

Abstract
Summary
Benchmark studies are widely used to compare and evaluate tools developed for answering various biological questions. Despite the popularity of these comparisons, the implementation is often ad hoc, with little consistency across studies. To address this problem, we developed SummarizedBenchmark, an R package and framework for organizing and structuring benchmark comparisons. SummarizedBenchmark defines a general grammar for benchmarking and allows for easier setup and execution of benchmark comparisons, while improving the reproducibility and replicability of such comparisons. We demonstrate the wide applicability of our framework using four examples from different applications.
Availability and implementation
SummarizedBenchmark is an R package available through Bioconductor (http://bioconductor.org/packages/SummarizedBenchmark).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
With the advancement of high-throughput technologies, data and computing have become key components of scientific discovery in biology. New computational methods to analyze genomic data are constantly being developed, with several methods often addressing the same biological question. As a result, researchers are now faced with the challenge of deciding between a plethora of tools, each leading to slightly different answers.
For several common analyses in computational biology, benchmark studies have been published to help users pick an appropriate tool from a subset of alternatives. This includes studies of methods for differential expression (DE) analysis in bulk RNA-seq (Schurch et al., 2016) and single-cell RNA-seq (Soneson and Robinson, 2018), peak calling in ChIP-seq (Wilbanks and Facciotti, 2010), genetic variant calling (Hwang et al., 2015), and transcript assembly (Steijger et al., 2013), among others. These studies often assess and compare the performance of different methods using several complementary data sets, and evaluate the methods according to carefully chosen metrics. As a result, users are able to make a decision based on the results presented for the conditions that most closely resemble their own study.
Given the importance of benchmark comparisons, a number of software packages have been developed for streamlining this process, including rnaseqcomp (Teng et al., 2016) and iCOBRA (Soneson and Robinson, 2016). These packages provide performance metrics, data visualizations, and curated datasets for comparing methods. However, these frameworks were designed to address specific problems or classes of problems, such as isoform quantification, and binary classification and feature ranking. Furthermore, these packages only provide resources for evaluating the results of computational methods, but they do not provide a framework to design and execute benchmark experiments. By excluding the early steps of the benchmark process, these frameworks lose important metadata, such as software version and parameters used, leaving much of the burden of benchmarking to the user.
To address these limitations, we developed SummarizedBenchmark, a general framework for designing, executing, and evaluating benchmark comparisons. In addition to providing a flexible grammar to structuring benchmark comparisons, the package facilitates reproduction and replication of benchmark studies by tying metadata to the benchmark results. In the following sections, we describe the main features of SummarizedBenchmark.
2 Materials and methods
The general framework of the SummarizedBenchmark package is shown in Figure 1, where a benchmarking experiment is formulated as the combination of methods, data, and performance metrics. Methods, which we define to include parameter settings, are applied to data and evaluated according to the set of performance metrics. In the SummarizedBenchmark package, actions to perform these tasks are split into two major steps: benchmark design and benchmark evaluation.
Fig. 1.
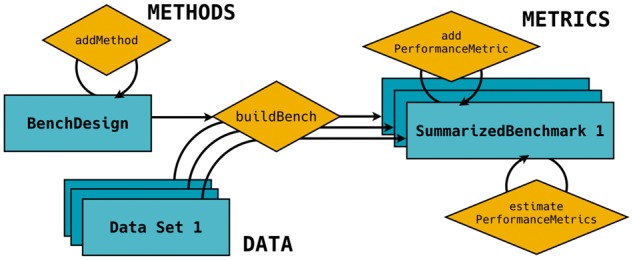
General structure of a benchmark experiment with SummarizedBenchmark. Objects are shown as rectangles and methods as diamonds. A BenchDesign object is first constructed with the methods of interest. The BenchDesign is executed with any number of data sets using buildBench, and the results are stored in a SummarizedBenchmark object along with relevant metadata. Performance metrics can be added and estimated for the SummarizedBenchmark
2.1 Benchmark design
The design of the benchmark experiment is stored in a BenchDesign object. This object contains methods to be benchmarked and, optionally, the data set to be analyzed. Methods not written in R can be passed as wrapper functions with system calls. Methods are added to the BenchDesign without being immediately run. This approach not only allows the user to delay evaluation until the entire design has been constructed, but it also enables the same design, i.e. methods, to be applied to various data sets, as is commonly done in benchmarking studies. The approach used for storing methods in the BenchDesign also helps extract useful metadata such as software version and parameter settings when the methods are evaluated.
After adding all methods, a simple call to buildBench executes each method on the data set included in the BenchDesign or passed to the function. The execution process can be parallelized, so that each method runs separately. Error handling is also implemented to prevent a single failed method from terminating the entire benchmarking process. Additionally, as benchmarking can be a iterative process, the software also includes features for updating experiments, such that after the first execution, methods can be further added or modified without rerunning all methods.
2.2 Benchmark evaluation
Once the benchmark is executed, the results are stored in a SummarizedBenchmark object. The SummarizedBenchmark class extends the SummarizedExperiment class, a core data structure of the Bioconductor ecosystem (Huber et al., 2015). The object contains the results as one or more matrices where each column corresponds to a method in the original BenchDesign. Metadata for columns, such as software version and method parameters, as well as metadata for rows, which may include ground truth or expected values when available, are also stored in the object and tied directly to the results.
The SummarizedBenchmark class contains a field to store functions that define performance metrics. Users can manually define functions relevant to their specific benchmark or use common metrics used to compare methods, which are available by default. After defining or selecting performance metrics, users can evaluate these metrics and either store them as column metadata or retrieve them as a long formatted data for downstream analysis. We implemented functions to visualize method performance as ROC curves and explore the method overlaps using upsetR plots (Conway et al., 2017). We also provide instructions to convert SummarizedBenchmark classes into data structures of other benchmarking packages, such as rnaseqcomp and iCOBRA.
3 Results
SummarizedBenchmark provides a flexible grammar to design benchmark experiments. We demonstrate this flexibility by designing benchmarks for four different analyses: DE testing, isoform quantification, single-cell RNA-seq simulation, and false discovery rate control (see http://bioconductor.org/packages/SummarizedBenchmark/). Parts of the BenchDesign and SummarizedBenchmark objects from the benchmark comparison of DE methods are shown in Supplementary Figure S1.
4 Discussion
The innovations of the SummarizedBenchmark framework can be summarized in three points. First, by integrating the design and execution of the benchmark experiment into the framework, SummarizedBenchmark allows tracking of important metadata such as software version and parameters. Storing such metadata is crucial for reproducibility as methods are constantly improved and results can vary with parameter settings. Second, it extends well-established Bioconductor infrastructure to store the results of benchmark experiments and to enable users to use metrics to summarize and visualize the performance of methods. Finally, SummarizedBenchmark provides a versatile framework to perform benchmark comparisons that is not tied to specific applications. We anticipate that SummarizedBenchmark will be useful for researchers performing benchmark analyses as well as bioinformatics facilities that often run several methods on many different datasets.
Supplementary Material
Acknowledgements
The authors would like to thank members of the Irizarry lab at the Dana-Farber Cancer Institute for providing feedback on earlier versions of the software.
Funding
This work was supported by the National Institutes of Health [grant numbers R01HG005220, R01GM083084, P41HG004059].
Conflict of Interest: none declared.
References
- Conway J.R. et al. (2017) Upsetr: an r package for the visualization of intersecting sets and their properties. Bioinformatics, 33, 2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W. et al. (2015) Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods., 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S. et al. (2015) Systematic comparison of variant calling pipelines using gold standard personal exome variants. Scientific Reports, 5, 17875.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch N.J. et al. (2016) How many biological replicates are needed in an rna-seq experiment and which differential expression tool should you use? RNA, 22, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., Robinson M.D. (2016) icobra: open, reproducible, standardized and live method benchmarking. Nat. Methods., 13, 283.. [DOI] [PubMed] [Google Scholar]
- Soneson C., Robinson M.D. (2018) Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods., 15, 255.. [DOI] [PubMed] [Google Scholar]
- Steijger T. et al. (2013) Assessment of transcript reconstruction methods for rna-seq. Nat. Methods., 10, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M. et al. (2016) A benchmark for rna-seq quantification pipelines. Genome Biol., 17, 74.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbanks E.G., Facciotti M.T. (2010) Evaluation of algorithm performance in chip-seq peak detection. PLoS One, 5, e11471.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.