

Abstract
Motivation
Recent advances in genomics and precision medicine have been made possible through the application of high throughput sequencing (HTS) to large collections of human genomes. Although HTS technologies have proven their use in cataloging human genome variation, computational analysis of the data they generate is still far from being perfect. The main limitation of Illumina and other popular sequencing technologies is their short read length relative to the lengths of (common) genomic repeats. Newer (single molecule sequencing – SMS) technologies such as Pacific Biosciences and Oxford Nanopore are producing longer reads, making it theoretically possible to overcome the difficulties imposed by repeat regions. Unfortunately, because of their high sequencing error rate, reads generated by these technologies are very difficult to work with and cannot be used in many of the standard downstream analysis pipelines. Note that it is not only difficult to find the correct mapping locations of such reads in a reference genome, but also to establish their correct alignment so as to differentiate sequencing errors from real genomic variants. Furthermore, especially since newer SMS instruments provide higher throughput, mapping and alignment need to be performed much faster than before, maintaining high sensitivity.
Results
We introduce lordFAST, a novel long-read mapper that is specifically designed to align reads generated by PacBio and potentially other SMS technologies to a reference. lordFAST not only has higher sensitivity than the available alternatives, it is also among the fastest and has a very low memory footprint.
Availability and implementation
lordFAST is implemented in C++ and supports multi-threading. The source code of lordFAST is available at https://github.com/vpc-ccg/lordfast.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
High throughput sequencing (HTS) technologies have been evolving since their inauguration (Margulies et al., 2005). Especially, recent advances in single molecule sequencing (SMS) technologies such as Pacific Biosciences (Eid et al., 2009; Korlach et al., 2010) and Oxford Nanopore (Cherf et al., 2012; Eisenstein, 2012; Manrao et al., 2012) are a breakthrough in this evolution. Although the next generation sequencing (NGS) technologies have proven their ability in detection of genetic variation (1000 Genomes Project Consortium, 2010, 2012), finding disease causing mutations (O’Roak et al., 2011) and building de novo genome assemblies (Gnerre et al., 2011), computational analysis based on NGS data is still far from being perfect due to their short read length relative to the lengths of common repeat sequences (Alkan et al., 2011; Hormozdiari et al., 2009).
The increase in the length of the reads generated by SMS technologies offers solutions to many unresolved questions in genomics. Unfortunately, SMS technologies introduce higher error rates (≃15% versus 1%) with different types of errors (insertions and deletions rather than substitutions) making it difficult to use them in standard genomic analysis pipelines. This promoted the development of new algorithms and pipelines specifically tailored for SMS technologies capable of handling very long, erroneous reads for various applications. These applications include de novo assembly (Berlin et al., 2015; Chin et al., 2013; Koren et al., 2013; Loman et al., 2015), hybrid de novo assembly (Goodwin et al., 2015; Koren et al., 2012) (where the long reads are mixed with more accurate short reads from NGS), gap filling in scaffolds (English et al., 2012), genome finishing (Bashir et al., 2012; Brown et al., 2014; Chin et al., 2013), reconstruction of GC-rich and complex regions (Huddleston et al., 2014; Scott and Ely, 2014; Shin et al., 2013), structural variation (SV) detection (Chaisson et al., 2015; Doi et al., 2014; Fan et al., 2017; Huddleston et al., 2017; Ummat and Bashir, 2014), haplotype phasing (Chaisson et al., 2017; Pendleton et al., 2015) and finding methylation sites (Rand et al., 2017; Simpson et al., 2017).
The very first step for most of the downstream analysis pipelines involves mapping the reads to a reference genome. On an Illumina-like short read with a low error rate, it is usually possible to find a ‘long’ substring that would exactly match its mapping locus on the reference genome. All existing tools for mapping short reads are based on this fundamental observation. They aim to find such exact matches by using either (i) BW Transform/FM Index (Burrows and Wheeler, 1994; Ferragina and Manzini, 2000) based methods (Langmead and Salzberg, 2012; Li et al., 2009; Li and Durbin, 2009), or (ii) substring hashing (Alkan et al., 2009; David et al., 2011; Gontarz et al., 2013; Hach et al., 2010, 2014; Lin et al., 2008; Weese et al., 2012; Xin et al., 2013), or (iii) hybrid methods that combine FM Index with hashing (Marco-Sola et al., 2012; Siragusa et al., 2013). Unfortunately, because of high error rates [up to 20% reported for PacBio (Travers et al., 2010; Thompson and Milos, 2011) and up to 15% reported for Oxford Nanopore (Rang et al., 2018)], this key observation is not valid for SMS technologies, this key observation is not valid for SMS technologies. Furthermore, even when the mapping locus for a read can be correctly found, it is quite difficult to differentiate sequencing errors from actual genomic variants.
There are a number of available methods for mapping long reads to the reference genome. BLASR (Chaisson and Tesler, 2012) is the first tool specifically designed for PacBio reads. It finds all sufficiently long exact matches between a long read and a reference genome using a suffix array index. Then it groups the matches into clusters and ranks them by a frequency weighted score. The top scored clusters which correspond to candidate genomic locations are used for performing sparse dynamic programing (SDP) followed by a banded alignment. BWA-MEM (Li, 2013) is another mapper that was originally designed to align short sequence reads as well as assembled contigs to a reference genome. It has been also extended to map long SMS reads by tuning its alignment parameters (via option -x pacbio or -x ont2d). BWA-MEM achieves this by finding the longest exact match covering each query position as a possible initial match, chaining these matches (and filtering out those chains ‘contained’ by others), ranking the initial matches by the length of the chains containing them, and finally extending the initial matches based on a specific score cutoff, to get a complete alignment. Another tool, rHAT (Liu et al., 2016), is a hash table based mapper that uses a heuristic to estimate the approximate location of the mapping for each read. This is done by finding potential mapping regions for the middle 1000 bp segment of the long read on the reference genome through an approximate k-mer counting scheme. Then, for each potential mapping region, a lookup table is built to find short seeds and a chain of these seeds using an SDP-based heuristic. The final alignment is formed from the selected chains. A fourth tool, GraphMap (Sović et al., 2016), uses gapped spaced seeds and performs an approximate alignment by clustering these seeds. It then constructs alignment anchors by finding an exact walk in their ‘alignment graph’ built from short k-mers of the target, chains these anchors, and finally refines the chain to generate the final alignment. Another tool, LAMSA (Liu et al., 2017), splits the long read to some ‘seeding fragments’ and finds all their approximate matches on the reference genome using GEM mapper (Marco-Sola et al., 2012). It then finds the ‘skeleton’ of the alignment using a directed acyclic graph based on SDP. Lastly, LAMSA prioritizes the candidate skeletons and fills the gaps within the skeletons while accounting for different possible structural differences (e.g. large deletions). Recently, two new mappers, NGMLR (Sedlazeck et al., 2018) and Minimap2 (Li, 2018), have been published. Similar to LAMSA, NGMLR starts by finding alignments of subsegments of a read that are aligned by a single linear alignment. For each pair of such subsegment alignments, it then performs a pairwise sequence alignment using a convex gap-cost model. It finally scans inside alignments to identify regions with low sequence identity that exist due to small SVs. Minimap2 uses the notion of minimizers (Roberts et al., 2004) for indexing the reference and finding seeds. It then performs chaining of the seeds and identifies the primary chains. At the end, it performs alignment between adjacent anchors of chains using its fast implementation based on SSE instructions.
Among the above tools, BLASR and BWA-MEM are sensitive but too slow in mapping large datasets. Speed is becoming a major issue since the delivery of Pacbio Sequel by Pacific Biosciences and the introduction of PromethION device by Oxford Nanopore, which promises higher throughput for long-read data at a lower cost. On the other hand, tools like rHAT and LAMSA are not sensitive enough to find the correct mapping locations for many reads. For instance, the candidate selection step of rHAT uses the seeds only from the middle 1000 bp segment of the long reads which could be problematic especially if that segment comes from repetitive regions.
In this paper, we introduce lordFAST, a novel long-read mapper that is especially designed for PacBio’s continuous long reads (CLR). lordFAST is a highly efficient and sensitive aligner that can tolerate high sequencing error rates observed in CLR reads, through its use of multiple short exact matches. lordFAST not only maps more reads in a PacBio dataset but also maps them more accurately than the available alternatives such as BLASR and BWA-MEM. It is worth mentioning that lordFAST is also capable of aligning reads generated by Oxford Nanopore Technology since the error models are somewhat similar. Our experimental results show that Minimap2 is the fastest tool among the above mappers. lordFAST achieves the highest sensitivity and precision on simulated data. This is especially due to the fact that it maps the highest number of bases correctly among all mappers we tested.
2 Materials and methods
2.1 Overview
lordFAST is a heuristic anchor-based aligner for long reads generated by third generation sequencing technologies. lordFAST aims to find a set of candidate locations (ideally, only one) per read before the costly step of base-to-base alignment to the reference genome. lordFAST works in two main stages. In stage one, it builds an index from the reference genome, which is used to find short exact matches. The index is a combination of a lookup table and an (uncompressed) FM index. In stage two, it maps the long reads to the reference genome in four steps: (i) on each read, it identifies a fixed number of evenly spaced k-mers (k = 14 in the default settings), which are matched to the reference genome through the use of the index. For each such match, it obtains the longest exact matching (prefix) extension. Among these extended matches of each k-mer identified in each read, it finally chooses the longest (there could be more than one) which acts as anchor matches; (ii) for each read, it then splits the reference genome into overlapping windows (of length twice that of the read) and identifies each such window as a candidate region if the number of anchor matches in that window is above a threshold value; (iii) for each candidate region, it identifies the longest chain of ‘concordant’ anchor matches (i.e. chain of anchor matches which have equal respective spacing in the read and the reference genome); (iv) it obtains the base-to-base alignment by performing dynamic programing between consecutive anchor matches in the selected chain. We provide a more detailed description for each step below.
2.2 Stage one: reference genome indexing
In order to build a (substring) index for the reference genome, we use a combination of a simple lookup table for initial short matches, and an (uncompressed) FM index for extending such initial matches. This combined index benefits from the speed of lookup table and the compactness of the Burrows–Wheeler transform (BWT) representation for the reference genome. The lookup table (with 4h entries for all possible h-mers) provides a constant time search capability for each h-mer’s position in the uncompressed FM index (Ferragina and Manzini, 2000) (in the default setting h = 12, but the user is given the option to pick any value). As is well known, the FM index provides a compact representation of a suffix array (Manber and Myers, 1993) which we use to find (exact matching) extensions of initial h-mer matches.
Note that in order to be able to perform efficient search on both strands of the reference genome, we use an extension to the FM index implemented in fermi (Li, 2012). Our combined index provides a 29% speed up over the standard uncompressed FM index (see Supplementary Figs S2 and S3 for speed gain and extra memory usage).
2.3 Stage two: read mapping
Given a set of long reads, lordFAST aligns one read at a time as follows:
2.3.1 Step 1: sparse extraction of anchor matches
For a given read with length ℓ, lordFAST identifies C (user defined, default 1000) evenly spaced anchoring positions on the read. For each anchoring position, it finds the longest prefix match(es) (of length at least k = 14) to the genome as follows. First, it extracts the first h-mer starting from the anchoring position and uses the lookup table of the genome index to obtain the interval that represents the initial set of matching locations on the FM index. It then uses the LF-mapping operation of the FM index to extend the initial set of matches and identify the longest match(es). Note that using longest matches reduces the total number of anchor matches significantly.
The longest matches are then added to the set of anchors, , as triplets (r, g, s) where r is the anchoring position on the read, g is the starting location of the longest match on the genome and s is the length of the match. At the end of this step, is partitioned into and based on the strand of the matching location on the genome. (Note that for reads that are ‘too short’, i.e. ℓ<C + k – 1, we use ℓ – k + 1 anchoring positions instead of C anchoring positions.)
2.3.2 Step 2: candidate region selection
In order to select the candidate regions for alignment, lordFAST splits the reference genome into overlapping windows of size 2ℓ (as illustrated in Fig. 1a). For each window, it calculates two scores for the forward and reverse strands from anchor matches of the respective strands ( and ). For each anchor match falling in a window, it adds s – k + 1 to the score of that window. lordFAST keeps all the windows with score >scoremax∕f where f is the factor defining the significance of the window score (default 4) and scoremax is the maximum window score. In other words, lordFAST keeps those windows whose score is not significantly worse than the maximum window score. In cases where two overlapping windows both meet the minimum window score requirement, lordFAST will keep the one with higher window score in the final list (ties are broken by choosing the window with smaller reference coordinate). Figure 1b depicts an example of the selection process.
Fig. 1.
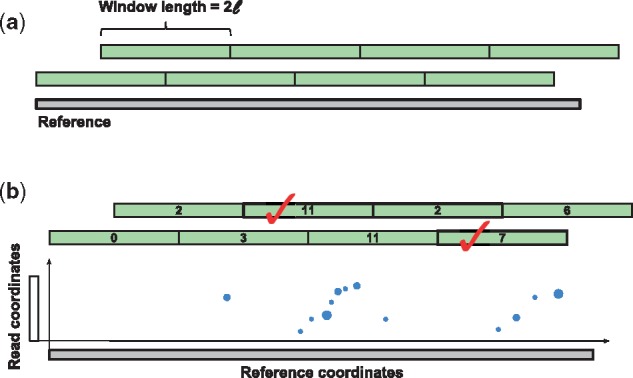
(a) The implicit windows considered on the reference genome for the candidate selection step. If the read length is ℓ, then the windows are of size 2ℓ and overlap by ℓ bases. (b) An example of the candidate selection step. Each dot represents an anchor and its size represents the weight of the anchor. In this example, f = 2, and since the maximum window score is 11, every window with score ≤5.5 will be ignored. In addition, the window with score 6 is not kept since it is overlapping with a window with score 7. Also, only one of the windows with score 11 will be in the final list of candidates since the other window is overlapping it
2.3.3 Step 3: chaining and anchor selection
Among all the anchor matches in a candidate region, lordFAST chooses a set of ‘concordant’ anchors using local chaining. The best local chain is a set of co-linear, non-overlapping anchors on the reference genome that has the highest score among all such sets (Ohlebusch and Abouelhoda, 2005). To calculate the best local chain, lordFAST assigns a weight to each anchor match equal to the length of the match. lordFAST supports two chaining algorithms. By default, it obtains the best chain using the dynamic programing based chaining algorithm (Ohlebusch and Abouelhoda, 2005). Note that the time complexity of this chaining algorithm is quadratic, but in practice, it is fast due to our small number of anchor matches per read. It is also possible for the user to select the alternative chaining algorithm based on clasp (Otto et al., 2011). The anchor matches in the best local chain form the basis of the alignment in that region.
2.3.4 Step 4: alignment
lordFAST prioritizes the candidate regions based on their best chaining score and performs the final alignment for the top N regions (default value for N is 10). In order to generate the base-to-base alignment of a region, it uses anchor matches from the top scoring chain and performs banded global alignment for gaps between pairs of consecutive anchor matches. Furthermore, the alignment between the prefix of the read and the reference prior to the first anchor can be performed by the use of an anchored global-to-local alignment and the alignment between the suffix of the read and the reference following the last anchor can be computed in an identical fashion. This strategy is a widely used technique to avoid computing the full alignment between long sequences as that needs huge memory and computational time. lordFAST uses Edlib (Šošić and Šikić, 2017) for computing the global alignments and ksw library (https://github.com/attractivechaos/klib) for computing the global-to-local alignments. Edlib is a library implementing a fast bit-vector algorithm devised by Myers (1999). ksw, on the other hand, provides alignment extension based on affine gap-cost model.
It is worth mentioning that lordFAST supports clipping as follows: if the prefix of the read before the first anchor (or, respectively, suffix of the read after the last anchor) has an alignment score/similarity which is lower than a threshold (thclip), lordFAST performs clipping of that prefix (or, respectively, suffix). This is done by using ksw library to extend the alignment as long as a significant drop in the alignment score/similarity is not observed (using a parameter similar to BLAST’s X-dropoff).
In addition, lordFAST supports split alignment as follows: Let Si,j denote the substring of S that starts at position i and ends at position j. Suppose we are mapping a long read R to the reference genome G. Consider two consecutive anchors A = (rA, gA, sA) and B = (rB, gB, sB), as per the definition above, in the best chain chosen for a candidate window. If the alignment between and has a score lower than a threshold (thsplit) we split the alignment and report one alignment as primary and another as supplementary (as the definition in the SAM format specification). One alignment corresponds to the substring before anchor A and the other alignment corresponds to the substring after anchor B. Furthermore, since the drop in alignment score/similarity could be due to the presence of an inversion, we check if the alignment between the reverse complement of and has a score higher than thsplit. In that case, such an alignment will be also reported as another supplementary alignment.
3 Results
We evaluated the performance of lordFAST-v0.0.9 against BLASR-v5.3.4323a52 (Chaisson and Tesler, 2012), BWA-MEM-v0.7.15-r1140 (Li, 2013), GraphMap-v0.5.1 (Sović et al., 2016), LAMSA-v1.0.0 (Liu et al., 2017), rHAT-v0.1.1 (Liu et al., 2016), NGMLR-v0.2.6 (Sedlazeck et al., 2018), Minimap2-v2.10-r761 (Li, 2018) and another recently available software minialign-v0.5.3 (https://github.com/ocxtal/minialign). Note that although GraphMap is specifically designed for Oxford Nanopore reads, we included it in our experiment as it is capable of mapping PacBio long reads with default parameters (Sović et al., 2016).
We compared the methods on both simulated and real datasets. We used the results on the simulated dataset for calculating the methods’ precision and recall. All experiments were performed on a server running Cenots 6.9 equipped with 4 twelve-core (2 threads per core) Intel(R) Xeon(R) CPU processors (E7-4860 v2 @ 2.60 GHz) and 1000 GB RAM. The commands and parameters used to run each tool are provided in the Supplementary Material.
Note that on real PacBio datasets, we observed that more than 99% of the sequence data are provided in reads of length 1000 bp or longer (see Supplementary Fig. S1 for details). Thus, we only focused on aligning reads that are 1000 bp or longer.
3.1 Experiment on a simulated dataset
3.1.1 Simulation without structural variations
To evaluate the precision and recall of lordFAST against the above mentioned tools, we simulated 25 000 long reads from hg38 using PBSIM (Ono et al., 2013) which infers the read length and error model from a real human read dataset (see Supplementary Material for details). Note that we did not introduce any SNPs, indels or structural variants in this experiment, i.e. the correct alignment between a read and the reference genome has mismatches and gaps only due to (simulated) read errors. For each read, PBSIM provides both the originating location on the reference genome and the ‘true’ base-to-base alignment of the read to the reference genome in that location. Because for any base on any read, its ‘true’ base pairing on the reference genome is known, we have been able to calculate the number of correctly mapped reads/bases.
We consider a read to be correctly mapped if (i) it gets mapped to the correct chromosome and strand; and (ii) the subsequence on the reference genome the read maps to, overlaps with the ‘true’ mapping subsequence by at least p bases. In order to compare the methods we tested with respect to the number of correctly mapped reads, we used two values of p: a fixed value of 1 bp and a variable value which is set to 90% of the length of the originating ‘true’ mapping subsequence. Note that, for most of the methods there is not a big difference between the results based on the two settings for p; however some methods cannot identify the ‘correct’ mapping subsequence in its entirety and report only a partial alignment—accordingly, those methods perform poorly for the variable setting of p.
We consider a base in a read to be correctly mapped if (i) the read is correctly mapped (as per the definition above) and (ii) the mapped location of the base is within 25 bp of the true alignment locus of the base (a smaller value for the second condition makes the definition of a correctly mapped base more stringent. Supplementary Tables S1 and S2 show the result when this threshold is increased to 50 bp and decreased to 5 bp, respectively). Sensitivity is thus defined as the fraction of correctly mapped bases (according to this notion of a correct mapping) out of the total number of bases in the reads. Similarly, precision is defined as the fraction of correctly mapped bases out of the total number of mapped bases in the reads.
Using the definitions above, we compared all of the above mentioned methods; a summary of the results are presented in Table 1. As can be seen, lordFAST not only maps more reads correctly than any other mapper, but also aligns about 98.9% of the total number of bases correctly, which is 0.9–9.4% more than its competitors. In addition, lordFAST achieves the highest base sensitivity and precision. It is important to note that for GraphMap, the precision value is much higher than the sensitivity because it leaves many of the bases unmapped. In that sense, we believe that sensitivity provides a much better measure to compare the tools—even though lordFAST is the best with respect to both measures. On this small dataset, Minimap2 is the fastest tool followed by minialign and lordFAST. However, on a larger simulated dataset, minialign performs better in terms of running time (see Supplementary Table S4). BWA-MEM, lordFAST and LAMSA show the lowest memory footprint.
Table 1.
Comparison between different tools capable of mapping PacBio long reads on the simulated human dataset
| Minimum | Correctly | Correct | Incorrect | Unmapped | Sensitivitya | Precisionb | Timec | Memoryc | |
|---|---|---|---|---|---|---|---|---|---|
| overlap (p) | Mapper | mapped | bases (Mb) | bases (Mb) | bases (Kb) | (%) | (%) | (s) | (GB) |
| 1 bp | BLASR | 24 642 | 164.52 | 18.39 | 698.22 | 89.61 | 89.95 | 9233 | 14.67 |
| BWA-MEM | 24 603 | 170.63 | 12.50 | 525.11 | 92.91 | 93.17 | 6842 | 5.22 | |
| GraphMap | 24 161 | 177.26 | 4.05 | 2297.27 | 96.55 | 97.77 | 17 546 | 42.56 | |
| LAMSA | 24 458 | 176.00 | 6.40 | 282.15 | 96.36 | 96.51 | 1277 | 5.85 | |
| rHAT | 24 409 | 177.59 | 5.63 | 391.52 | 96.72 | 96.93 | 1044 | 13.95 | |
| NGMLR | 24 194 | 170.50 | 8.86 | 4246.51 | 92.86 | 95.06 | 2970 | 5.45 | |
| Minimap2 | 24 745 | 180.06 | 3.34 | 223.46 | 98.06 | 98.18 | 154 | 6.50 | |
| minialign | 24 567 | 178.25 | 4.73 | 621.60 | 97.08 | 97.41 | 201 | 12.70 | |
| lordFAST | 24 751 | 181.68 | 1.89 | 29.35 | 98.95 | 98.97 | 696 | 5.43 | |
| 90% | BLASR | 24 563 | 164.46 | 18.47 | 675.95 | 89.57 | 89.90 | ||
| BWA-MEM | 24 485 | 170.23 | 12.98 | 417.84 | 92.70 | 92.91 | |||
| GraphMap | 24 161 | 177.26 | 4.05 | 2297.27 | 96.55 | 97.77 | |||
| LAMSA | 24 371 | 176.87 | 6.59 | 208.22 | 96.30 | 96.41 | |||
| rHAT | 24 372 | 177.55 | 5.98 | 80.98 | 96.70 | 96.74 | |||
| NGMLR | 23 769 | 169.66 | 10.44 | 3508.56 | 92.40 | 94.20 | |||
| Minimap2 | 24 740 | 180.04 | 3.35 | 223.20 | 98.05 | 98.17 | |||
| minialign | 24 469 | 177.84 | 5.53 | 233.74 | 96.86 | 96.98 | |||
| lordFAST | 24 747 | 181.68 | 1.90 | 29.10 | 98.95 | 98.97 | |||
Note: This dataset contains 25 000 reads and 183.61 million bases. Best result in each column is marked with bold typeface. A read is considered to be mapped correctly if its aligned subsequence in the reference overlaps with the ‘correct’ mapping subsequence by at least p bases. On the other hand, a base in a read is considered to be correctly mapped if the read is correctly mapped and the mapping location of the base is within a 25 bp vicinity of the correct alignment locus of the base.
The sensitivity is defined as the number of correctly mapped bases/the total number of bases.
The precision is defined as the number of correctly mapped bases/the number of mapped bases.
The running time and peak memory usage are measured using /usr/bin/time -v Unix command.
We also evaluated the ability of different tools to distinguish between unique and repetitive hits in terms of the assigned mapping quality (MAPQ) per Li (2018). For this evaluation, a read is considered as correctly mapped if its best mapping aligns to a region of the reference that (i) overlaps with 10% of the ‘true’ mapping region (Fig. 2a), and (ii) 90% of the ‘true’ mapping region (Fig. 2b). In general, Minimap2 and lordFAST map higher portion of reads with high MAPQ to correct location compared to other tools, especially with the more stringent definition of the correct mapping (see Fig. 2b).
Fig. 2.
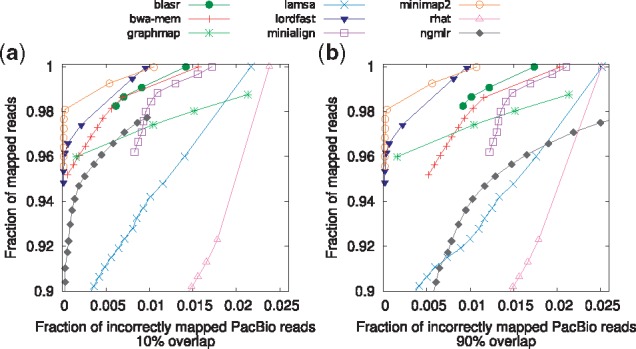
Read mappings are sorted based on their mapping quality in descending order. Then for each mapping quality threshold, the fraction of mapped reads with mapping quality above the threshold (out of total number of reads) and the fraction of incorrectly mapped read (out of the number of mapped reads) are plotted along the curve
3.1.2 Simulation in presence of structural variations
In order to evaluate the capability of lordFAST for mapping reads that span SVs, we performed another experiment to detect simulated SVs using Sniffles (Sedlazeck et al., 2018). Sniffles requires minimum of 15× coverage to have a good accuracy. Therefore, for this experiment, we focused on only chr1 and generated a simulated dataset by inserting 21 SVs from DGV (nine insertions, nine deletions and three inversions) of different sizes (See Supplementary Material for details).
Here, we provide the results of SV calling using Sniffles based on the mappings from different tools. We define a call as ‘exact’ if (i) its start and end coordinates are at most 25 bp away from the actual simulated breakpoints; and (ii) it overlaps with one simulated SV of the same type. However, if the first condition is not satisfied, the call is considered as ‘inexact’. If the second condition is not satisfied the call is considered as ‘mis-classified’. If none of the conditions is satisfied the call is considered as ‘wrong’. Among all mappers, Sniffles generated SV calls only for NGMLR, BWA-MEM, rHAT and lordFAST. As it can be seen in the Table 2, all calls based on rHAT mappings are wrong. Also, Sniffles finds more ‘exact’ calls with lordFAST and NGMLR mappings in comparison to mappings provided by BWA-MEM. This suggests that lordFAST does not generate misalignments around SV breakpoints.
Table 2.
Structural variations called by Sniffles based on mappings from different tools
| Mapper | # calls | # exact | # inexact | # mis-classified | # wrong |
|---|---|---|---|---|---|
| NGMLR | 19 | 17 | 1 | 0 | 1 |
| BWA-MEM | 18 | 12 | 5 | 0 | 1 |
| rHAT | 35 | 0 | 0 | 0 | 35 |
| lordFAST | 17 | 16 | 1 | 0 | 0 |
3.2 Experiment on a real dataset
We evaluated the above methods on a real dataset, containing 23 155 reads sequenced from a human genome (CHM1; Supplementary Material contains details related to this dataset).
Since the true mapping locations of the reads are not known a priori, we compared methods based on the quality of their reported alignments. For each mapping of a read, we count the number of its bases that are aligned to the identical bases in the reference (matched bases). In addition, we calculated the alignment score by adding up +1 for every matching base and –1 for every mis-matching, inserted, deleted or unmapped/clipped base. For each tool, we reported the sum of alignment scores of all the reads in the dataset. Although the number of matched bases per se may not be the best comparison measure (since one could match all the bases in the read without paying attention to the gaps created in the reference), it is complementary to the alignment score. If a program tries to greedily maximize the number of matched bases, it will very likely produce a low alignment score. Table 3 shows the result of this experiment. lordFAST has the highest total alignment score. More precisely, lordFAST reports 2.79 million higher alignment score and 1.74 million higher number of matched bases compared to the closest competitor.
Table 3.
Evaluation of the performance of various long-read mappers on a real human dataset
| Mapped | Mapped | Matched | Alignmenta | Timeb | Memoryb | |
|---|---|---|---|---|---|---|
| Mapper | reads | bases (Mb) | bases (Mb) | score | (s) | (GB) |
| BLASR | 22 866 | 163.11 | 148.58 | 108 002 225 | 12 243 | 14.96 |
| BWA-MEM | 22 913 | 170.76 | 154.15 | 119 117 389 | 8810 | 5.25 |
| GraphMap | 22 159 | 169.57 | 151.93 | 113 717 041 | 17 745 | 42.56 |
| LAMSA | 23 154 | 173.90 | 155.68 | 122 035 697 | 2040 | 6.29 |
| rHAT | 23 136 | 159.99 | 142.40 | 92 824 214 | 1769 | 13.95 |
| NGMLR | 21 295 | 155.83 | 143.06 | 97 830 317 | 4629 | 5.43 |
| Minimap2 | 22 818 | 170.97 | 154.78 | 119 673 199 | 262 | 6.57 |
| minialign | 23 006 | 152.61 | 139.22 | 89 538 289 | 207 | 12.70 |
| lordFAST | 22 961 | 176.18 | 157.42 | 124 826 081 | 765 | 5.43 |
Note: This dataset includes 23 155 reads and 178.45 million bases. Best result in each column is marked with bold typeface.
The alignment score is calculated by adding up +1 for every matching base and −1 for every mis-matching, inserted, deleted or unmapped/clipped base.
The running time and peak memory usage are measured using /usr/bin/time -v Unix command.
We also measured the agreement between various methods based on their alignment of the reads. For a given read, an alignment x covers another alignment y if and only if the subsequence on the reference genome covered by x overlaps with at least 90% of the subsequence on the reference genome covered by y. Figure 3 shows examples of covering and non-covering alignments. Table 4 shows how best alignments from different methods cover each other. More specifically, each row contains the percentage of mappings reported by the corresponding tool that cover mappings of other tools. For instance, among all reads for which both lordFAST and BLASR report an alignment, 90.84% of the alignments reported by BLASR are covered by lordFAST, while only 88.28% of the alignments reported by lordFAST are covered by BLASR. As can be observed, lordFAST alignments provide a high coverage of the alignments obtained by the alternative tools.
Fig. 3.
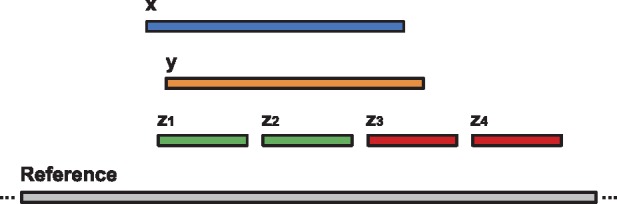
Examples of covering and non-covering alignments. Suppose x, y, z1, z2, z3 and z4 are different alignments of the same read. In this figure, alignments x and y cover each other as they span the subsequences on the reference genome that have at least 90% overlap. The alignments x and y cover alignments z1 and z2 but not the alignments z3 and z4. On the other hand, the alignments z1, z2, z3, and z4 do not cover either alignment x or y
Table 4.
Agreement of different methods in reporting alignments
| BLASR | BWA | GraphMap | LAMSA | rHAT | NGMLR | Minimap2 | minialign | lordFAST | |
|---|---|---|---|---|---|---|---|---|---|
| BLASR | N/A | 92.38 | 90.13 | 89.40 | 89.57 | 97.10 | 93.04 | 91.82 | 88.28 |
| BWA | 90.10 | N/A | 87.45 | 87.25 | 86.99 | 95.11 | 90.98 | 91.09 | 86.34 |
| GraphMap | 92.47 | 92.55 | N/A | 89.06 | 90.76 | 96.69 | 92.54 | 91.71 | 91.59 |
| LAMSA | 85.74 | 87.06 | 83.91 | N/A | 84.12 | 91.02 | 86.13 | 88.11 | 83.89 |
| rHAT | 90.51 | 89.87 | 90.62 | 87.85 | N/A | 93.84 | 89.96 | 89.41 | 88.21 |
| NGMLR | 86.33 | 87.02 | 84.88 | 83.72 | 83.67 | N/A | 86.93 | 86.99 | 82.47 |
| Minimap2 | 92.89 | 93.45 | 89.83 | 89.17 | 89.35 | 97.48 | N/A | 93.01 | 88.25 |
| minialign | 79.97 | 81.54 | 77.40 | 78.49 | 77.57 | 84.92 | 80.88 | N/A | 77.11 |
| lordFAST | 90.84 | 91.76 | 91.96 | 89.20 | 88.77 | 94.44 | 91.01 | 91.79 | N/A |
Note: Each row shows the percentage of best alignments from the corresponding mapper that cover alignments from other mappers. Note that this table is not symmetric.
In addition, in Table 5, we compared the performance of the tools on reads for which their alignments do not agree. To give an example, there are 2930 reads for which BLASR does not cover alignments of lordFAST. For those reads, BLASR reports alignments with an average of 28.84% lower identity. In contrast, there are 2094 reads for which lordFAST does not cover BLASR’s alignments. For those reads, on average, lordFAST’s alignments have only 7.40% lower identity than BLASR’s. With a lack of the true mappings for the real dataset, the information in Tables 4 and 5 are some extra support for the fact that lordFAST’s alignments are reliable.
Table 5.
The performance of different methods on reads for which their alignments do not agree
| BLASR | BWA | GraphMap | LAMSA | rHAT | NGMLR | Minimap2 | minialign | lordFAST | |
|---|---|---|---|---|---|---|---|---|---|
| BLASR | N/A | −22.85 | −29.05 | 6.47 | −8.57 | 10.61 | −20.92 | −7.92 | −28.84 |
| (1747) | (2187) | (2454) | (2414) | (617) | (1585) | (1882) | (2930) | ||
| BWA | −4.78 | N/A | −25.10 | 14.25 | −2.58 | −2.16 | −11.00 | 5.89 | −20.97 |
| (2264) | (2780) | (2951) | (3010) | (1041) | (2059) | (2049) | (3074) | ||
| GraphMap | −16.66 | −34.19 | N/A | 5.78 | −10.97 | 3.05 | −30.42 | −16.13 | −36.39 |
| (1721) | (1708) | (2533) | (2137) | (704) | (1702) | (1907) | (2033) | ||
| LAMSA | −25.88 | −30.42 | −38.40 | N/A | −22.07 | −29.02 | −31.68 | −18.04 | −37.96 |
| (3261) | (2964) | (3566) | (3673) | (1913) | (3166) | (2735) | (4047) | ||
| rHAT | −17.68 | −25.53 | −41.22 | 0.17 | N/A | −22.59 | −25.16 | −12.33 | −37.63 |
| (2171) | (2320) | (2079) | (2814) | (1312) | (2291) | (2436) | (2723) | ||
| NGMLR | −37.90 | −46.62 | −43.60 | −21.69 | −34.93 | N/A | −46.60 | −36.96 | −44.03 |
| (3126) | (2973) | (3351) | (3770) | (3778) | (2983) | (2992) | (4024) | ||
| Minimap2 | −0.11 | −13.12 | −25.17 | 15.10 | −1.46 | 9.39 | N/A | 3.23 | −16.81 |
| (1626) | (1501) | (2253) | (2508) | (2464) | (537) | (1608) | (2697) | ||
| minialign | −26.03 | −28.14 | −34.78 | −9.67 | −22.32 | −29.23 | −30.00 | N/A | −33.90 |
| (4579) | (4229) | (5007) | (4981) | (5190) | (3211) | (4366) | (4530) | ||
| lordFAST | −7.40 | −17.31 | −29.20 | 17.64 | −2.82 | −14.90 | −17.58 | 1.84 | N/A |
| (2094) | (1887) | (1781) | (2500) | (2598) | (1183) | (2051) | (1889) |
Note: Each row shows the performance superiority of the corresponding method over other methods for the inconsistent alignments, in terms of the average identity difference. The numbers in parentheses (for each row) show the number of reads for which the corresponding method reports alignments that do not cover alignments of other methods. Note that this table is not symmetric.
We also compared the speed and memory requirement of all tools we tested on this dataset. Supplementary Figures S4 and S5 show this comparison when using multiple threads.
4 Discussion
The main performance/running time bottleneck in most genome sequence analysis pipelines is mapping. Since the introduction of the first HTS platforms, the bioinformatics research community has developed many mapping methods to address this issue. After the emergence of PacBio and Oxford Nanopore technologies that produce long but noisy reads mapping has again become a central bioinformatics challenge.
In this paper, we present lordFAST, a fast and highly sensitive mapping tool for long noisy reads. Its sparse anchor extraction strategy has an important impact on the speed of its chaining step. Our experiment on the simulated data shows that despite using a small number of anchors, lordFAST not only maps more reads to their true originating region compared to its competitors but also is highly accurate in base-level alignment (see Table 1).
In addition, lordFAST provides both clipped and split alignments of the reads. This makes lordFAST appropriate for aligning reads originating from regions with long SVs, so that downstream analysis of its alignments would be simpler for the task of variation discovery.
Funding
This work was supported in part by NIH grant GM108348, NSF grant 1619081, the Indiana University Grand Challenges Precision Health Initiative and NSERC Discovery Frontiers Grant on the Cancer Genome Collaboratory to S.C.S. and NSERC Discovery Grant to F.H..
Conflict of Interest: none declared.
Supplementary Material
References
- 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium (2012) An integrated map of genetic variation from 1, 092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C. et al. (2009) Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet., 41, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C. et al. (2011) Genome structural variation discovery and genotyping. Nat. Rev. Genet., 12, 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir A. et al. (2012) A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol., 30, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K. et al. (2015) Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol., 33, 623–630. [DOI] [PubMed] [Google Scholar]
- Brown S.D. et al. (2014) Comparison of single-molecule sequencing and hybrid approaches for finishing the genome of clostridium autoethanogenum and analysis of CRISPR systems in industrial relevant clostridia. Biotechnol. Biofuels, 7, 40.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows M., Wheeler D.J. (1994) A block-sorting lossless data compression algorithm. Technical Report. DEC Labs.
- Chaisson M.J., Tesler G. (2012) Mapping single molecule sequencing reads using basic local alignment with successive refinement (blasr): application and theory. BMC Bioinformatics, 13, 238.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M.J. et al. (2015) Resolving the complexity of the human genome using single-molecule sequencing. Nature, 517, 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M.J. et al. (2017) Resolving multicopy duplications de novo using polyploid phasing In: International Conference on Research in Computational Molecular Biology, pp. 117–133. Springer, Cham. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherf G.M. et al. (2012) Automated forward and reverse ratcheting of dna in a nanopore at 5-a precision. Nat. Biotechnol., 30, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C.-S. et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- David M. et al. (2011). Shrimp2: sensitive yet practical short read mapping. Bioinformatics, 27, 1011–1012. [DOI] [PubMed] [Google Scholar]
- Doi K. et al. (2014) Rapid detection of expanded short tandem repeats in personal genomics using hybrid sequencing. Bioinformatics, 30, 815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J. et al. (2009) Real-time dna sequencing from single polymerase molecules. Science, 323, 133–138. [DOI] [PubMed] [Google Scholar]
- Eisenstein M. (2012) Oxford nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol., 30, 295–296. [DOI] [PubMed] [Google Scholar]
- English A.C. et al. (2012) Mind the gap: upgrading genomes with pacific biosciences rs long-read sequencing technology. PLoS One, 7, e47768.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X. et al. (2017) Hysa: a hybrid structural variant assembly approach using next-generation and single-molecule sequencing technologies. Genome Res., 27, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferragina P., Manzini G. (2000) Opportunistic data structures with applications. In: Proceedings 41st Annual Symposium on Foundations of Computer Science (FOCS'00), pp. 390–398. IEEE Computer Society, Redondo Beach, California, USA. [Google Scholar]
- Gnerre S. et al. (2011) High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA, 108, 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontarz P.M. et al. (2013) SRmapper: a fast and sensitive genome-hashing alignment tool. Bioinformatics, 29, 316–321. [DOI] [PubMed] [Google Scholar]
- Goodwin S. et al. (2015) Oxford nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res., 25, 1750–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F. et al. (2010) mrsfast: a cache-oblivious algorithm for short-read mapping. Nat. Methods, 7, 576–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F. et al. (2014) mrsfast-ultra: a compact, snp-aware mapper for high performance sequencing applications. Nucleic Acids Res., 42, gku370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F. et al. (2009) Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res., 19, 1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J. et al. (2014) Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res., 24, 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J. et al. (2017) Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res., 27, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol., 30, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2013) Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol., 14, R101.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J. et al. (2010) Real-time dna sequencing from single polymerase molecules. Methods Enzymol., 472, 431–455. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2012) Exploring single-sample snp and indel calling with whole-genome de novo assembly. Bioinformatics, 28, 1838–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with bwa-mem. arXiv, 1303, 3997. [Google Scholar]
- Li H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics, 25, 1966–1967. [DOI] [PubMed] [Google Scholar]
- Lin H. et al. (2008) Zoom! zillions of oligos mapped. Bioinformatics, 24, 2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. et al. (2016) rhat: fast alignment of noisy long reads with regional hashing. Bioinformatics, 32, 1625–1631. [DOI] [PubMed] [Google Scholar]
- Liu B. et al. (2017) Lamsa: fast split read alignment with long approximate matches. Bioinformatics, 33, 192–201. [DOI] [PubMed] [Google Scholar]
- Loman N.J. et al. (2015) A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods, 12, 733–735. [DOI] [PubMed] [Google Scholar]
- Manber U., Myers G. (1993) Suffix arrays: a new method for on-line string searches. SIAM J. Comput., 22, 935–948. [Google Scholar]
- Manrao E.A. et al. (2012) Reading dna at single-nucleotide resolution with a mutant MsPa nanopore and phi29 dna polymerase. Nat. Biotechnol., 30, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Sola S. et al. (2012) The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods, 9, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Margulies M. et al. (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature, 437, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers G. (1999) A fast bit-vector algorithm for approximate string matching based on dynamic programming. JACM, 46, 395–415. [Google Scholar]
- Ohlebusch E., Abouelhoda M.I. (2005) Chaining Algorithms and Applications in Comparative Genomics. Chapman & Hall/CRC. [Google Scholar]
- Ono Y. et al. (2013) PBSIM: PacBio reads simulator toward accurate genome assembly. Bioinformatics, 29, 119–121. [DOI] [PubMed] [Google Scholar]
- O'Roak B.J. et al. (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet., 43, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C. et al. (2011) Fast local fragment chaining using sum-of-pair gap costs. Algorithms Mol. Biol., 6, 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M. et al. (2015) Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods, 12, 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A.C. et al. (2017) Mapping dna methylation with high-throughput nanopore sequencing. Nat. Methods, 14, 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang F.J. et al. (2018) From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol., 19, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts M. et al. (2004) Reducing storage requirements for biological sequence comparison. Bioinformatics, 20, 3363–3369. [DOI] [PubMed] [Google Scholar]
- Scott D., Ely B. (2014) Comparison of genome sequencing technology and assembly methods for the analysis of a GC-rich bacterial genome. Curr. Microbiol., 70, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck F.J. et al. (2018) Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods, 15, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.C. et al. (2013) Advantages of single-molecule real-time sequencing in high-GC content genomes. PLoS One, 8, e68824.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T. et al. (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods, 14, 407–410. [DOI] [PubMed] [Google Scholar]
- Siragusa E. et al. (2013) Fast and accurate read mapping with approximate seeds and multiple backtracking. Nucleic Acids Res., 41, e78.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šošić M., Šikić M. (2017) Edlib: a c/c++ library for fast, exact sequence alignment using edit distance. Bioinformatics, 33, 1394–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sović I. et al. (2016) Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun., 7, 11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.F., Milos P.M. (2011) The properties and applications of single-molecule DNA sequencing. Genome Biol., 12, 217.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K.J. et al. (2010) A flexible and efficient template format for circular consensus sequencing and snp detection. Nucleic Acids Res., 38, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ummat A., Bashir A. (2014) Resolving complex tandem repeats with long reads. Bioinformatics, 30, 3491–3498. [DOI] [PubMed] [Google Scholar]
- Weese D. et al. (2012) Razers 3: faster, fully sensitive read mapping. Bioinformatics, 28, 2592–2599. [DOI] [PubMed] [Google Scholar]
- Xin H. et al. (2013) Accelerating read mapping with fastHASH. BMC Genomics, 14 (Suppl. 1), S13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.