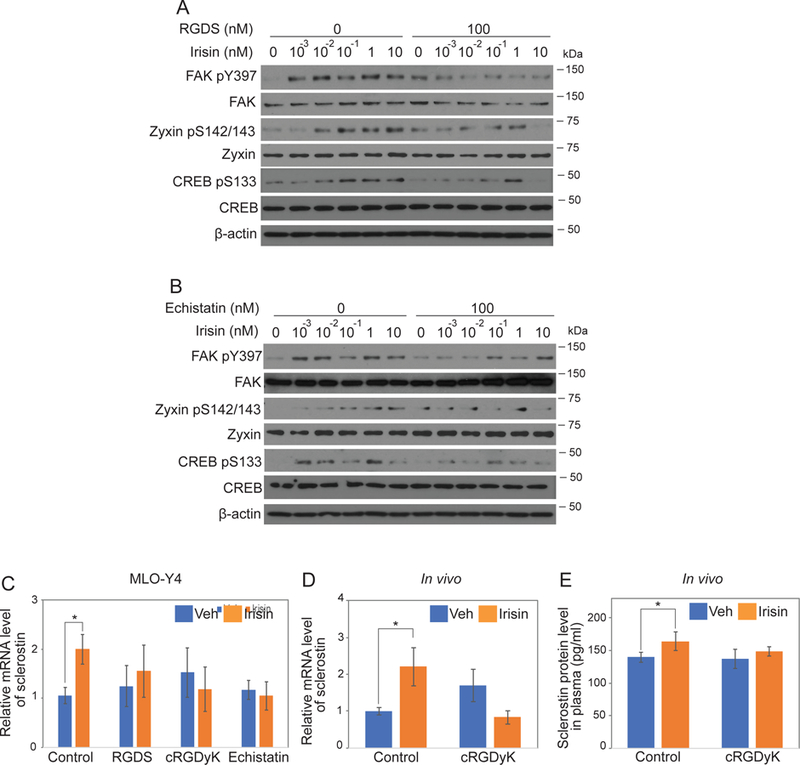
(A-B) MLO-Y4 cells were treated as described in Fig. 4E with addition of pretreatment of integrin inhibitors, RGDS peptide or echistatin. Cells were lysed to detect the indicated protein level using immunoblot analysis. (C) MLO-Y4 cells were treated as described in Fig. 1B except with addition of the pretreatment of integrin inhibitors for 10 minutes. (D-E) 8-week-old wild- type C57BL/6J mice were treated as described in Fig. 1C-D except co-injection of 1mg/kg cyclo RGDyK (cRGDyK). Data are represented as mean ± SEM. For C and D, n = 9–12 animals/group. *; p<0.05.
We also tested whether cyclo RGDyK blocked the irisin-integrin αV/β5 signaling in a dose- dependent manner. After forced expression of integrin αV/β5 in HEK293T cells, cyclo RGDyK was co-treated with irisin. Immunoblot data showed that 10nM cyclo RGDyK prevented phosphorylation of FAK significantly and 100nM cyclo RGDyK blocked the phosphorylation completely, indicating that IC50 is 10~50nM in the presence of irisin (Figure S6B). We then extended these observations to the level of gene expression: MLO-Y4 cells were treated with irisin in the presence of a negative control RGD peptide, RGD peptide or cyclo RGDyK and echistatin (Figure 6C). In the presence of control RGD peptide, irisin raised the mRNA level of sclerostin, while these inhibitors all prevented sclerostin induction. We also injected the irisin peptide into wild-type mice, in combination with control RGD peptide or cyclo RGDyK, an integrin inhibitor that is widely used for in vivo studies (Chen et al., 2004; Guo et al., 2014) (Figure 6D-E). Cyclo RGDyK prevented the irisin-induced gene expression of sclerostin in osteocyte-enriched bones, as well as the protein level in plasma. Additionally, SB273005, which has a higher affinity to integrin αV/β5 than integrin αV/β3, was also employed. As shown in Fig. S6C, SB273005 significantly prevented the irisin-induced gene expression in vivo. These results together strongly suggest that irisin acts on integrin αV family and integrin αV/β5 is particularly important in the functions of irisin on osteocyte cells.
Integrins mediate the irisin-induced thermogenic gene program.
It has been shown that irisin raised the expression of Ucp1 and other thermogenic genes in fat cells. (Bostrom et al., 2012; Huh et al., 2014; Lee et al., 2014). Furthermore, thermogenic gene expression was also elevated when FNDC5 was expressed from the liver with adenoviral vectors and irisin was released in the circulation (Bostrom et al., 2012). To examine whether recombinant irisin induced the thermogenic gene expression in vivo, we injected recombinant irisin into wild-type mice for one week; irisin treatment increased the mRNA level of Ucp1 more than 2-fold (Figure 7A). The protein level in whole tissue, as detected by western blots, was also increased by the irisin injections (Figure 7B). To test whether integrins mediate these effects, we injected the irisin peptide with control RGD peptide or cyclo RGDyK. As shown in Fig. 7C and D, cyclo RGDyK blocked the irisin-induced gene expression of Ucp1 and Dio2 as well as the induction of the protein level of Ucp1. We also observed that recombinant irisin treatment increased the gene expression of Ucp1 in primary inguinal fat cells (Figure 7E). Proteomic data showed that in primary inguinal fat cells, integrin β1 is the most abundant followed by integrin β6, integrin α1, integrin β5, and integrin αV. Integrin β3 wasn’t detectable in these cells (Table S6). Cyclo RGDyK treatment prevented irisin-induced gene expression (Figure 7E), indicating that irisin also works on fat cells directly via integrin αV family. Thus, integrin αV complexes also appear to act as receptors for irisin in fat tissue, and mediate the irisin-induced thermogenic gene program.
Figure 7. Integrin mediates irisin-induced thermogenesis.
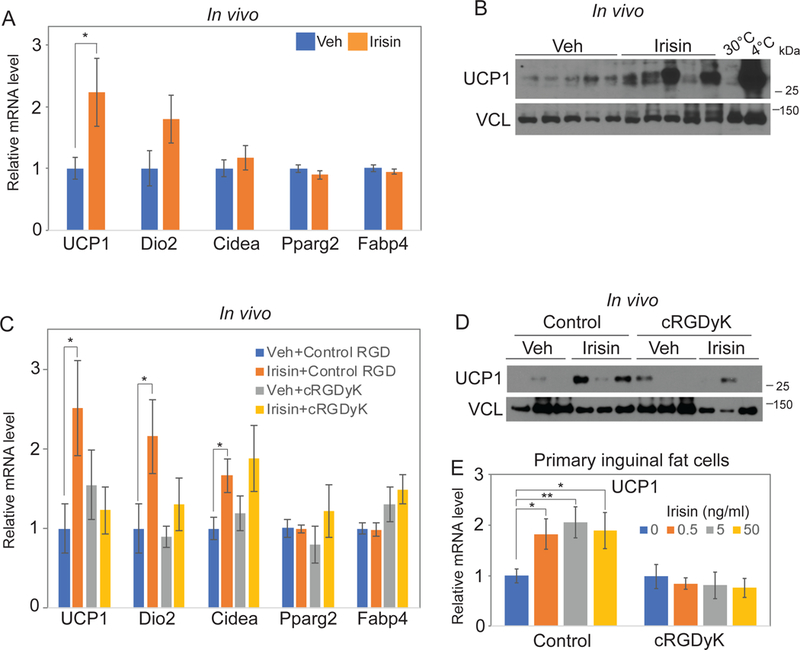
(A-B) 1mg/kg irisin was injected to 8-week-old wild-type C57BL/6J mice every other day for a week. mRNA levels of indicated genes in inguinal fat were analyzed by qRT-PCR. Cyclophilin was used for house-keeping gene (A). Inguinal fats were also lysed to detect the indicated protein level using immunoblot analysis (B). (C-D) Mice were treated and analyzed as (A-B) with addition of co-injection of 1m/kg control RGD peptide or cyclo RGDyK (cRGDyK). mRNA levels of indicated genes in inguinal fat were analyzed by qRT-PCR. Cyclophilin was used for house-keeping gene (C). Inguinal fats were also lysed to detect the indicated protein level using immunoblot analysis (D). (E) Primary inguinal fat cells were treated with indicated concentration of irisin with 10µM control peptide or cyclo RGDyK (cRGDyK) every other day during 6 days differentiation. Ucp1 mRNA level was analyzed by qRT-PCR. Cyclophilin was used as a control house-keeping gene. Data are represented as mean ± SEM. For A and B, n = 12–13 animals/group. For C and D, n = 11–13 animals/group. *; p<0.05, **; p<0.01.
Discussion
αV integrin complexes are irisin receptors.
Since its discovery in 2012, irisin has been reported to have various functions in many organs (Perakakis et al., 2017; Polyzos et al., 2018). These effects are related mainly to known benefits of exercise, such as strengthening bones, increasing energy expenditure and improving cognition (Bostrom et al., 2012; Colaianni et al., 2015; Colaianni et al., 2017; Lee et al., 2014; Wrann et al., 2013; Zhang et al., 2017). However, the mechanisms underlying these benefits were unclear, in large measure because the irisin receptor(s) had not been identified. Here we describe the irisin receptor as a subset of integrin complexes. Importantly, this conclusion is drawn from several independent lines of evidence. First, our quantitative proteomic analysis showed that irisin binds to osteocyte cells in a way that allows chemical cross-linking to integrin β1. Second, protein-protein binding assay using purified irisin and integrin complexes showed that irisin binds to several integrin complexes, including integrin α1/β1; however, integrin αVβ5 has the highest apparent affinity in these experiments. Third, HDX/MS also demonstrated that irisin binds to integrin αV/β5 and this analysis allowed mapping of binding motifs on both irisin and the integrin complex. Fourth, irisin activates signaling characteristic of integrin receptors. One of the main features of integrin signaling is the Y397 phosphorylation of FAK upon ligand binding; irisin treatment of osteocytes raised the phosphorylation level of FAK within one minute. Irisin is also incredibly potent in that 10pM irisin triggers this phosphorylation and other phosphorylation events known to occur with integrin signaling. Fifth, ectopic expression of integrin αV/β1 or integrin αV/β5 in cultured HEK293T cells showed that irisin can trigger elevated integrin signaling compared to cells transfected with empty vectors. Lastly, it is notable that well-characterized integrin inhibitors or an antagonistic antibody directed against integrin αV/β5 suppressed nearly all irisin-mediated signaling and its downstream gene expression. Taken together, these data prove that a subset of integrins, especially those involving αV integrin, are functional irisin receptors, at least in osteocytes and fat tissues.
Irisin binds to integrin αV/β5 in structural homology to other integrin ligands
The αV family of integrins has previously been reported to contribute to bone remodeling (Duong et al., 2000; Duong and Rodan, 1998; Thi et al., 2013). Interactions of the αV family of integrins with extracellular matrix proteins such as osteopontin and vitronectin lead to adhesion of osteoclasts to the bone surface followed by bone resorption (Duong et al., 2000; Duong and Rodan, 1998; Flores et al., 1992; Horton et al., 1991). HDX/MS experiment determined here that regions proximal to the RGD-like loop of irisin is involved in the interaction with integrin αV/β5. Interestingly, this loop (amino acids 55 to 57), was predicted as a potential receptor binding loop based on the structural similarity with an RGD-sequence containing loop in fibronectin (Schumacher et al., 2013). In addition, within integrin β5 subunit, the HDX/MS method identified putative binding motifs in the βA domain, which are also reported as the interaction site for RGD-containing ligands (Marinelli et al., 2004; Van Agthoven et al., 2014). Based on these data, the ability of RGD-mimetics to block both irisin-induced signaling and irisin-induced gene expression (Figure 6 and 7) is predictable from a mechanistic perspective.
Irisin regulates bone remodeling, targeting osteocytes.
Our studies reveal for the first time that osteocytes are direct targets of irisin, acting via the integrin αV family. Osteocytes use both mechanical and chemical sensation to maintain bone homeostasis (Bonewald, 2011) by directly controlling skeletal remodeling. With respect to the bone resorption component of skeletal remodeling, osteocytes regulate osteoclasts in two ways: First, by directly secreting RANKL, the most potent inducer of osteoclastogenesis, and second, by secreting sclerostin, an inhibitor of bone formation that also suppresses osteoprotogerin (OPG) a decoy receptor for RANKL. In the most common animal model of osteoporosis, OVX, the loss of estrogen triggers RANKL production and suppresses OPG, leading to greater RANKL bioactivity, increased bone resorption and ultimately bone loss, (Komori, 2015). Histologically this is manifested by greater numbers of osteoclasts on the bone surface and enhanced osteocytic osteolysis (Alameida, 2017). In our experiments, deletion of FDNC5 suppressed bone resorption, by blocking the increase in osteoclast number and eroded surfaces, thereby preventing bone loss after OVX. Furthermore, deficiency of FNDC5 inhibited OVX-induced perilacunar enlargement. a manifestation of osteocytic osteolysis, indicating that the phenotype is at least mediated partly through an inactivation of osteocyte function(s), as well as through inhibition of osteoclast number and function. In addition, we demonstrated that sclerostin was directly induced by irisin in vitro and in vivo. Of course, it is entirely possible that irisin has additional effects on other bone cells in the remodeling unit, as demonstrated by a previous literature (Colaianni et al., 2014).
These data and previous results from others (Colaianni et al., 2015; Colaianni et al., 2017) suggest that irisin could be a useful target for the treatment of osteoporosis. Although irisin targets bone resorption, intermittent treatment with irisin has been shown to improve bone density and strength (Colaianni et al., 2015; Colaianni et al., 2017). Considered within the light of our data, this may seem counter-intuitive. However, a comparable example of a peptide that both stimulates resorption and is anabolic when administered intermittently, is parathyroid hormone (i.e. PTH). Chronically high PTH levels drive bone resorption to maintain eucalcemia. Moreover, Kohrt et al recently demonstrated that during an acute bout of physical activity, serum calcium rapidly decreased and this drove a secondary increase in PTH. Yet it has been well established that intermittent PTH treatment is anabolic to the skeleton, at least over the first twelve months of therapy (Dempster et al., 2001; Lane et al., 1998). Therefore, it seems likely that irisin could both target bone resorption but also act on remodeling in a favorable manner with intermittent pulse dosing. On the other hand, the striking data that OVX-induced osteoporosis is entirely prevented in the FNDC5 KO mice, suggests another more conventional therapeutic approach: inhibition/neutralization of irisin or its receptors, the αV integrins.
Irisin induces a thermogenic program in fat through integrins.
Ucp1 and Dio2 are key proteins contributing to mitochondrial proton leak and thermogenesis in adipose tissues. Here we show that treatment of mice with recombinant irisin protein raised the expression of Ucp1 and Dio2 in subcutaneous (inguinal) adipose tissues, despite the very short half-life of irisin in vivo. Importantly, irisin’s effects on these thermogenic genes are also sensitive to simultaneous administration of the αV integrin inhibitor. This suggests the generality of the integrins, especially the αV integrins, as irisin receptors.
Irisin receptors, signaling and therapeutics
The identification of the irisin receptors as integrins in osteocytes and thermogenic fat suggests that the αV family of integrins complexes are likely to be the major irisin receptors in all tissues. However, it is important to note that nothing presented here rules out the possibility of other receptors for irisin within the integrin family or even outside of the integrins. Importantly, the identification of an irisin receptor and its signaling systems can be very useful as both a quality control for irisin preparations and for the development of irisin inhibitors. It is also likely that these data might allow for the development of irisin ligands that have altered activity, including proteins that can bind to different integrins among subsets of these receptors. Our findings should also contribute to the study of irisin in other organs where FNDC5 is also highly expressed, including brain, skeletal muscle itself and heart. Healthy humans have levels of circulating irisin in the 3–5ng/ml range and they are, on average, increased with exercise (Jedrychowski et al., 2015). As shown here, these are the levels of irisin that are quite sufficient to activate irisin receptors. Exercise brings well-known improvements in mood and cognition and there are already data suggesting that irisin might mediate some of these effects in the brain (Wrann et al., 2013). Data presented here should accelerate these and other studies of irisin.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bruce M. Spiegelman (bruce_spiegelman@dfci.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Animal experiments were performed per procedures approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. All strains were on a C57BL/6J background. Unless otherwise stated, mice were housed in a temperature-controlled (20–22°C) room on a 12-hour light/dark cycle. Mice were fed with chow diet. Fndc5 floxed mice were developed with the Texas A&M Institute for Genomic Medicine and crossed with EIIa-cre mice to generate germline deletion of Fndc5 (Exon 2 and 3). Experiments were performed with sex- and age-matched global FNDC5 knockout and littermate wild-type control mice. 9-month- old female mice were used for OVX experiment and analysis of vertebrae and femurs. 5-month- old female mice were used for µCT analysis of femurs and gene expression analysis of tibia. 8- week-old C57BL/6J wild-type female mice were ovariectomized and sacrificed after 2 weeks of OVX to measure irisin level in plasma. 8-week-old C57BL/6J wild-type male mice were used for irisin injection experiments.
Cell lines
MLO-Y4 cells were established from female mice and cultured as previously described (Kato et al., 1997). The cells were seeded on type I collagen-coated 6 well plates under MEMα medium (Thermo Fisher Scientific, 12571–063), 2.5% (v/v) Fetal Bovine Serum (Hyclone, SH30396.03,Lot AB217307), 2.5% (v/v) calf serum (Hyclone, SH30072.03, AAL11105), penicillin- streptomycin (P/S) 100 U/ml. Cells were treated with irisin or other reagents at 60% cell density. HEK293T cells were set up for experiments at 1 × 105 cells per well in 6 well plate under DMEM (Thermo Fisher Scientific, MT10017CV), 10% (v/v) Fetal Bovine Serum (Gemini BioProducts), penicillin-streptomycin (P/S) 100 U/ml. All cell lines were maintained at 37°C in 5% CO2
Primary white adipocyte cultures
Inguinal fat tissues from 6-week-old wild-type C57BL/6J female mice were dissected and washed with PBS, minced and digested for 1 hour at 37°C in PBS containing 10mM CaCl2, 2.4 U/ml dispase II (Roche) and 10 mg/ml collagenase D (Roche). After adding warm DMEM/F12 (1:1) with 10% FCS, digested tissue was filtered through a 70μm cell strainer and centrifuged at 600×g for 10 minutes. Pellet was resuspended by 40ml DMEM/F12 (1:1) with 10% FCS and filtered through a 40μm cell strainer followed by centrifugation at 600×g for 10 minutes. Pelleted inguinal stromal vascular cells were grown to confluence and split onto type I collagen-coated coated 12 well plates.
The cells were induced to differentiate by treatment with 1µ M rosiglitazone, 5µ M dexamethasone, 0.5µ M isobutyl methyl xanthine for 2 days. After that, cells were maintained in 1µM rosiglitazone for 4 days with medium change every other day. Cells were maintained at 37°C in 10% CO2
METHOD DETAILS
Expression and purification of human/mouse recombinant his-tag irisin
His-tag recombinant irisin was generated by transfection of an irisin (human/mouse)-10 his tag DNA plasmid. This protein with a C-terminal his tag was produced and purified from mammalian HEK293 cells after transient DNA transfection. The protein was purified from 250 ml conditioned media using IMAC column, followed by Superdex200 in 50mM HEPES pH7.2, 150mM NaCl. The protein was diluted in sterilized PBS to use in cell culture experiments and in vivo injection.
Cell culture experiments
MLO-Y4 cells were cultured as previously described (Kato et al., 1997). The cells were seeded on type I collagen-coated 6 well plates under MEMα medium (Thermo Fisher Scientific, 12571– 063), 2.5% Fetal Bovine Serum (Hyclone, SH30396.03, Lot AB217307), 2.5% calf serum (Hyclone, SH30072.03, AAL11105), penicillin-streptomycin (P/S) 100 U/ml. At 60% cell density, medium was switched to FreeStyle293 Expression medium after washing with warm PBS. After 4 hours incubation, the cells were treated with indicated doses of irisin for indicated times. For integrin inhibitor treatment, cells were treated with indicated concentration of the inhibitors for 10 minutes before irisin treatment. For antagonistic antibody treatment, cells were treated with 0.9μg/ml antagonistic antibodies against integrin αV/β3 or integrin αV/β5 οr monoclonal mouse IgG as a negative control for 10 minutes before irisin treatment. After treatments, medium was aspirated on ice and cold PBS was added to the cells. RIPA buffer for lysis was added after aspiration of cold PBS for immunoblot analysis.
For primary inguinal fat cells, the cells were induced to differentiate by treatment with 1 µ M rosiglitazone, 5 µM dexamethasone, 0.5 µM isobutylmethyl xanthine in the presence of 0, 0.5, 5 or 50ng/ml recombinant 10 his-tag irisin protein for 2 days. After that, cells were maintained in 1µM rosiglitazone in the presence of 0, 0.5, 5 or 50ng/ml recombinant 10 his-tag irisin protein for 4 days with medium change every other day. mRNA levels were analyzed as described in gene expression analysis.
Transient transfection
On day 0, HEK293T cells were set up for experiments at 1 × 105 cells per well in 6 well plate. On day 2, cells were transiently transfected with the indicated plasmids with FuGENE6 reagent (Roche Applied Science) according to the manufacturer’s protocol. After 24 hours of incubation, Freestyle 293 medium were added and the cells were incubated for 3 hours followed by treatment of indicated concentration of irisin for 5 minutes or by pre-treatment of 10μM cyclo RGDyK for 10 minutes and treatment of 0.3nM irisin for 5 minutes. After treatments, medium was aspirated on ice and cold PBS was added to the cells. RIPA buffer for lysis was added after aspiration of cold PBS for immunoblot analysis.
Animal studies
Animal experiments were performed per procedures approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. Experiments were performed with sex- and age-matched global FNDC5 knockout and littermate wild-type control mice. Female mice were initially ovariectomized to deplete ovarian hormones and induce osteoporosis. Mice were sacrificed after 3 weeks of OVX at the age of 36~38 weeks. 8-week-old C57BL/6J wild type mice were ovariectomized and sacrificed after 2 weeks of OVX to measure irisin level in plasma. The remaining uterine fundus, cervical region and vaginal vault were removed as a whole from the mice and weighed to ensure shrinkage from the ovariectomy procedure.
C57BL/6J wild-type male mice for recombinant irisin injection were acquired from The Jackson Laboratory (000664). Mice were mock injected with sterilized PBS for at least three days. We excluded mice if the mice lost more than 5% body weight during mock or irisin injections. For bone studies, the mice were injected with 1mg/kg irisin by daily intraperitoneal (IP) injection for 6 days. Plasma was collected to analyze sclerostin protein level and tibia was collected to analyze mRNA level in osteocyte-enriched bones. To get osteocyte-enriched bones, the bones were flushed with HBSS and then cut longitudinally by surgical blade in α-MEM without phenol red (Gibco, 41061–029). The bones were incubated with α-MEM containing 250u/m collagenase (Sigma-Aldrich, C9891) for 30 minutes followed by 30 minutes incubation with 5mM EDTA with 0.1% BSA, pH 7.4 after washing the bones with HBSS three times. The bones were incubated with α-MEM containing 250u/m collagenase (Sigma-Aldrich, C9891) for 30 minutes additionally after washing the bones with HBSS three times. After aspiration of the medium, the osteocyte-enriched bones were homogenized by a mechanical homogenizer in cold room (4°C) with metal beads and TRIzol for gene expression analysis.
For inguinal fat, the mice were injected IP with 1mg/kg irisin every other day for a week. Inguinal fats were homogenized by a mechanical homogenizer in cold room (4°C) with metal beads and TRIzol for gene expression analysis. For immunoblot analysis, the fats were homogenized with metal beads and 2% SDS, 150mM NaCl, 50mM HEPES pH 8.8, 5mM DTT.
To test the effect of cyclo RGDyK, the mice were co-injected with 1mg/kg cyclo RGDyK or same amount of control RGD peptide. For the injection of SB273005, the compound dissolved in 5% DMSO+2% Tween 80+30% PEG 300+ddH2O.
Bone histomorphometric analysis for trabecular bone
Mice were subcutaneously injected with 20mg/kg of calcein (Sigma Aldrich, St. Louis, MO, USA) and 40mg/kg of demeclocycline (Sigma Aldrich, St. Louis, MO, USA) 9 and 2 days prior to the sacrifice, respectively. Lumbar vertebrae (L3-L5) were harvested and immediately fixed in 70% ethanol for 3 days. The fixed bone samples were dehydrated and embedded in methylmethacrylate. Undecalcified 4-µm-thick sections were obtained using a motorized microtome (RM2255, Leica, Nussloch, Germany) and stained with Von Kossa method for showing the mineralized bone. Consecutive second section was left unstained for the analysis of fluorescence labeling and the third section was stained with 2% Toluidine Blue (pH 3.7) for the analysis of osteoblasts, osteoid, osteoclasts. The bone histomorphometric analysis was performed under 200× magnification in a 1.8mm high x 1.3mm wide region located 400µm away from the upper and lower growth plate using OsteoMeasure analyzing software (Osteometrics Inc., Decatur, GA, USA). The structural parameters [bone volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N) and trabecular separation (Tb.Sp)] were obtained by taking an average from 2 different measurement of consecutive sections. The structural, dynamic and cellular parameters were calculated and expressed according to the standardized nomenclature (Dempster et al., 2013). The total bone area was subtracted by the blood vessel area to get the mineralized bone area.
Osteocyte analysis
The residual methylmethacrylate embedded tibia sample blocks from bone histomorphometry were used for the osteocyte analysis. Blocks were trimmed and the bone surface was sequentially ground with silicon carbide sandpaper of increasing grid number (Scientific Instrument Services Inc., NJ, USA). The sample surface was then carbon coated by vacuum evaporation (Auto 306 Vacuum Coater, Boc Edwards, UK) followed by fixation on the specimen mount with aluminum conductive tape (Ted Pella Inc., CA, USA). A digital scanning electron microscope (SEM, Supra 55 VP, Zeiss, Oberkochen, Germany, Center for Nanoscale Systems in Harvard University, Cambridge, MA) was employed with an accelerating voltage of 20kV, a working distance of 10mm and 500× magnification for taking backscattered electron images of a standardized tibial midshaft area located 4.5mm distal from the tibia-fibula junction. Images were analyzed with the Image J software (NIH, MD) for measuring osteocyte lacunae area and density (Qing et al., 2012). The total osteocyte lacunae area was measured in the number of pixels and then converted to the metric system. The total area was divided by the number of osteocytes to get the lacunae area. The number of osteocyte was divided by mineralized bone area to get the lacunae density.
Analysis of femur using µCT
We used high-resolution desktop microcomputed tomography imaging (µCT40, Scanco Medical, Brüttisellen, Switzerland) for analysis as previously reported (Spatz et al., 2013). We assessed trabecular and cortical bone microstructure in the distal femur and femoral diaphysis, respectively. Scans were acquired using a 10 µ m3 isotropic voxel size, 70kVP peak x-ray tube potential, 114mAs tube current, 200ms integration time, and were subjected to Gaussian filtration and segmentation. Image acquisition and analysis protocols adhered to the JBMR guidelines for the assessment of rodent bones by µCT (Bouxsein et al., 2010). In the distal femur, transverse µCT slices were evaluated in a region of interest beginning 200µm superior to the distal growth plate and extending proximally 1500 µm. The trabecular bone region was identified by semi-manually contouring the trabecular bone in the ROI with the assistance of an auto-thresholding software algorithm. Morphometric variables were computed from the binarized images using direct, 3D techniques we assessed the bone volume fraction (Tb.BV/TV, %), trabecular bone mineral density (Tb.BMD, mgHA/cm3), trabecular thickness (Tb.Th, µ m), trabecular number (Tb.N, mm−1), trabecular separation (Tb.Sp, µm), and connectivity density (mm−3). Cortical bone was analyzed in 50 transverse µCT slices (ROI length = 500µm) at the femoral mid-diaphysis. The region of interest included the entire outer most edge of the cortex. Images were subjected to Gaussian filtration and segmented using a fixed threshold of 700mgHA/cm3 to measure the following variables total cross-sectional area (Tt.Ar, mm2), cortical bone area (Ct.Ar, mm2), medullary area (Ma.Ar, mm2), bone area fraction (Ct.Ar/Tt.Ar, %), cortical tissue mineral density (Ct.TMD, mgHA/cm3), cortical thickness (Ct.Th, mm), cortical porosity (%), and the polar moment of inertia (pMOI, mm4).
Gene expression analysis
RNA was extracted from cultured cells or frozen tissues using TRIzol (Thermo Fischer Scientific) and purified with RNeasy mini kit (QIAGEN 74106). RNA was extracted from osteocyte-enriched tibia as described above (Qing et al., 2012). To perform qRT-PCR analysis, normalized RNA was reverse transcribed using a high-capacity cDNA reverse-transcription kit (Applied Biosystems). cDNA was analyzed by qRT-PCR with indicated primers. Relative mRNA levels were calculated using the comparative CT method and normalized to cyclophilin mRNA. Primer sequences used are listed in Table S7.
Immunoblot analysis
Cells were harvested in RIPA buffer containing protease-inhibitor cocktail and phosphatase- inhibitor cocktail. Whole-cell lysates were homogenized by 10 times passages through a 22G needle fitted to a 1ml syringe. Homogenized samples were rotated gently in cold room for 20 minutes followed by 15,000×g centrifugation for 10 minutes. 10µ l supernatant were used for normalization using BCA assay and remaining supernatants were mixed with 4×NuPAGE LDS sample buffer and 2.5% β-mercaptoethanol. The samples were incubated at 98°C for 5 minutes. The samples were separated by SDS-PAGE, and transferred to ImmobilonP membranes (Millipore). Protein levels were analyzed via western blot using indicated antibody. Inguinal fat pads were homogenized by a mechanical homogenizer in cold room (4°C) with 800µl of 2% SDS, 150mM NaCl, 50mM HEPES pH 8.8, 5mM DTT containing protease- inhibitor cocktail and phosphatase-inhibitor cocktail in cold room followed by incubation at 60°C for 30 minutes. 100µl of the homogenized samples were mixed with 300µ l methanol, 200µl chloroform and 250µl sterilized H2O. After centrifugation at 4000×g for 10 minutes at room temperature, upper and lower phases were removed by aspiration and interphase were washed with 1ml cold methanol three times. After drying at 37°C, the interphase was solubilized by 8M Urea and 50mM HEPES pH 8.5. After normalization of the protein using BCA assay, the samples were separated by SDS-PAGE, and transferred to ImmobilonP membranes (Millipore). Protein levels were analyzed using western blot against indicated antibody.
Protein-protein binding assays
100nM flag-tagged mammalian irisin was incubated with 5nM of the indicated his-tag integrins in a final volume of 600µ l in 1.5ml Protein LoBind Tubes (Eppendorf, 022431081) for 5 minutes at room temperature under rotation. After rotation, 60µl Ni-NTA agarose (ThermoFisher Scientific, R901–01) was applied to immunoprecipitated integrins. Precipitated integrins were detected by immunoblot analysis against his tag. Co-precipitated irisin was detected by immunoblot analysis against flag-tag.
Anti-apoptosis assay
MLO-Y4 cells were seeded in type-I collagen coated 96 well plate (3000 cells/well) in 1% FBS, 1% CS, α-MEM without phenol red (Gibco, 41061–029) on day 0. The medium was aspirated and 1% FBS, 1% CS, α-MEM without phenol red containing the indicated concentration of irisin was added to the wells. After 24 hours incubation, 0.5% FBS, 0.5% CS, α-MEM without phenol red containing the indicated concentration of irisin and 0.3mM H2O2 were added and the cells were incubated for 4 hours. The cells were stained with 2μM ethidium Homodimer-1 (ThermoFisher Scientific, E1169) to detect dead cells. The cell images were taken using Nikon Eclipse TE300 inverted fluorescence microscope with a Photometrics Coolsnap EZ cooled CCD camera and analyzed using ImageJ. Percentage of cell death was calculated as EthD-1 positive cells divided by the total number of cells stained with 5µg/mL Hoechst 33342 (ThermoFisher Scientific, H3570) as a nuclear counterstain (Kitase et al., 2018).
Identification of irisin receptor using quantitative proteomics Co-immunoprecipitation of candidates of irisin receptors
MLO-Y4 cells were seeded on 30×150mm type-I collagen coated dishes as described in cell culture experiment. At 60% cell density, medium was switched to FreeStyle293 Expression medium. After 4 hours incubation, the cells were chilled on ice for 10 minutes, followed by treatment of 10nM his-tag irisin or his-tag adipsin for 20 minutes. The cells were then incubated with 1.5mM DTSSP for 30 minutes on ice to do cross-linking, after washing with 15ml cold PBS twice. The cross-linking was quenched by addition of a final concentration of 20mM Tris-pH 7.5. The cells were then harvested and homogenized in 1ml RIPA buffer containing protease- inhibitor cocktail and phosphatase-inhibitor cocktail. Whole-cell lysates were homogenized by 10 times passages through a 22G needle fitted to a 3ml syringe. Homogenized samples were rotated gently in cold room for 20 minutes followed by 15,000×g centrifugation for 10 minutes. After addition of a final concentration of 10mM imidazole, supernatants were incubated with 100µl Ni-NTA agarose for 1 hour. After centrifugation at 500×g for 1 minute, the supernatants were aspirated and 1ml cold RIPA buffer containing 10mM imidazole were added to the agarose. After 10 minutes rotation in cold room, the supernatants were aspirated and 1ml cold RIPA buffer containing 30mM imidazole were added to the agarose. After repeating the washing 3 times, 0.8ml RIPA buffer containing 250mM imidazole was added and the agarose were gently rotated in a cold room for 20 minutes. After centrifugation at 1000×g for 2 minutes, the supernatants were transferred to 1.5ml tubes and incubated with 100µ l 0.2% sodium deoxycholate and 100µl 10% trichloroacetic acid in ice for 1 hour. After centrifugation at 12,000×g for 10 minutes at 4°C, the supernatants were removed and 1ml cold acetone were added to pellet followed by vortexing for 10 seconds. After 1 more wash with cold acetone, the pellets were dried at 37°C and 39µ l PBS and 13µl 4×NuPAGE LDS were added to pellet with a final concentration of 5mM DTT. Solubilized proteins were incubated at 65°C for 20 minutes followed by incubation with a final concentration of 14mM iodoacetamide for 45 minutes in the dark. 38µ l samples were loaded to 4–12% gradient SDS-PAGE for separation followed by Coomassie Blue staining. The gels were submitted to quantitative proteomics.
Protein digestion and isobaric tag peptide labeling
For in-gel digestions, gels were stained with Coomassie Blue and were excised into 8 equal segments for control and irisin lanes. Gel pieces were destained and dehydrated with 100% acetonitrile, vacuumed dried, and digested in 25mM HEPES (pH 8.5) with 500ng sequencing grade trypsin (Promega) for an overnight incubation at 37°C (Shevchenko et al., 1996). Digests were with 1% formic acid and purified using C18 Stage-Tips as previously described (Rappsilber et al., 2007). Peptides were eluted with 70% acetonitrile and 1% formic acid, then dried using a speedvac.
Isobaric labeling of digested peptides was accomplished using 6-plex tandem mass tag (TMT) reagents (Thermo Fisher Scientific, Rockford, IL). The reagents, 5.0mg, were dissolved in 252µl acetonitrile (ACN) and 5µl of the solution were added to the digested peptides dissolved in 25µl of 200mM HEPES, pH 8.5. After 1 hour at room temperature, the reaction was quenched by adding 1µl of 5% hydroxylamine. Labeled peptides were combined and acidified prior to C18 Stage-Tips desalting.
Liquid chromatography separation and tandem mass spectrometry (LC-MS/MS)
All LC-MS/MS experiments were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 100µm inner diameter microcapillary column packed with 35cm of Accucore C18 resin (1.8µm, 100Å, Thermo Fisher Scientific). Peptides were separated using a 2 hour gradient of 6–33% acetonitrile in 0.125% formic acid with a flow rate of ~400nL/min. Each analysis used an MS3-based TMT method as described previously29. MS1 data was acquired at a mass range of m/z 350 – 1350, resolution 120,000, AGC target 5 × 105, maximum injection time 150ms, and with a dynamic exclusion of 120 seconds for the peptide measurements in the Orbitrap.
Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 2.2 × 104 and a maximum injection time of 120ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 5.5 × 105, maximum injection time of 200ms, resolution at 15,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
Data processing and spectra assignment
In-house developed software was used to convert mass spectrometric data (.raw files) to an mzXML format, as well as to correct monoisotopic m/z measurements. All experiments used the Mouse UniProt database (downloaded 10 April 2017) where reversed protein sequences and known contaminants such as human keratins and albumin were appended. SEQUEST searches were performed using a 20ppm precursor ion tolerance, while requiring each peptide’s amino/carboxy terminus to have trypsin protease specificity and allowing up to two missed cleavages. Six-plex TMT tags on peptide N termini and lysine residues (+ 229.162932Da) and carbamidomethylation of cysteine residues (+57.02146Da) were set as static modifications while methionine oxidation (+ 15.99492Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of less than 1% was achieved by applying the target- decoy database search strategy (Elias and Gygi, 2007) Filtering was performed using an in-house linear discrimination analysis method to create one combined filter parameter from the following peptide ion and MS2 spectra metrics: Sequest parameters XCorr and ∆Cn, peptide ion mass accuracy and charge state, peptide length and mis-cleavages. Linear discrimination scores were used to assign probabilities to each MS2 spectrum for being assigned correctly and these probabilities were further used to filter the dataset with an MS2 spectra assignment FDR of smaller than a 1% at the protein level (Huttlin et al., 2010).
Determination of TMT reporter ion intensities and quantitative data analysis
For quantification, a 0.03m/z window centered on the theoretical m/z value of each the two reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were further de-normalized based on their ion accumulation time for each MS2 or MS3 spectrum and adjusted based on the overlap of isotopic envelopes of all reporter ions (as per manufacturer specifications). The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially sample handling variance.
Quantification of irisin in plasma and integrins in cells using quantitative proteomics In-gel digestion and TMT labeling
Blood was collected 2 weeks after OVX and plasma was separated by centrifugation. Plasma specimens (35µl) were depleted of albumin and IgG using the ProteoExtract kit and subsequently concentrated using 3kDa molecular weight cut-off spin-filter columns (Millipore). Deglycosylation of plasma was performed using Protein Deglycosylation Mix (NEB) as per the manufacturer’s denaturing protocol. Deglycosylated murine plasma samples were reduced with 5 mM DTT and alkylated with 75 mM iodoacetamide prior to being resolved by SDS-PAGE using 4–12% Bis-Tris precast gels (Life Technologies). Gels were coomassie stained and fragments were excised and cut into smaller fragments from the 10–15 kDa region. Gel pieces were destained and dehydrated with 100% acetonitrile, vacuumed dried, and 25mM HEPES (pH 8.5) with 500 ng sequencing grade trypsin (Promega) was added for an overnight incubation at 37°. Digests were quenched after 12hr with 70% acetonitrile/1% formic acid, dried and desalted using in-house stage tips as previously described (Rappsilber et al., 2007). Peptides were eluted with 70% acetonitrile/1% formic acid, dried using a speedvac, and resuspended in 12µl of 5% formic acid and 5% acetonitrile containing the heavy valine synthesized irisin peptides (Table S8, 1 femtomole). Aqua peptides of FNDC5 amino acid 32–43 (DSPSAPVNVTVR, underline is heavy amino acid, light mass (Da) is 1240.631 and heavy mass (Da) is 1246.655) and FNDC5 amino acid 79–88 (FIQEVNTTTR, underline is heavy amino acid, light mass (Da) is 1207.609 and heavy mass (Da) is 1213.634) were used.
Isobaric labeling of the digested peptides for receptor quantitative proteomics from MLO-Y4 cells or primary inguinal fat cells was accomplished using 6-plex tandem mass tag (TMT) reagents (Thermo Fisher Scientific, Rockford, IL). Reagents (5.0 mg) were dissolved in 252µl acetonitrile (ACN) and 1/4 of the solution were added to digested peptides dissolved in 100µl of 200mM HEPES, pH 8.5. After 1hr at room temperature, the reaction was quenched by adding 3µl of 5% hydroxylamine. Labeled peptides were combined and acidified prior to desalting.
Mass spectrometry and liquid chromatography
Mass spectrometry data was collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled with µHPLC (EASY-nLC™ 1200 system, Thermo Scientific). Peptides were separated onto a 75µm inner diameter microcapillary column packed with ∼40cm of Accucore C18 resin (2.6µm, 150 Å, Thermo Fisher Scientific). For each analysis, we loaded ~4µl onto the column. Peptides were separated using a 60-minute gradient of 8 to 30% acetonitrile in 0.125% formic acid with a flow rate of ∼400nL/min.
Parallel reaction monitoring acquisition
Parallel reaction monitoring (PRM) analyses were performed using an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific). A full MS scan from 575–700m/z at an Orbitrap resolution of 120,000 (at m/z 200), AGC target 1 × 106 and a 1000ms maximum injection time. Full MS scans were followed by 25–50 PRM scans at 30,000 resolution (AGC target 1 × 106, 2000ms maximum injection time) as triggered by a scheduled inclusion list (see above). The PRM method employed an isolation of target ions by a 1.6 Th isolation window, fragmented with normalized collision energy (NCE) of 35, MS/MS scans were acquired with a starting mass range of 110m/z and acquired as a profile spectrum data type. Fragment ions for all peptides were quantified using Skyline version 3.5 (MacLean et al., 2010).
Peptide and protein identification
Liquid chromatography separation and tandem mass spectrometry (LC-MS/MS)
All LC-MS/MS receptor proteomic experimental data was collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 75µm inner diameter microcapillary column packed with 35 cm of Accucore C18 resin (2.6µm, 100Å, ThermoFisher Scientific). Peptides were separated using a 3hr gradient of 6–27% acetonitrile in 0.125% formic acid with a flow rate of 400nL/min. Each analysis used an MS3- based TMT method as described previously (McAlister et al., 2014). The data were acquired using a mass range of m/z 350 – 1350, resolution 120,000, AGC target 1 × 106, maximum injection time 100 ms, dynamic exclusion of 120 seconds for the peptide measurements in the Orbitrap.
Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104 and a maximum injection time of 120ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 1.5 × 105, maximum injection time of 150ms, resolution at 15,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
Data processing and spectra assignment
A compendium of in-house developed software was used to convert mass spectrometric data (Raw file) to the mzXML format, as well as to correct monoisotopic m/z measurements and erroneous assignments of peptide charge state (Elias and Gygi, 2007). All experiments used the Mouse UniProt database (downloaded 10 April 2014) where reversed protein sequences and known contaminants such as human keratins were appended. SEQUEST searches were performed using a 20ppm precursor ion tolerance, while requiring each peptide’s amino/carboxy (N/C) terminus to have trypsin protease specificity and allowing up to two missed cleavages. Six-plex TMT tags on peptide N termini and lysine residues (+ 229.162932 Da) and carbamidomethylation of cysteine residues (+57.02146 Da) were set as static modifications while methionine oxidation (+ 15.99492 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of less than 1% was achieved by applying the target- decoy database search strategy (Elias and Gygi, 2007). Filtering was performed using a in-house linear discrimination analysis method to create one combined filter parameter from the following peptide ion and MS2 spectra metrics: Sequest parameters XCorr and ∆Cn, peptide ion mass accuracy and charge state, in-solution charge of peptide, peptide length and mis-cleavages. Linear discrimination scores were used to assign probabilities to each MS2 spectrum for being assigned correctly and these probabilities were further used to filter the dataset with an MS2spectra assignment FDR of smaller than a 1% at the protein level (Huttlin et al., 2010).
Determination of TMT reporter ion intensities and quantitative data analysis
For quantification, a 0.03 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were further denormalized based on their ion accumulation time for each MS2 or MS3 spectrum and adjusted based on the overlap of isotopic envelopes of all reporter ions (as determined by the manufacturer). The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially uneven TMT labeling and/or sample handling variance.
HDX/MS
Differential HDX-MS experiments were conducted as previously described with a few modifications (Chalmers et al., 2006).
Peptide Identification:
Protein samples were injected for inline pepsin digestion and the resulting peptides were identified using tandem MS (MS/MS) with an Orbitrap mass spectrometer (Fusion Lumos, ThermoFisher). Following digestion, peptides were desalted on a C8 trap column and separated on a 1 hour linear gradient of 5–40% B (A is 0.3% formic acid and B is 0.3% formic acid 95% CH3CN). Product ion spectra were acquired in data-dependent mode with a one second duty cycle such that the most abundant ions selected for the product ion analysis by higher-energy collisional dissociation between survey scan events occurring once per second. Following MS2 acquisition, the precursor ion was excluded for 16 seconds. The resulting MS/MS data files were submitted to Mascot (Matrix Science) for peptide identification. Peptides included in the HDX analysis peptide set had a MASCOT score greater than 20 and the MS/MS spectra were verified by manual inspection. The MASCOT search was repeated against a decoy (reverse) sequence and ambiguous identifications were ruled out and not included in the HDX peptide set.
HDX-MS analysis:
Apo proteins (irisin and integrin αV/β5) were analyzed at 10µ M each. For differential HDX, integrin αV/β5 (10µ M) was concentrated 3X using an Amicon Ultra Centrifugal Filter Unit with a 50K membrane (Part #: UFC505008) and the protein complex was formed by incubating irisin (10μM) with integrin αV/β5 (30uM) for 1 hour at room temperature. Next, 5μl of sample was diluted into 20μl D2O buffer (20mM Tris-HCl, pH 7.4, 150mM NaCl, 2mM DTT) and incubated for various time points (0, 10, 60, 300, 900 and 3600 s) at 4°C. The deuterium exchange was then slowed by mixing with 25μl of cold (4°C) 3M urea and 1% trifluoroacetic acid. Quenched samples were immediately injected into the HDX platform. Upon injection, samples were passed through an immobilized pepsin column (2mm × 2cm) at 200μl min−1 and the digested peptides were captured on a 2mm × 1cm C8 trap column (Agilent) and desalted. Peptides were separated across a 2.1mm × 5cm C18 column (1.9μl Hypersil Gold, ThermoFisher) with a linear gradient of 4% - 40% CH3CN and 0.3% formic acid, over 5 minutes. Sample handling, protein digestion and peptide separation were conducted at 4°C. Mass spectrometric data were acquired using an Orbitrap mass spectrometer (Q Exactive, ThermoFisher). HDX analyses were performed in triplicate, with single preparations of each protein ligand complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation. This is accomplished determining the observed averages of the undeuterated and fully deuterated spectra and using the conventional formula described elsewhere (Zhang and Smith, 1993). Statistical significance for the differential HDX data is determined by an unpaired t-test for each time point, a procedure that is integrated into the HDX Workbench software (Pascal et al., 2012).
Corrections for back-exchange were made on the basis of an estimated 70% deuterium recovery, and accounting for the known 80% deuterium content of the deuterium exchange buffer.
Data Rendering:
The HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach. Briefly, for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily and longer peptides were weighted less. Each of the weighted deuterium incorporation values were then averaged to produce a single value for each amino acid. The initial two residues of each peptide, as well as prolines, were omitted from the calculations. This approach is similar to that previously described (Keppel and Weis, 2015). HDX analyses were performed in triplicate, with single preparations of each purified protein/complex. Statistical significance for the differential HDX data is determined by t-test for each time point, and is integrated into the HDX Workbench software (Pascal et al., 2012).
Generation of docking model with ZDOCK
A model for irisin-integrin αV/β5 was generated using homology modeling. The models for integrin β5 and irisin were generated using Modeller (Sali and Blundell, 1993) based on a model of Fibronectin-αV/β3 (PDB 4MMX). Irisin was docked to integrin β5 using the ZDOCK server (http://zdock.umassmed.edu/) according to the guide line (Pierce et al., 2014). The resulting model that agreed with the observed HDX-MS data was used to generate the Irisin-integrin αV/β5 model.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values in graphs are presented as mean +/− S.E.M. Two-way ANOVA for multiple comparison were used to analyze the data. Significant differences between two groups were evaluated using a two-tailed, unpaired Student’s t-test as the samples groups displayed a normal distribution and comparable variance (* p < 0.05, ** p < 0.01, *** p < 0.001). For analysis of histomorphometry, the structural parameters [bone volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N) and trabecular separation (Tb.Sp)] were obtained by taking an average from 2 different measurement of consecutive sections. The structural, dynamic and cellular parameters were calculated and expressed according to the standardized nomenclature (Dempster et al., 2013).
For osteocyte analysis, the total bone area was subtracted by the blood vessel area to get the mineralized bone area. Images were analyzed with the Image J software (NIH, MD) for measuring osteocyte lacunae area and density (Qing et al., 2012). The total osteocyte lacunae area was measured in the number of pixels and then converted to the metric system. The total area was divided by the number of osteocytes to get the lacunae area. The number of osteocyte was divided by mineralized bone area to get the lacunae density.
For analysis of femur using µ CT, morphometric variables were computed from the binarized images using direct, 3D techniques we assessed the bone volume fraction (Tb.BV/TV, %), trabecular bone mineral density (Tb.BMD, mgHA/cm3), trabecular thickness (Tb.Th, µm), trabecular number (Tb.N, mm−1), trabecular separation (Tb.Sp, µm), and connectivity density (mm−3). Cortical bone was analyzed in 50 transverse µCT slices (ROI length = 500µm) at the femoral mid-diaphysis. The region of interest included the entire outer most edge of the cortex. Images were subjected to Gaussian filtration and segmented using a fixed threshold of 700mgHA/cm3 to measure the following variables total cross-sectional area (Tt.Ar, mm2), cortical bone area (Ct.Ar, mm2), medullary area (Ma.Ar, mm2), bone area fraction (Ct.Ar/Tt.Ar, %), cortical tissue mineral density (Ct.TMD, mgHA/cm3), cortical thickness (Ct.Th, mm), cortical porosity (%), and the polar moment of inertia (pMOI, mm4).
For anti-apoptosis assay, Percentage of cell death was calculated as EthD-1 positive cells divided by the total number of cells stained with 5µg/mL Hoechst 33342 (ThermoFisher Scientific, H3570) as a nuclear counterstain (Kitase et al., 2018).
For identification of irisin receptor using quantitative proteomics, a 0.03m/z window centered on the theoretical m/z value of each the two reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were further de-normalized based on their ion accumulation time for each MS2 or MS3 spectrum and adjusted based on the overlap of isotopic envelopes of all reporter ions (as per manufacturer specifications). The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially sample handling variance. For quantification of irisin in plasma and integrins in cells using quantitative proteomics, a 0.03 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were further denormalized based on their ion accumulation time for each MS2 or MS3 spectrum and adjusted based on the overlap of isotopic envelopes of all reporter ions (as determined by the manufacturer). The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were adjusted to account for potentially uneven TMT labeling and/or sample handling variance.
For data rendering of HDX/MS, the HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach. Briefly, for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily and longer peptides were weighted less. Each of the weighted deuterium incorporation values were then averaged to produce a single value for each amino acid. The initial two residues of each peptide, as well as prolines, were omitted from the calculations. This approach is similar to that previously described (Keppel and Weis, 2015). HDX analyses were performed in triplicate, with single preparations of each purified protein/complex. Statistical significance for the differential HDX data is determined by t-test for each time point, and is integrated into the HDX Workbench software (Pascal et al., 2012).
Supplementary Material
Highlights.
The αV class of integrins are irisin receptors in osteocytes and adipose tissues.
Irisin increases sclerostin expression in osteocytes to induce bone resorption.
Genetic deletion of FNDC5/Irisin completely blocks OVX-induced trabecular bone loss.
Acknowledgements
We thank members of the Spiegelman lab for very helpful discussions. We also thank Eric N. Olson at UT Southwestern Medical Center for his generous help with transgenic mice early in this project. We are grateful to Richard O. Hynes at Massachusetts Institute of Technology for helpful discussions. This work was supported by NIH grants (4 R01DK54477–19, DK61502, B.M.S.) and the JPB Foundation (B.M.S.). H.K. was supported by cardiovascular T32 grant (5T32HL007374–38). C.D.W. was supported by K99NS087096. (PO1AG039335, L.F.B.). L.F.B. was supported by NIH grant (PO1AG039335). C.J.R. was supported by NIH grant (NIDDK RC2 092759).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
B.M.S., H.K., L.F.B., C.J.R., and R.B. have a pending patent on irisin.
References
- Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, Vanderschueren D, and Manolagas SC (2017). Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiological reviews 97, 135–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, and Kneissel M (2013). WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nature medicine 19, 179–192. [DOI] [PubMed] [Google Scholar]
- Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, and Jilka RL (2005). Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146, 4577–4583. [DOI] [PubMed] [Google Scholar]
- Bonewald LF (2011). The amazing osteocyte. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 26, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, and Doolittle RF (1992). Proposed acquisition of an animal protein domain by bacteria. Proceedings of the National Academy of Sciences of the United States of America 89, 8990–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, et al. (2012). A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, and Muller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 25, 1468–1486. [DOI] [PubMed] [Google Scholar]
- Braam LA, Knapen MH, Geusens P, Brouns F, and Vermeer C (2003). Factors affecting bone loss in female endurance athletes: a two-year follow-up study. The American journal of sports medicine 31, 889–895. [DOI] [PubMed] [Google Scholar]
- Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, and Tarone G (2006). Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovascular research 70, 422–433. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, and Griffin PR (2006). Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Analytical chemistry 78, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Chen X, Park R, Tohme M, Shahinian AH, Bading JR, and Conti PS (2004). MicroPET and autoradiographic imaging of breast cancer alpha v-integrin expression using 18F- and 64Cu-labeled RGD peptide. Bioconjugate chemistry 15, 41–49. [DOI] [PubMed] [Google Scholar]
- Colaianni G, Cuscito C, Mongelli T, Oranger A, Mori G, Brunetti G, Colucci S, Cinti S, and Grano M (2014). Irisin enhances osteoblast differentiation in vitro. International journal of endocrinology 2014, 902186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, Liu P, Lu P, Sartini L, Di Comite M, Mori G, et al. (2015). The myokine irisin increases cortical bone mass. Proceedings of the National Academy of Sciences of the United States of America 112, 12157–12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaianni G, Mongelli T, Cuscito C, Pignataro P, Lippo L, Spiro G, Notarnicola A, Severi I, Passeri G, Mori G, et al. (2017). Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Scientific reports 7, 2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico M, Hulit J, Amanatullah DF, Zafonte BT, Albanese C, Bouzahzah B, Fu M, Augenlicht LH, Donehower LA, Takemaru K, et al. (2000). The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. The Journal of biological chemistry 275, 32649–32657. [DOI] [PubMed] [Google Scholar]
- Dechantsreiter MA, Planker E, Matha B, Lohof E, Holzemann G, Jonczyk A, Goodman SL, and Kessler H (1999). N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. Journal of medicinal chemistry 42, 3033–3040. [DOI] [PubMed] [Google Scholar]
- Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, and Parfitt AM (2013). Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 28, 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, Shane E, Plavetic K, Muller R, Bilezikian J, et al. (2001). Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 16, 1846–1853. [DOI] [PubMed] [Google Scholar]
- Duckham RL, Peirce N, Bailey CA, Summers G, Cameron N, and Brooke-Wavell K (2013). Bone geometry according to menstrual function in female endurance athletes. Calcified tissue international 92, 444–450. [DOI] [PubMed] [Google Scholar]
- Duong LT, Lakkakorpi P, Nakamura I, and Rodan GA (2000). Integrins and signaling in osteoclast function. Matrix biology : journal of the International Society for Matrix Biology 19, 97–105. [DOI] [PubMed] [Google Scholar]
- Duong LT, and Rodan GA (1998). Integrin-mediated signaling in the regulation of osteoclast adhesion and activation. Frontiers in bioscience : a journal and virtual library 3, d757–768. [DOI] [PubMed] [Google Scholar]
- Eatemadololama A, Karimi MT, Rahnama N, and Rasolzadegan MH (2017). Resistance exercise training restores bone mineral density in renal transplant recipients. Clinical cases in mineral and bone metabolism : the official journal of the Italian Society of Osteoporosis, Mineral Metabolism, and Skeletal Diseases 14, 157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Flores ME, Norgard M, Heinegard D, Reinholt FP, and Andersson G (1992). RGD-directed attachment of isolated rat osteoclasts to osteopontin, bone sialoprotein, and fibronectin. Experimental cell research 201, 526–530. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, and Ruoslahti E (1999). Integrin signaling. Science (New York, NY) 285, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Guo L, Ding W, and Zheng LM (2014). The C(RgdyK)-conjugated Fe3O4 nanoparticles with high drug load for dual-targeting integrin alpha(v)beta3-expressing cancer cells. Journal of nanoscience and nanotechnology 14, 4858–4864. [DOI] [PubMed] [Google Scholar]
- Horton MA, Taylor ML, Arnett TR, and Helfrich MH (1991). Arg-Gly-Asp (RGD) peptides and the anti-vitronectin receptor antibody 23C6 inhibit dentine resorption and cell spreading by osteoclasts. Experimental cell research 195, 368–375. [DOI] [PubMed] [Google Scholar]
- Hu DD, Lin EC, Kovach NL, Hoyer JR, and Smith JW (1995). A biochemical characterization of the binding of osteopontin to integrins alpha v beta 1 and alpha v beta 5. The Journal of biological chemistry 270, 26232–26238. [DOI] [PubMed] [Google Scholar]
- Huh JY, Dincer F, Mesfum E, and Mantzoros CS (2014). Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. International journal of obesity (2005) 38, 1538–1544. [DOI] [PubMed] [Google Scholar]
- Humphries JD, Byron A, and Humphries MJ (2006). Integrin ligands at a glance. Journal of cell science 119, 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO (1973). Alteration of cell-surface proteins by viral transformation and by proteolysis. Proceedings of the National Academy of Sciences of the United States of America 70, 3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idris AI (2012). Ovariectomy/orchidectomy in rodents. Methods in molecular biology (Clifton, NJ) 816, 545–551. [DOI] [PubMed] [Google Scholar]
- Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, Nair KS, Gygi SP, and Spiegelman BM (2015). Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell metabolism 22, 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Windle JJ, Koop BA, Mundy GR, and Bonewald LF (1997). Establishment of an osteocyte- like cell line, MLO-Y4. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 12, 2014–2023. [DOI] [PubMed] [Google Scholar]
- Keller C, Steensberg A, Pilegaard H, Osada T, Saltin B, Pedersen BK, and Neufer PD (2001). Transcriptional activation of the IL-6 gene in human contracting skeletal muscle: influence of muscle glycogen content. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 15, 2748–2750. [DOI] [PubMed] [Google Scholar]
- Keller H, and Kneissel M (2005). SOST is a target gene for PTH in bone. Bone 37, 148–158. [DOI] [PubMed] [Google Scholar]
- Keppel TR, and Weis DD (2015). Mapping residual structure in intrinsically disordered proteins at residue resolution using millisecond hydrogen/deuterium exchange and residue averaging. Journal of the American Society for Mass Spectrometry 26, 547–554. [DOI] [PubMed] [Google Scholar]
- Kirwan JP, Sacks J, and Nieuwoudt S (2017). The essential role of exercise in the management of type 2 diabetes. Cleveland Clinic journal of medicine 84, S15–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov VV, Skladchikova G, Hinsby AM, Jensen PH, Kulahin N, Soroka V, Pedersen N, Tsetlin V, Poulsen FM, Berezin V, et al. (2003). Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure (London, England : 1993) 11, 691–701. [DOI] [PubMed] [Google Scholar]
- Kitase Y, Vallejo JA, Gutheil W, Vemula H, Jahn K, Yi J, Zhou J, Brotto M, and Bonewald LF (2018). beta-aminoisobutyric Acid, l-BAIBA, Is a Muscle-Derived Osteocyte Survival Factor. Cell reports 22, 1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrt WM, Wherry SJ, Wolfe P, Sherk VD, Wellington T, Swanson CM, Weaver CM, and Boxer RS (2018). Maintenance of Serum Ionized Calcium During Exercise Attenuates Parathyroid Hormone and Bone Resorption Responses. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 33, 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T (2015). Animal models for osteoporosis. European journal of pharmacology 759, 287–294. [DOI] [PubMed] [Google Scholar]
- Krolner B, Toft B, Pors Nielsen S, and Tondevold E (1983). Physical exercise as prophylaxis against involutional vertebral bone loss: a controlled trial. Clinical science (London, England : 1979) 64, 541–546. [DOI] [PubMed] [Google Scholar]
- Kumar CC, Nie H, Rogers CP, Malkowski M, Maxwell E, Catino JJ, and Armstrong L (1997). Biochemical characterization of the binding of echistatin to integrin alphavbeta3 receptor. The Journal of pharmacology and experimental therapeutics 283, 843–853. [PubMed] [Google Scholar]
- Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, and Arnaud CD (1998). Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. Results of a randomized controlled clinical trial. The Journal of clinical investigation 102, 1627–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lark MW, Stroup GB, Dodds RA, Kapadia R, Hoffman SJ, Hwang SM, James IE, Lechowska B, Liang X, Rieman DJ, et al. (2001). Antagonism of the osteoclast vitronectin receptor with an orally active nonpeptide inhibitor prevents cancellous bone loss in the ovariectomized rat. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 16, 319– 327. [DOI] [PubMed] [Google Scholar]
- Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, Perron RM, Werner CD, Phan GQ, Kammula US, et al. (2014). Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell metabolism 19, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Thabane L, Papaioannou A, Ioannidis G, Levine MA, and Adachi JD (2017). An overview of osteoporosis and frailty in the elderly. BMC musculoskeletal disorders 18, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, et al. (2009). Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 24, 578–588. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics (Oxford, England) 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli L, Gottschalk KE, Meyer A, Novellino E, and Kessler H (2004). Human integrin alphavbeta5: homology modeling and ligand binding. Journal of medicinal chemistry 47, 4166–4177. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Analytical chemistry 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung MR (2017). Sclerostin antibodies in osteoporosis: latest evidence and therapeutic potential. Therapeutic advances in musculoskeletal disease 9, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WH, Alberts DP, Bhatnagar PK, Bondinell WE, Callahan JF, Calvo RR, Cousins RD, Erhard KF, Heerding DA, Keenan RM, et al. (2000). Discovery of orally active nonpeptide vitronectin receptor antagonists based on a 2-benzazepine Gly-Asp mimetic. Journal of medicinal chemistry 43, 22–26. [DOI] [PubMed] [Google Scholar]
- Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, et al. (2010). Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 25, 948–959. [DOI] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, and Griffin PR (2012). HDX workbench: software for the analysis of H/D exchange MS data. Journal of the American Society for Mass Spectrometry 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, and Febbraio MA (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nature reviews Endocrinology 8, 457–465. [DOI] [PubMed] [Google Scholar]
- Pekkala S, Wiklund PK, Hulmi JJ, Ahtiainen JP, Horttanainen M, Pollanen E, Makela KA, Kainulainen H, Hakkinen K, Nyman K, et al. (2013). Are skeletal muscle FNDC5 gene expression and irisin release regulated by exercise and related to health? The Journal of physiology 591, 5393–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perakakis N, Triantafyllou GA, Fernandez-Real JM, Huh JY, Park KH, Seufert J, and Mantzoros CS (2017). Physiology and role of irisin in glucose homeostasis. Nature reviews Endocrinology 13, 324– 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering ME, Simon M, Sornay-Rendu E, Chikh K, Carlier MC, Raby AL, Szulc P, and Confavreux CB (2017). Serum Sclerostin Increases After Acute Physical Activity. Calcified tissue international 101, 170–173. [DOI] [PubMed] [Google Scholar]
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, and Weng Z (2014). ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics (Oxford, England) 30, 1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, and Smith JW (2000). Ligand binding to integrins. The Journal of biological chemistry 275, 21785–21788. [DOI] [PubMed] [Google Scholar]
- Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie G, and Ginsberg MH (1987). Arginyl-glycyl- aspartic acid sequences and fibrinogen binding to platelets. Blood 70, 110–115. [PubMed] [Google Scholar]
- Polyzos SA, Anastasilakis AD, Efstathiadou ZA, Makras P, Perakakis N, Kountouras J, and Mantzoros CS (2018). Irisin in metabolic diseases. Endocrine 59, 260–274. [DOI] [PubMed] [Google Scholar]
- Potts JR, and Campbell ID (1994). Fibronectin structure and assembly. Current opinion in cell biology 6, 648–655. [DOI] [PubMed] [Google Scholar]
- Prince RL, Smith M, Dick IM, Price RI, Webb PG, Henderson NK, and Harris MM (1991). Prevention of postmenopausal osteoporosis. A comparative study of exercise, calcium supplementation, and hormone-replacement therapy. The New England journal of medicine 325, 1189–1195. [DOI] [PubMed] [Google Scholar]
- Qiao X, Nie Y, Ma Y, Chen Y, Cheng R, Yin W, Hu Y, Xu W, and Xu L (2016). Irisin promotes osteoblast proliferation and differentiation via activating the MAP kinase signaling pathways. Scientific reports 6, 18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, Wysolmerski J, and Bonewald LF (2012). Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 27, 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H, and Bonewald LF (2009). Osteocyte remodeling of the perilacunar and pericanalicular matrix. International journal of oral science 1, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, Jedrychowski MP, Ruas JL, Wrann CD, Lo JC, et al. (2014). Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157, 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre- fractionation and storage of peptides for proteomics using StageTips. Nature protocols 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Sali A, and Blundell TL (1993). Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, and Parsons JT (1994). Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Molecular and cellular biology 14, 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Chinnam N, Ohashi T, Shah RS, and Erickson HP (2013). The structure of irisin reveals a novel intersubunit beta-sheet fibronectin type III (FNIII) dimer: implications for receptor activation. The Journal of biological chemistry 288, 33738–33744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield KL, and Hecht S (2012). Bone health in endurance athletes: runners, cyclists, and swimmers. Current sports medicine reports 11, 328–334. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Smith JW, Vestal DJ, Irwin SV, Burke TA, and Cheresh DA (1990). Purification and functional characterization of integrin alpha v beta 5. An adhesion receptor for vitronectin. The Journal of biological chemistry 265, 11008–11013. [PubMed] [Google Scholar]
- Spatz JM, Ellman R, Cloutier AM, Louis L, van Vliet M, Suva LJ, Dwyer D, Stolina M, Ke HZ, and Bouxsein ML (2013). Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 28, 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler A, Paz S, Berman A, Lucero E, Contino N, Penalba A, Tirado S, Santana M, and Zeballos AC (1997). Muscular strength and bone mineral density in haemodialysis patients. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 12, 128–132. [DOI] [PubMed] [Google Scholar]
- Thi MM, Suadicani SO, Schaffler MB, Weinbaum S, and Spray DC (2013). Mechanosensory responses of osteocytes to physiological forces occur along processes and not cell body and require alphaVbeta3 integrin. Proceedings of the National Academy of Sciences of the United States of America 110, 21012–21017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Agthoven JF, Xiong JP, Alonso JL, Rui X, Adair BD, Goodman SL, and Arnaout MA (2014). Structural basis for pure antagonism of integrin alphaVbeta3 by a high-affinity form of fibronectin. Nature structural & molecular biology 21, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, and Lowik CW (2004). Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. The Journal of experimental medicine 199, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent KR, and Braith RW (2002). Resistance exercise and bone turnover in elderly men and women. Medicine and science in sports and exercise 34, 17–23. [DOI] [PubMed] [Google Scholar]
- Wrann CD, White JP, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, Lin JD, Greenberg ME, and Spiegelman BM (2013). Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell metabolism 18, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, West AP Jr., Allendorph GP, Choe S, and Bjorkman PJ (2008). Neogenin interacts with hemojuvelin through its two membrane-proximal fibronectin type III domains. Biochemistry 47, 4237– 4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YP, Wang Q, Liu YC, and Xie Y (2014). Molecular basis for the targeted binding of RGD- containing peptide to integrin alphaVbeta3. Biomaterials 35, 1667–1675. [DOI] [PubMed] [Google Scholar]
- Zhang J, Valverde P, Zhu X, Murray D, Wu Y, Yu L, Jiang H, Dard MM, Huang J, Xu Z, et al. (2017). Exercise-induced irisin in bone and systemic irisin administration reveal new regulatory mechanisms of bone metabolism. Bone research 5, 16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Smith DL (1993). Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein science : a publication of the Protein Society 2, 522– 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.