

Abstract
Pancreatic cancer is one of the leading causes of cancer death, with 5-year survival of 8.5%. The lack of significant progress in improving therapy reflects our inability to overcome the desmoplastic stromal barrier in pancreatic ductal adenocarcinoma (PDAC) as well as a paucity of new approaches targeting its genetic underpinnings. RNA interference holds promise in targeting key mutations driving PDAC; however, a nucleic acid delivery vehicle that homes to PDAC and breaches the stroma does not yet exist. Noting that the cyclic peptide iRGD mediates tumor targeting and penetration through interactions with αvβ3/5 integrins and neuropilin-1, we hypothesized that “tandem” peptides combining a cell-penetrating peptide and iRGD can encapsulate siRNA to form tumor-penetrating nanocomplexes (TPNs) capable of delivering siRNA to PDAC. The use of directly-conjugated iRGD is justified by receptor expression patterns in human PDAC biopsies. In this work, we optimize iRGD TPNs with polyethylene glycol (PEG)-peptide conjugates for systemic delivery to sites of disease. We show that TPNs effectively knockdown siRNA targets in PDAC cell lines and in an immunocompetent genetically-engineered mouse model of PDAC. Furthermore, we validate their tumor-penetrating ability in three-dimensional organoids and autochthonous tumors. In murine therapeutic trials, TPNs delivering anti-Kras siRNA significantly delay tumor growth. Thus, iRGD TPNs hold promise in treating PDAC by not only overcoming physical barriers to therapy, but by leveraging the stroma to achieve knockdown of the gold standard genetic target. Moreover, the modular construction of this delivery platform allows for facile adaptation to future genetic target candidates in pancreatic cancer.
Keywords: Pancreatic cancer, antisense oligonucleotides, oncogenes, tumor suppressor genes, gene products as targets for therapy, cancer nanotechnology
1. Introduction
Pancreatic cancer is a devastating disease that kills over 40,000 people in the U.S. annually, with only 8.5% of patients surviving five years past their initial diagnosis (1). Even in localized disease treated with surgical resection, 5-year survival remains around 20-25% (2). Thus, there is a pressing need for improved therapeutic approaches for all stages of pancreatic cancer. Standard first-line treatment for advanced or metastatic pancreatic ductal adenocarcinoma (PDAC), which comprises 85% of pancreatic cancers (3), generally consists of either a gemcitabine combination (such as gemcitabine plus nab-paclitaxel) (4) or FOLFIRINOX (5). Clinical trials of targeted therapies including anti-VEGF (6–8) and anti-EGFR (9–10) have failed to demonstrate additional benefit, and limited studies on immunotherapy have thus far shown lackluster response to single-agent checkpoint blockade (11). These treatment outcomes reflect the importance of both directing therapy at the genetic targets that drive pancreatic cancer progression and developing effective delivery strategies for disease-relevant therapeutic agents. PDAC is characterized by dense and poorly vascularized stromal layers that functionally fortify the tumor cells, preventing penetration of even small molecule therapeutics (12–13). Several strategies for dismantling or reorganizing the pancreatic cancer stroma for improved small molecule drug penetration have been explored, most prominently hedgehog inhibition (IPI-926/saridegib) (13) and PEGylated hyaluronidase (PEGPH20) (12,14). These approaches have demonstrated improved delivery to animal models of PDAC, and PEGPH20 has advanced to Phase II trials in combination with standard chemotherapy, with predominantly musculoskeletal side effects (15). On the other hand, saridegib has not shown any therapeutic benefit in Phase II clinical trials for pancreatic cancer (16); indeed, long-term hedgehog inhibition and stromal depletion has been shown to actually accelerate pancreatic cancer growth by allowing increased vascularization (17). As a more focused approach, we considered targeting and transiently penetrating the stromal barrier rather than frankly abolishing it. Specifically, we hypothesized that PDAC penetration could be achieved by harnessing the CendR class of internalizing peptides, which share a consensus C-terminal motif that actively mediates transcytosis and endocytosis by engaging neuropilin-1 (18), a semaphorin receptor present on endothelial cells, fibroblasts, and cancer cells. In particular, the internalizing RGD peptide (iRGD) operates by first binding αvβ3/5 integrins specifically expressed on tumor vasculature before proteolytic cleavage reveals an otherwise veiled CendR domain (19). iRGD has been conjugated to or coadministered with compounds and particles to enhance tumor-penetrating delivery in a number of contexts, including diagnostics such as FEB fluorescent dye (20) and therapeutics such as the toxic peptide sTRAIL to models of gastric cancer (21), silicasome-based chemotherapy to pancreatic cancer (22), and pH-sensitive polymer-based siRNA vehicles to models of prostate cancer (23).
The ideal therapeutic approach to pancreatic cancer treatment is targeted at the key signaling pathways and mutations that drive PDAC growth. PDAC is characterized by nearly ubiquitous mutations in KRAS and CDKN2A, each present in over 90% of tumors (24). Oncogenic KRAS mutations are of particular interest due to their important role in PDAC formation and maintenance, including growth and stromal interactions (25). Unfortunately, oncogenic KRAS is notoriously difficult to target because of a lack of well-defined surface pockets for drug binding, and promising strategies such as farnesyltransferase inhibition and membrane docking inhibition have failed in clinical trials (26–27). RNA interference via siRNA delivery presents an attractive, versatile strategy for specifically knocking down otherwise “undruggable” targets such as KRAS (28–29), and tumor-targeted delivery of siRNA can avoid the toxicity associated with global inhibition. However, siRNA delivery in tumors has been challenging in general and even moreso in PDAC by the stromal barriers to drug delivery. While many successful delivery vehicles have been developed to protect siRNA cargo and target it to tissues or tumors while facilitating cytoplasmic delivery (30–33), siRNA carriers to date have relied upon passive distribution once at the site of disease. Indeed, local delivery of siRNA against KRAS leads to therapeutic responses in PDAC xenograft models (34); however, effective systemic delivery of RNAi to organs aside from the liver and lungs, especially the pancreas, remains a challenge. As such, there does not yet exist an RNAi carrier designed to specifically overcome the tumor penetration challenges presented by PDAC. Towards the goal of achieving tumor-penetrating siRNA delivery, our group has developed the modular tandem peptide platform for credentialing genetic targets in ovarian cancer through intraperitoneal delivery, in which siRNA is encapsulated by a multifunctional peptide possessing both targeting and cell-penetrating domains (35–36).
In this work, we address both the delivery and gene target challenges facing pancreatic cancer therapy by developing iRGD-guided tumor penetrating nanocomplexes (TPNs) for systemic delivery of siRNA therapy to pancreatic cancer, with the tandem peptide framework as a starting point. Incorporating iRGD in combination with particle optimization to maintain function and stability in the bloodstream, we create nanoparticles that mediate robust knockdown of gene targets in pancreatic tumor cells in vitro and in vivo. Mechanistically, the iRGD TPNs achieve tumor penetration in patient-derived primary organoid models in vitro as well as in a variety of mouse models of pancreatic tumors, including autochthonous tumors in the genetically-engineered KPC model that recapitulates chemoresistance patterns seen in human tumors and is fully immunocompetent (13). Moreover, treatment of a murine model of PDAC with iRGD TPNs carrying KRAS siRNA leads to a decrease of oncogene expression and inhibition of tumor growth in vivo. Thus, we demonstrate both the mechanistic tumor-penetrating delivery and functional therapeutic aspects of these new RNAi nanoparticles in a comprehensive collection of PDAC model systems. These results indicate that effective delivery of therapeutic RNAs is possible in an autochthonous solid rodent tumor model of PDAC, transforming the stromal barrier into a “back-door” conduit for delivery. Given the elevated expression of iRGD receptors in patient biopsies, iRGD TPNs offer promise in translating our growing understanding of pancreatic cancer as a genetic disease into viable therapies, while effectively bypassing the delivery barriers that have stymied current systemic therapies in the clinic.
2. Materials & Methods
Peptide & siRNA synthesis:
pTP-TAMRA-iRGD (CH3(CH)15-[GWTLNSAGYLLGKINLKALAALAKKIL-GGK(TAMRA)GGCRGDKGPDC, Cys-Cys bridge]) used in all figures except Fig. S1 was synthesized by CPC Scientific. The experiments in Fig. S1 used the identical peptide except with myristic acid CH3(CH)13 in place of palmitic acid. All siRNAs were synthesized by Dharmacon (GE Healthcare) with ON-TARGETplus specificity enhancement. The sequences used were as follows (given as the sense strand without overhangs): siLuc against firefly luciferase: 5’-CUUACGCUGAGUACUUCGA-3’, siGFP: 5’-GGCUACGUCCAGGAGCGCACC-3’, siKras.476 against murine and human KRAS: 5’-ACCAUUAUAGAGAACAAAUUA-3’, siKras.476 seed-matched control: 5’-ACCAUUAUUCUGAACAAAUUA-3’, siNC non-targeted control: 5’-UUCUCCGAACGUGUCACGUUU-3’.
pTP-PEG-iRGD synthesis:
We used the same approach as the synthesis of pTP-PEG-LyP-1 described in J. H. Lo et al, 2016 (37). Briefly, orthopyridyl disulfide-PEG-succinimidyl valeric acid (OPSS−PEG−SVA) 5K (Laysan Bio) was reacted with 5 equivalents of N-[(1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl]-1,8-diamino-3,6-dioxaoctane (Sigma) for 3 hours at RT. The resulting conjugate was dialyzed using a 3500 MWCO membrane and lyophilized; the product was then dissolved in DMF and reacted with 1.2 equivalents of palmitoyl-transportan bearing a C-terminal cysteine for 3 hours at RT followed by addition of 1.2 equivalents of azidoacetyl-GGG-iRGD (N3-CH2-CO-[peptide: GGGCRGDKGPDC, Cys-Cys bridge]) with the reaction proceeding overnight. The final product was purified via dialysis with a 3500 MWCO membrane into water. This was again lyophilized and resuspended shortly before use. The final sequence is (CH3(CH)15-[GWTLNSAGYLLGKINLKALAALAKKILC]-S-S-(OCH2CH2)n (avg MW 5000 kDa)-X-[GGGCRGDKGPDC, Cys-Cys bridge]), where X is the product of the reaction between the cycloalkyne and azidoacetyl groups with structural formula as depicted in the bottom panel of Figure S1 of Reference 37.
Cell culture:
All stabilized cell lines including KPC-derived cell lines and MIA PaCa-2 cells (ATCC) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin, with the exception of PANC-1 cells (ATCC), which were grown in DMEM + 20% FBS + penicillin/streptomycin. KP A13, B22, and D8-175 murine KPC cell lines were derived from KPC tumors harvested in the lab of Tyler Jacks. Cell lines were most recently tested for mycoplasma on May 31, 2017.
Antibody staining:
For quantification of surface receptor expression, cells were trypsinized and brought to single-cell suspension in FACS buffer (1× PBS + 2% FBS). Primary antibody was added at 1 μg/million cells in 100 μL total solution (for mouse cells: rat anti-mouse αv integrin (BD Pharmingen 551380) or rat IgG isotype control (Invitrogen); for human cells: mouse anti-αvβ3 integrin, direct PE conjugate (BioLegend 304406) or mouse IgG κ chain isotype control, direct PE conjugate (BioLegend 400112); for neuropilin-1 staining in all cells, rabbit anti-NRP-1 (Novus Biologicals NBP1-40666) or normal rabbit IgG isotype control (R&D)) and incubated for one hour on ice. For direct fluorophore-conjugated primary antibodies, cells were washed with PBS and resuspended in FACS buffer. Otherwise, after washing the cells 2× in PBS, cells were incubated with secondary fluorescently-tagged antibody (Invitrogen) for 45 minutes and washed 1× in PBS. Cells were analyzed on BD LSR-II or Fortessa HTS flow cytometers. Data were analyzed in FlowJo (TreeStar Software).
Immunohistochemistry:
PDAC tumor microarrays (US Biomax, slide PA242c) were stained with anti-NRP-1 (Abcam ab81321) or anti-alpha v integrin (Abcam ab179475) primaries in accordance with manufacturer instructions, followed by HRP secondaries (BioCare Rabbit-on-Rodent RMR622 and Mouse-on-Mouse MM620L polymers). Slides were digitized using an Aperio slide scanner and quantified using standard DAB and hematoxylin deconvolution functions in ImageJ. Grading was objective and based on linearly spaced bins by DAB to hematoxylin ratio (Grade 1: 0-5; Grade 2: 5-10, Grade 3: 10-15, Grade 4: 15+).
Electrophoretic mobility shift assay:
TPNs were formed at 5-30:1 peptide (pTP-iRGD):siRNA ratios for a final concentration of 200 nM siRNA (DyLight 677-siLuc) in 1× PBS. 10 μL of each TPN sample or free siRNA was mixed with 2 uL of 30% glycerol and loaded into a 2% agarose gel. The gel was run at 100 V for 45 minutes in 1× TAE buffer and siRNA fluorescence was imaged on a LI-COR Odyssey infrared scanner (LI-COR Biosciences). Signal was quantified using ImageJ.
Transfection:
For all in vitro transfection assays, TPNs were formed at the specified ratios by adding peptide diluted in Opti-MEM (Gibco, Life Technologies) to an equal volume of siRNA diluted in Opti-MEM, combining to form a final concentration of 100 nM siRNA. Cells were dosed in multi-well plates by removing growth media and adding TPN solution at 100 nM siRNA. The volumes used were as follows: 96-well plate (luciferase knockdown): 100 μL/well; 24-well plate: 500 µL; 12-well plate: 1 mL; 6-well plate (GFP knockdown): 2 mL. After 4-6 hours of incubation at 37 °C, media was replaced with normal growth media.
Fluorescence microscopy:
Cells were transfected as described above. At the specified timepoints, cells were imaged live on a Nikon Eclipse Ti inverted microscope using a 20× Plan Apo objective. Images were collected in NIS-Elements AR software (Nikon), with individual channels combined in Photoshop CS5 (Adobe) with linear level adjustments applied identically to all images within an experiment.
Luciferase knockdown:
48 hours after transfection of KP A13 or B22 cells with siLuc, luciferase function was quantified by lysing cells with Cell Culture Lysis Reagent (Promega); 10 μL of lysate was then mixed thoroughly with 40 μL of luciferin (Promega Luciferase Assay System) and loaded into a white 96-well plate (Corning 3600). Luciferase bioluminescence was quantified using a Centro LB 960 Microplate Luminometer (Berthold Technologies). Knockdown of destabilized GFP in HeLa dGFP cells was assessed at 24 hours post-transfection using flow cytometry, quantified using Flow-Jo.
Quantitative PCR:
mRNA was isolated by lysing cells with Buffer RLT (Qiagen), filtering out debris using the Qiashredder homogenizer (Qiagen), and then purifying mRNA using an RNeasy kit (Qiagen) according to manufacturer’s instructions. mRNA concentration was quantified via NanoDrop 2000 Spectrophotometer (Thermo). cDNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). qPCR was performed on a C1000 Touch Thermal Cycler with CFX96 Touch Real-Time PCR Detection System (Bio-Rad) using the following primer pairs: mouse Kras: Forward 5’-ACAGTGCAATGAGGGACCAG-3’ and Reverse 5’-ATCGTCAACACCCTGTCTTGT-3’; mouse Hprt as loading control: Forward 5’-GTCAACGGGGGACATAAAAG-3’ and Reverse: 5’-CAACAATCAAGACATTCTTTCCA-3’; human KRAS: Forward 5’-ACTGGGGAGGGCTTTCTTTG-3’ and Reverse 5’-GCATCATCAACACCCTGTCT-3’; human TBP as loading control Forward 5’-GGAGAGTTCTGGGATTGTAC-3’ and Reverse 5’-CTTATCCTCATGATTACCGCAG-3’.
Western blotting:
Protein was isolated by lysing cells in 1× RIPA buffer with protease inhibitors for 30 minutes. Protein was quantified using the bincinchoninic acid (BCA) assay (Pierce, Thermo) against bovine serum albumin standards and standardized to 2 mg/mL. Samples were then mixed 1:1 with Laemmli loading buffer and run on a Novex 4-12% Bis-Tris gel (Life Technologies) following manufacturer protocol, along with MagicMark XP and Kaleidoscope ladders. Bands were transferred to nitrocellulose membranes at 375 mA for 1 hour. The membrane was cut at the 30 kDa marker in order to stain for K-Ras (21 kDa) and α-tubulin (50 kDa) separately. The membranes were blocked with 5% skim milk in TBS-Tween (TBST) for 1 hour at 4 °C and then incubated with primary antibody diluted in 5% skim milk overnight at 4 °C: for K-Ras, F234 mouse monoclonal antibody (Santa Cruz) was used at a 1:100 dilution; for tubulin, mouse monoclonal anti-tubulin (Invitrogen 32-2500) was used at a 1:1000 dilution. Membranes were washed 2× in TBST for 5 min. shaking, then incubated with secondary antibody: goat anti-mouse (sc-2005, Santa Cruz) at a 1:2000 dilution in TBST. Following final 2× TBST washes, blots were imaged using the SuperSignal West Pico chemiluminescent substrate (Pierce, Thermo).
Transmission electron microscopy:
7 µL of TPN solution (15:7.5:1 peptide:PEG-peptide:siRNA, 1 µM siRNA concentration, 0.1× PBS buffer) was dropped onto a carbon film/200 copper mesh grid, with excess solution wicked off after 1 minute. The grid was negatively stained with phosphotungstic acid (1% aqueous solution), again wicked off, and the grid was allowed to air-dry. The sample was imaged on an JEOL 2100 FEB microscope operated at 200 kV, with images captured on a Gatan 2kx2k UltraScan CCD camera.
Organ biodistribution:
Swiss Webster mice were intravenously injected under isoflurane anesthesia with non-PEGylated iRGD TPNs or 15:10:1 iRGD TPNs at 0.5 nmol siRNA dose per mouse, n=3 per condition. VivoTag-S750 siRNA was used to minimize interference from autofluorescent background. After 3 hours, mice were euthanized and necropsy was performed to remove the lungs, heart, kidneys, liver, and spleen. Organs were scanned using a LI-COR Odyssey near-infrared scanner (LI-COR Biosciences) and analysis of average fluorescence intensity was performed in ImageJ.
Organoids:
Trp53fl/fl, Kras+/LSL-G12D, and Pdx1-Cre strains in C57Bl/6 background were interbred to obtain Pdx1-Cre; Kras+/LSL-G12D; Trp53 fl/fl (KPC) mice (38). The breeding strains were a kind gift from the Tyler Jacks laboratory at MIT. All animal experiments were conducted in accordance with procedures approved by the DCM at MIT. To isolate primary tumor cell, sliced tumor tissues were immediately digested in HBSS media (Sigma) with 4 mg/mL collagenase/Dispase (Roche) and 0.05% Trypsin-EDTA for over 1 hour and were seeded in growth-factor-reduced (GFR) Matrigel (BD). Human pancreatic tumor organoids were embedded in GFR Matrigel, and cultured in human complete medium (Advanced DMEM/F12 medium supplemented with HEPES [1×, Invitrogen], Glutamax [1×, Invitrogen], penicillin/streptomycin [1×, Invitrogen], B27 [1×, Invitrogen], Primocin [1 mg/ml, InvivoGen], N-acetyl-L-cysteine [1 mM, Sigma], Wnt3a-conditioned medium [50% v/v, derived from Wnt3A-expressing L cells from ATCC], R-Spondin 1-conditioned medium [10% v/v, derived from Rspo1-Fc expressing 293T cells from Dr. Calvin Kuo, Stanford University], recombinant murine noggin [100 ng/ml, Peprotech], recombinant murine epidermal growth factor [EGF, 50 ng/ml, Peprotech], Leu15-Gastrin I [10 nM, Sigma], recombinant human fibroblast growth factor 10 [FGF10, 100 ng/ml, Peprotech], Nicotinamide [10 mM, Sigma], and A83-01 [0.5 μM, Tocris]). PEGylated TPNs carrying 100 nM Alexa 647 labeled Kras-siRNAs were added into culture media and incubated overnight prior to fixation in 4% PFA for 1 hr. Fixed mouse organoids were imaged with Olympus FV1200 Laser Scanning Confocal Microscope. Human primary tumor organoids were first incubated with PEGylated TPNs carrying 100 nM Alexa 647 labeled Kras-siRNAs and further incubated with CellMask™ Green plasma membrane stain for 20 min prior to 4% PFA fixation.
Organoid quantitative analysis:
Analysis was performed in MATLAB 2017a (Mathworks). Please refer to Figure S4 and subsequent annotated MATLAB script.
Confocal Microscopy and Immunofluorescence:
Seeded in a Nunc™ Lab-Tek™ Chambered Coverglass system (Thermo Fisher Scientific), primary mouse and human PDAC organoids were rinsed with PBS, fixed in 4% paraformaldehyde (PFA) in PBS for 60 min cells after TPN incubation, and imaged under a Zeiss LSM 700 inverted confocal scanning microscope. The excitation wavelength of Alexa 647 labeled Kras-siRNAs was 633 nm, and the corresponding emission filter was 660-710 nm. To track the localization of TPNs in vivo, subcutaneously transplanted tumor tissues were extracted, embedded in optimum cutting temperature (O.C.T.) compound and sectioned into 6 µm slices. After blocking with 5% goat serum, 2% BSA, 0.1% Triton-X 100 in PBS for 1 h, sections were stained with a primary antibody against αv integrin (Abcam) at 2 μg/mL (1% BSA in PBS) or primary antibody against TAMRA (Thermo Fisher Scientific) at 5 μg/mL (1% BSA in PBS) overnight at 4 °C. To address the intratumoral biodistribution of TPNs, 10 min prior to euthanasia, 50 µg fluorescein-conjugated Lycopersicon Esculentum (tomato) lectin was intravenously injected into mice bearing orthotropic pancreatic tumors. O.C.T.-embedded tissue sections were stained with anti-NRP-1 (Abcam ab81321) or anti-alpha smooth muscle actin (Abcam ab5694) primaries in accordance with manufacturer instructions. Fluorescently-labeled secondary antibodies [Invitrogen, Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L), Alexa Fluor 647 Goat Anti-Rabbit IgG (H+L)] were incubated at 1 μg/mL (1% BSA in PBS) for 30 min at RT. The excitation wavelength of the secondary antibody was 488/633 nm, and the corresponding emission filter was 500-550/663-738 nm.
Intravital imaging:
NCR/nude mice were implanted with bilateral flank grafts of MIA PaCa-2 (ATCC) xenografts and were imaged when tumors reached a major-axis diameter of ~1 cm. Mice were injected intravenously with TAMRA-labled TPNs at a 0.3 nmol siRNA dose, alongside fluorescently-tagged dextran (70 kDa FITC dextran, Life Technologies). Surgical exteriorization of the flank tumor was performed with the mice adequately anesthetized with inhaled isoflurane, and the tumor was mounted against a glass slide over the microscope objective for imaging. Second-harmonic generation microscopy was used to visualize collagen fibrils, while TAMRA fluorescence was used to track TPNs. Images were collected every 3 minutes over the course of 15 minutes, beginning 5 minutes post-injection due to the time elapsed to perform the surgery and locate suitable imaging regions. Sequences were captured as z-stacks with 21 layers, with z layers separated by 10 μm.
Orthotopic pancreatic cancer transplant model:
Intrapancreatic tumor xenografts were generated as described in Kim et al (39). Briefly, NCR/nude mice were anesthetized and the surgical site was sterilized. A ~5 mm incision was made in the left mid-abdomen and the spleen and pancreas were exteriorized with forceps. 100 μL of KP D8-175 (firefly luciferized KPC mouse-derived cell line) suspension at 10 million cells/mL, diluted in Opti-MEM + 10% growth factor-reduced, phenol red-free Matrigel (BD), was injected using a sterile syringe and needle. After 1 minute to allow solidification of Matrigel, the spleen and pancreas were returned to the abdominal cavity; the peritoneum was then sutured together and the skin approximated by wound clips. Tumors were allowed to mature for approximately 4 weeks prior to injection of particles and/or staining as described above.
Knockdown in KPC tumors:
Pdx1-Cre; Kras+/LSL-G12D; Trp53 fl/fl (KPC) mice were divided into three treatment groups (n=3) and administrated with (1) D5W i.v. injection and 1× PBS i.p. injection, (2) D5W i.v. injection and 15:2.5:1 pTP-TAM-iRGD:pTP-PEG(5 kDa)-iRGD:siNon-targeting control (siNC) 0.5 mg/kg, (3) 15:2.5:1 pTP-TAM-iRGD:pTP-PEG(5 kDa)-iRGD:siKras 923 0.5 mg/kg i.v. injection. 48 hours after injection, tumors were isolated and total RNA were extracted immediately using RNeasy Mini Kit (Qiagen). KRAS mRNA levels were quantified by RT-qPCR using KRAS TaqMan® Gene Expression Assays (Thermo Fisher Scientific) against reference gene TBP.
Therapeutic trial:
NCR/nude mice were implanted with bilateral flank allografts of KP D8–175 luciferized cells, each seeded with 5×105 cells in 100 μL Opti-MEM. Mice were divided into groups of nearly equal average tumor burden (~170 mm3 total tumor burden per mouse at the start of treatment) and standard deviation of tumor burden. The three treatment groups, n=6 each, were as follows: (1) D5W i.v. injection, (2) 15:2.5:1 pTP-TAM-iRGD:pTP-PEG(5 kDa)-iRGD:siNC (non-targeted control) 0.5 mg/kg i.v. injection in D5W, and (3) 15:2.5:1 pTP-TAM-iRGD:pTP-PEG(5 kDa)-iRGD:siKras 923 0.5 mg/kg i.v. injection in D5W. Mice were dosed on days 1, 4, 7 ,11,15, 18, 21, 25, 30, and 35. Doses based on body weight were administered such that a 25g mouse would receive a 100 μL injection, with the actual administered volume scaled according to each mouse’s individual weight. Tumor volumes were measured every three days by calipers by an independent researcher, with tumor burden per mouse computed as the sum of the volumes of the two flank tumors, these in turn calculated as half of the product of the major axis and minor axis squared. Survival curves were generated using a threshold aggregate tumor burden per mouse of 1500 mm3, which was used as a proxy for survival. Data from tumors exceeding this burden were not censored from the tumor growth curves. Curves were fit to the equation Y=Yo*exp(k*X) using GraphPad Prism, where the doubling time is computed as ln(2)/k. This experiment was repeated two additional times, with minor variation in dosing schedule. The effect of siKras treatment relative to untargeted siRNA control was always statistically significant at six weeks. Mouse experiments were performed in accordance with MIT Division of Comparative Medicine and Committee on Animal Care (CAC) policies and corresponding protocol 0414–025-20.
3. Results
3.1. Design and in vitro function of iRGD TPNs for pancreatic cancer
To develop tandem peptides for RNAi delivery to PDAC, we built upon the tumor-penetrating nanocomplex platform, where siRNA is complexed with a tandem peptide containing both a C-terminal tumor-penetrating domain and N-terminal cell-penetrating peptide (CPP) (Fig. 1A). Based on prior studies, we used transportan as the CPP, capped by an N-terminal saturated fatty acid chain to aid in endosomal escape (36). In contrast to our prior work, we selected iRGD (Fig. 1B) as the tumor-penetrating domain, on the basis of compelling immunohistological evidence of robust overexpression of its primary receptor, αv integrins, in a pancreatic cancer tissue microarray (TMA) (Fig. 1C, left and Fig. 1D) as well as expression of its secondary receptor, neuropilin-1, in stromal and immune cell components (Fig. 1E). Indeed, objective grading showed that nearly all tumor cores in the TMA overexpressed αv integrin to some degree (Fig. 1C, right), with many (classified as Grade 3 or 4) showing homogenously or heterogeneously intense staining; expression in healthy pancreas was limited to vessels and interstitial tissue (Fig. 1D, right).
Figure 1: Design, suitability, and in vitro function of iRGD TPNs for PDAC.
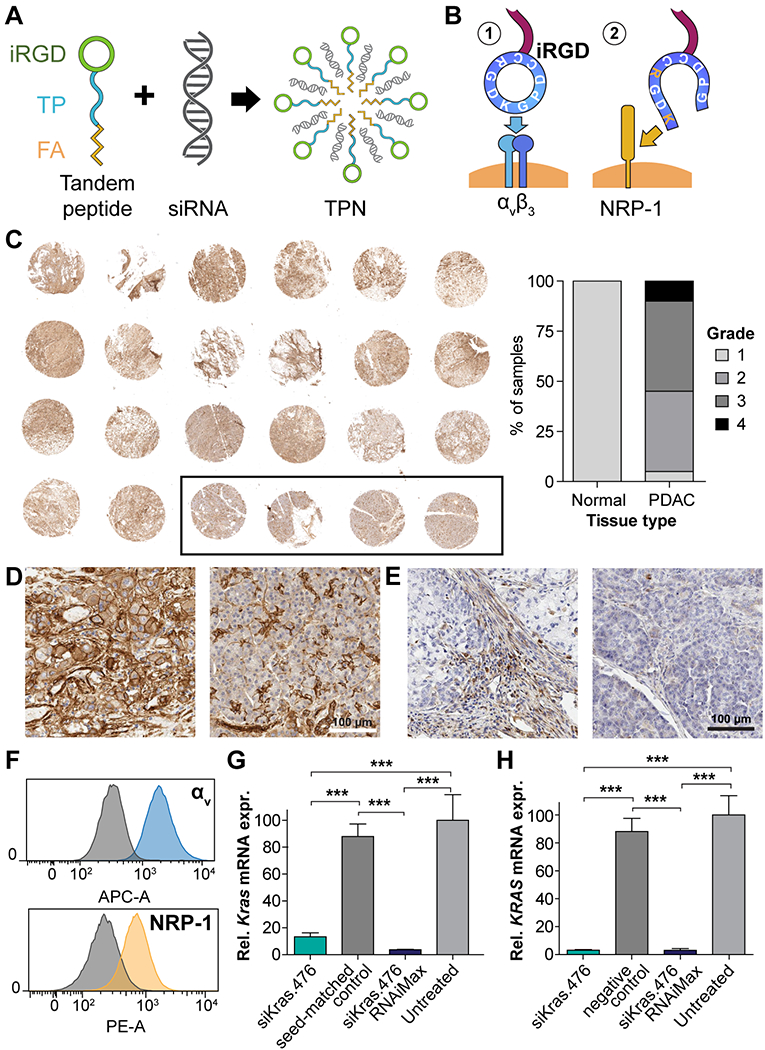
(A) Schematic depicting spontaneous formation of iRGD-based tumor-penetrating nanocomplexes (TPNs) by mixing tandem peptides and siRNA solutions (B) iRGD functions by binding to αvβ3/5 integrins when cyclized and by binding neuropilin-1 (NRP-1) following proteolytic cleavage. (C) Left: Immunohistochemical stain of PDAC tissue microarray (TMA) at low magnification, showing distribution of αv integrins (brown), with hematoxylin counterstain (purple). Black outline designates 4 normal pancreatic samples. Core diameter: 1.5 mm. Right: Grading of TMA overall staining intensity via objective digital quantification. (D) Micrograph of PDAC (left) and normal pancreas (right) from the above TMA showing detail of αv integrin distribution. Scale bar: 100 µm. (E) Micrograph of NRP-1 distribution in PDAC (left) and normal pancreas (right). Scale bar: 100 µm. (F) αv integrin and neuropilin-1 surface expression on murine Kras-p53 PDAC cell line B22, quantified by live-cell flow cytometry, compared to IgG control plus secondary antibody (gray histograms). (G) Kras mRNA knockdown in KP B22 cells using siKras.476, versus seed-matched control, as measured by qPCR. (H) KRAS mRNA knockdown in the human PANC1 PDAC cell line.
To ensure that our chosen mouse models mirrored this human expression pattern, we employed flow cytometry to confirm the presence of surface αv integrins and neuropilin-1 in a cell line (luciferized KP B22) derived from the KPC (KrasLSL-G12D/+; Trp53fl/fl; Pdx-1-Cre) genetically engineered mouse (GEM) model (Fig. 1F). We observed intracellular delivery of siRNA into B22 cells at an arbitrary starting ratio (Fig. S1A). Subsequently, we optimized the peptide (palmitoyl-transportan-iRGD or pTP-iRGD):siRNA stoichiometric ratio for cargo encapsulation (Fig. S1B) and optimized function based on luciferase marker knockdown in vitro, both with respect to the stoichiometric ratio (Fig. S1C) as well as the dose (Fig. S1D). We identified optimal function at ≥10:1 ratios, corresponding to a ~1.7:1 N/P (nitrogen/phosphate) ratio and siRNA concentrations ≥~50 nM.
Moving toward therapeutic applications, we anticipated that Kras knockdown would be the cornerstone of any potential therapeutic RNAi regimen for PDAC, given the ubiquity of Kras mutations and studies showing that PDAC cell lines stably expressing shRNA against KRAS grow much more slowly as tumor grafts (40), and that local, prolonged intratumoral delivery of siRNA against mutant KrasG12D in established PDAC tumors inhibited tumor cell proliferation (34). In B22 mouse cells, TPNs mediated ~90% knockdown of Kras mRNA at 48 hours, while a seed-matched siRNA control that accounted for miRNA-like seed sequence effects did not result in significant knockdown (Fig. 1G). In the PANC-1 human cell line, we observed profound knockdown of >95% with the same siRNA sequence (Fig. 1H). Lastly, as a proof-of-concept, we demonstrated in vitro knockdown of two independent siRNA targets simultaneously (Fig. S2A-B). In summary, we created iRGD TPNs to take advantage of NRP-1 and αv integrins as targeting and penetration receptors, and effected robust siRNA knockdown in both human and murine PDAC cell lines.
3.2. PEG formulations of iRGD TPNs improve pharmacokinetics
TPNs are not natively stable in the bloodstream, so we applied knowledge from our systematic comparison of modular reformulations for LyP-1 TPNs in ovarian cancer (37) to improve the properties of iRGD TPNs for in vivo applications. Applying the preferred chemical framework, we introduced a third component consisting of a 5 kDa polyethylene glycol (PEG) chain inserted linearly between the transportan and iRGD (“pTP-PEG-iRGD”), separating the CendR domain from the CPP-siRNA particle core so as to leave it accessible for target binding (Fig. 2A). Particles containing various ratios of pTP-iRGD:pTP-PEG-iRGD:siRNA were constructed to encapsulate siRNA cargo, and increasing the proportion of PEG-peptide in the TPNs yielded smaller particles based on dynamic light scattering (DLS) (Fig. 2B), which we observed to also be more stable over time. Since DLS gives only an effective hydrodynamic diameter, we confirmed the particles’ size using TEM with negative staining, which revealed particles of similar average diameter, but an intriguing sub-nanoparticle structure suggesting assemblies of smaller sub-units (Fig. S3A).
Figure 2. In vitro and in vivo characterization of iRGD TPNs formulated with PEG.
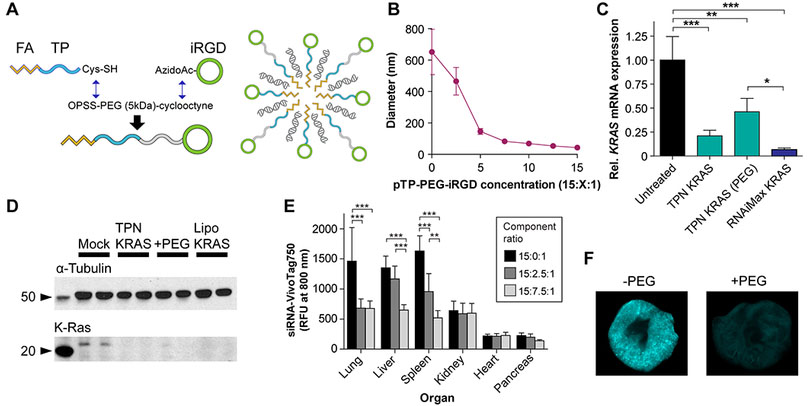
(A) Summary of chemical synthesis of transportan-PEG-iRGD and schematic of PEGylated iRGD TPN. (B) iRGD TPN hydrodynamic diameter as a function of PEG content, determined by dynamic light scattering. (C) in vitro mRNA knockdown in MiaPaCa-2 cells by non-PEGylated and PEGylated iRGD TPNs, with lipofectamine siKRAS as positive control; expression relative to TBP (TATA-binding protein) housekeeping control. (D) Western blot depicting knockdown of K-Ras protein in PANC-1 cells by non-PEGylated and PEGylated iRGD TPNs, with lipofectamine siKRAS as positive control. (E) Organ biodistribution of siRNA delivered by systemically-injected PEGylated vs. plain iRGD TPNs, performed in healthy wildtype mice (n=5 per condition). **: p<0.01 and ***: p<0.001 by two-way ANOVA. Unmarked comparisons within each organ are non-significant. (F) Comparison between lungs of animals dosed with PEGylated TPNs (above) and plain TPNs (bottom), pseudocolored based on near-infrared siRNA intensity.
The PEGylated particles demonstrated similar cellular delivery of both the siRNA and peptide components while eliminating extracellular aggregation in cell culture experiments (Fig. S3B-C). Furthermore, in MIA PaCa-2 human PDAC cells, we quantified KRAS knockdown efficacy of iRGD-TPNs with and without PEG incorporation and showed that they are equally potent in knockdown of KRAS at both the mRNA and protein levels (Fig 2C and D). To study the ramifications of PEG stabilization on iRGD TPN pharmacokinetics, we first injected PEGylated TPNs (15:2.5:1 or 15:7.5:1 ratios) into mice to contrast the organ biodistribution with non-PEGylated TPNs (Fig. 2E), noting marked, statistically significant reductions in lung, spleen, and liver accumulation. A representative near-infrared scan of lungs extracted from mice injected with PEGylated and non-PEGylated TPNs carrying fluorescently-tagged siRNA qualitatively demonstrates the difference in intensity and distribution due to particle formulation (Fig. 2F). Of note, the pancreas in this experiment is the healthy native pancreas rather than a tumor, with low accumulation in the native pancreatic tissue being favorable for selectively targeting pancreatic cancer tissue. This contrast is in agreement with the differences in receptor expression seen in Fig. 1D-E. Overall, these results demonstrated PEG-induced pharmacokinetic changes in organ distribution, particularly reduced off-target accumulation in reticuloendothelial system (liver and spleen) and first-pass entrapment in the lung.
3.3. iRGD mediates penetrating siRNA delivery in 3D organoid models
Satisfied with the nanocomplex formulation, we next set out to determine whether these particles could overcome the physical delivery barriers partly responsible for poor therapeutic outcomes of current PDAC treatment. To investigate whether the tumor penetration and trafficking mechanisms of iRGD persist in the context of covalent tethering to TPNs, we derived pancreatic organoids from primary mouse and human tumors to model tumor responses to nanocomplex exposure in a three dimensional, multi-cellular context (Fig 3 A&B) (41). Primary PDAC organoids cultured in matrigel maintain the histoarchitecture and phenotypic heterogeneity of the primary tumor while being more accessible to imaging than in vivo structures. We assessed the function and behavior of PEGylated iRGD TPNs in primary tumor organoids by tracking their distribution and penetration depth through the entire organoid. High-resolution optical imaging allows cellular-level analyses, enabling identification and quantification of cellular and behavioral heterogeneity within organoids. TPNs formulated with scrambled iRGD peptide bound only to the organoids’ periphery, lacking tumor-penetrating capacity (Fig. 3C). In contrast, fluorescently-tagged siRNAs in TPN formulation penetrated deeply in large (diameter > 200 µm) primary human tumor organoids, qualitatively reflected in the fluorescent siRNA observed in the center of the organoid (Fig. 3D). This interpretation was confirmed through image quantification in MATLAB; based on the assumption that siRNA can only enter the organoids from their outer edge, we segmented the organoid into “tree rings” of fixed depth from the edge, quantifying siRNA intensity over background (Fig. S4, Supplemental Code). This analysis revealed siRNA penetration beyond the outer rim of cells and indeed persistent siRNA intensity even as the cell density decreases towards the center (Fig. 3E). In murine PDAC organoids derived from KPC tumors (effective genotype: KrasG12D/+; p53−/−), we observed that free siRNA was hardly uptaken (Fig. 3F), whereas organoids treated with PEGylated iRGD TPNs displayed robust cytoplasmic siRNA distribution (Fig. 3G). Because these organoids were largely hollow, the quantified penetration profile is less revealing, but demonstrates siRNA uptake in proportion to cytoplasmic content at any depth (Fig. 3H). Thus, in human and mouse 3D organoid cultures, iRGD TPNs achieved robust penetrating siRNA delivery, often through cell layers hundreds of microns thick.
Figure 3. siRNA penetration modeled in 3D organoids.
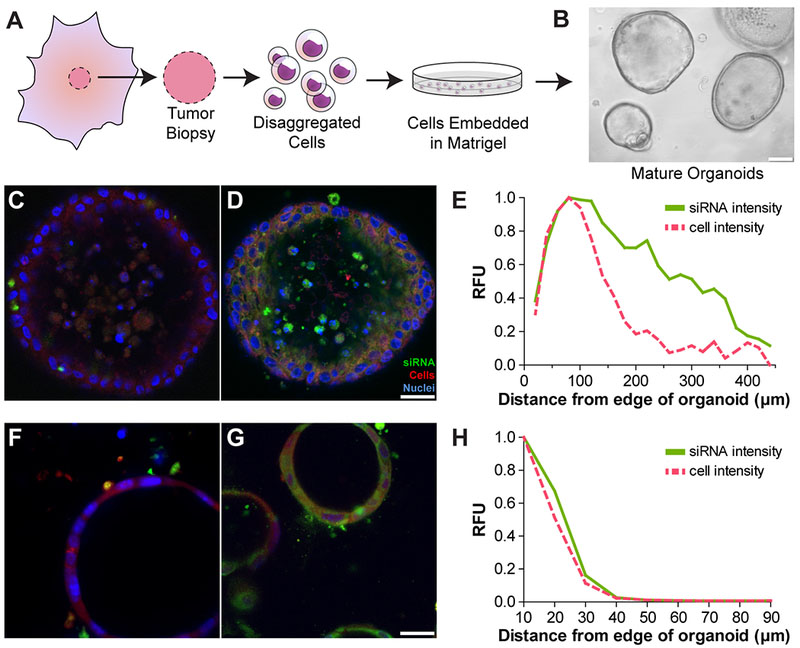
(A) Schematic of organoid production from human tumors. (B) Brightfield micrograph of mature organoids at 10× magnification; scale bar: 100 µm. (C and D) 20× fluorescent micrographs of human organoids after incubation with (C) non-targeted PEG TPNs and (D) PEG iRGD TPNs. Fluorescently-tagged siRNA shown in green, cytoplasmic dye in red, and nuclei in blue; scale bar: 25 µm. (E) Quantification of siRNA intensity in a human organoid as a function of distance from the outer edge, representing penetration of the siRNA. Cytoplasmic dye intensity reflects the reference density of cells. (F and G) 20× fluorescent micrographs of mouse cell line-derived organoids after incubation with (F) siRNA only and (G) PEG iRGD TPNs. Fluorescently-tagged siRNA shown in green, constitutive tdTomato in red, and nuclei in blue; scale bar: 25 µm. (H) Quantification of siRNA intensity in a murine organoid as a function of distance from the outer edge, representing penetration of the siRNA.
We performed several follow-up experiments to investigate the in vivo implications of these organoid findings. In a subcutaneous xenograft pancreatic tumor model, we performed intravital imaging of iRGD TPN accumulation in tumors in real-time following intravenous administration, with collagen visualized through second-harmonic generation as a marker of stromal elements. This timelapse imaging at various depths in the tumor demonstrated arrival of the particles via vasculature and rapid extravasation and penetration into surrounding tumor tissue, including the interstices between collagen bands (Supplemental Video 1; representative still image shown in Fig. S5). Going further, we generated orthotopic allograft tumor models via intrapancreatic transplantation of pancreatic tumor cells (39). These tumors robustly expressed NRP-1, in contrast with nearby normal pancreas tissue (Fig. S6A). Simultaneous fluorescent staining of microvessels (using tomato lectin) and NRP-1 in an orthotopic tumor section following intravenous administration of fluorescently-tagged iRGD TPNs revealed robust TPN accumulation in NRP-1-expressing cells, with extension beyond the microvessel networks (Fig. S6B). Finally, alpha-SMA staining in a stroma-rich portion of the tumor showed intact stromal networks with TPN accumulation in between (Fig. S7). Together, these findings suggest that the dynamics and characteristics of tumor penetration seen in the organoid models have in vivo correlates.
3.4. PEGylated iRGD TPNs mediate Kras knockdown and slow PDAC growth in vivo
Finally, we sought to characterize the performance of PEGylated iRGD TPNs in animal models. For delivery and knockdown studies, we utilized the KPC mouse model of pancreatic cancer (genotype: KrasLSL-G12D/+; Trp53fl/fl; Pdx-1-Cre), where Cre recombinase expressed under the control of a pancreas-specific promoter, Pdx-1, leads to Kras and p53 mutations that drive aggressive autochthonous tumors bearing close histological resemblance (including the desmoplastic stroma) to human PDACs, albeit at an accelerated pace with multiple foci of disease (38). Following intravenous administration of particles bearing near-infrared fluorescently-dyed siRNA to KPC mice, we observed widely distributed fluorescent uptake at the macroscopic level, with particularly intense fluorescence near the core of the representative cross-section shown (Fig. 4A). We also noted significant particle uptake in an orthotopic PDAC xenograft model, shown at the microscopic level (Fig. S6B). At the posttranscriptional level, we determined that mRNA was knocked down in response to TPN treatment with siKras in KPC mice 48 hours after a single dose, while Kras expression in tumors of mice that received non-targeted siRNA delivered in the same fashion was indistinguishable from controls (Fig. 4B).
Figure 4. In vivo function of PEGylated iRGD TPNs.
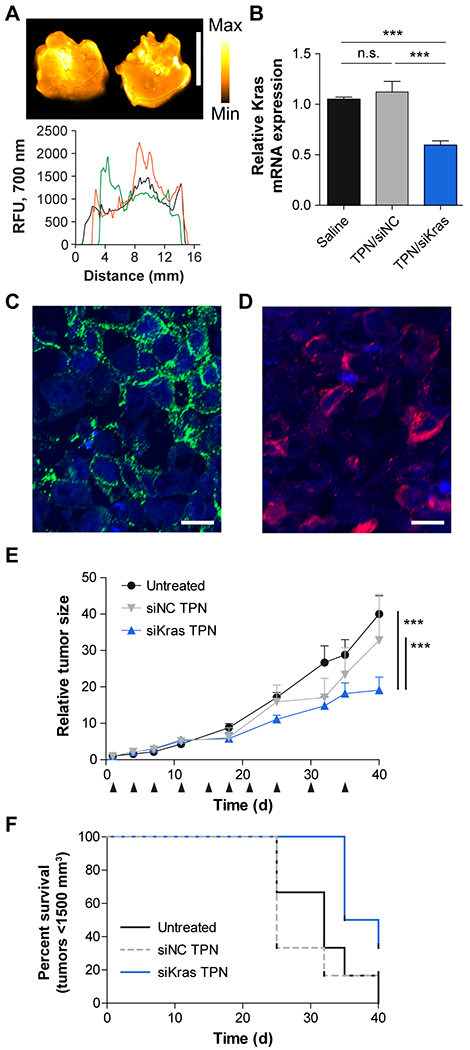
(A) PEGylated iRGD TPN delivery of fluorescently-tagged siRNA to a Kras-p53 (KPC) GEM model of PDAC, with representative tumor cross-sections shown above and linear intensity traces shown below. Scale bar: 1 cm. (B) 48-hour Kras mRNA knockdown in KPC tumors in vivo, n=3 per condition. ***: p<0.001, n.s.: not significant by one-way ANOVA. (C) Immunofluorescent staining of αv integrin (green) distribution in a PDAC isolated from the KPC model. Nuclei are blue. Scale bar: 25 µm. (D) Immunofluorescent staining of TAMRA-tagged tandem peptide distribution in a PDAC isolated from the KPC model after injection with PEG iRGD TPNs (red). Scale bar: 25 µm. (E) Tumor growth curves of mice bearing KPC-derived allograft tumors; mice were treated with diluent only (“Untreated”) or PEGylated iRGD TPNs containing siRNA against Kras (“siKras”) or a non-targeted siRNA (“siNC”), n=6 per group. Black arrows indicate dates of dosing. ***: p<0.001 by two-way ANOVA. Relative tumor size was computed as the current tumor volume divided by the starting tumor volume for each given mouse. Absolute starting tumor volumes were closely matched between treatment groups. (F) Kaplan-Meier plot of tumor growth of the cohorts shown in panel E, with a standard threshold absolute tumor volume used as a surrogate metric for survival.
With evidence of functional tumor-penetrating delivery of siRNA to pancreatic tumors, we then conducted therapeutic trials in a subcutaneous model of pancreatic cancer using KP D8–175 cells (another KPC cell line). Histological characterization of these subcutaneous tumors demonstrated that most cells exhibited surface αv integrins (Fig. 4C). After systemic dosing with PEGylated iRGD TPNs, we observed robust cytoplasmic presence of TAMRA-labeled pTP-iRGD as a proxy of intracellular siRNA delivery, as the siRNA itself was unlabeled (Fig. 4D). To test the impact of Kras knockdown in this model, we compared systemic treatment with TPNs carrying siRNA against Kras (siKras) versus negative controls of TPNs carrying untargeted siRNA, TPNs carrying untargeted siRNA (siNC), and saline injections, dosed twice a week over 5 weeks for a cumulative siRNA dose of 5 mg/kg. (Fig. 4E). siKras treatment delivered via TPNs resulted in statistically significant slowing of tumor growth relative to both negative controls (by two-way ANOVA), with tumor burden increasing to only 20 times the original volume over 40 days after the start of treatment, compared to 42 times in the untreated group and 38 times in the non-targeting siRNA-treated group (Fig. 4E). Meanwhile, the untargeted siRNA and saline injection controls were not statistically different from each other. Notably, while the tumors initially grew at approximately the same rate for the first week, we subsequently observed that from days 11 to 40, the growth rate (slope) of the siKras-treated tumors was 4.0 times lower than that of the untreated tumors, in agreement with the penetrating ability of the particles in larger tumors and the expected timeline of angiogenesis as a source of integrin targets. Overall, when we fit the growth curves to an exponential growth function, we found that the tumor doubling time was extended to 13.3 days in the siKras-treated group, as compared to 10.1 days in the untreated and 10.3 days in the non-targeted siRNA-treated group. Using an absolute tumor volume threshold as a surrogate marker for survival, the siKras treatment also resulted in extended survival relative to the two control groups (Fig. 4F). Thus, PEGylated iRGD TPNs deliver siRNA to tumors in a highly distributed fashion throughout the tumor tissue, and our therapeutic trial data indicate that treatment with iRGD TPNs delivering siRNA against KRAS slows tumor growth.
4. Discussion
We have designed a nanoparticle-based approach for nucleic acid delivery to pancreatic cancers through the development of PEGylated iRGD TPNs, which show promise in conveying gene-specific therapies to PDAC in vitro and in vivo. This work extends the modular concept behind TPNs on multiple fronts. In addition to interchanging the targeting domain, we successfully applied our prior work on optimizing TPN formulations with modular PEG compounds to a new tandem peptide, which, while still CendR-based, has a distinct chemical character, owing to its different net charge and updated receptor specificity (Fig. 2).
Since pancreatic cancer cells are typically surrounded by a thick, poorly perfused stroma that renders much of the tumor inaccessible to drugs, one of our priorities was developing particles that could actively engage this barrier. In contrast to stroma-depleting strategies (12–13), we considered that transient access mediated by iRGD might present an alternate strategy that would allow delivery without interfering with the tumor growth- and migration-constraining properties of these physical barriers (as suggested in Figs. S5 and S7). For the first time, we directly tested the capacity of TPNs to mediate 3D structure penetration using both mouse- and patient-derived human tumor organoids. Tumor tissue-derived organoids mimic the in vivo tumor microenvironment better than monolayer cell lines and reinstate barriers that are encountered by macromolecules, such as siRNAs. Patient-derived organoids also represent an important resource for developing clinical screens for drugs and for predicting treatment outcomes (41,42). Here, we demonstrated the utility of organoids to model interactions between TPNs and the tumor cells in a 3D context. In combination with high resolution microscopy, we tracked TPN penetration in organoids over time to assess cellular uptake of the nanoparticles and performed detailed characterization of the heterogeneity within the structures. Targeted TPNs exhibited efficient cellular uptake and penetration from the periphery to the center of the organoids, whereas naked siRNA and untargeted (scrambled) TPNs did not travel beyond the outer layers of the tumor (Fig. 3). The ability to quantify penetration – potentially in real time – opens up new avenues for understanding and improving CendR-guided nanocomplex delivery. Our first TEM micrographs characterizing the sub-nanocomplex structure of TPNs in a salt/buffer solution offer complementary clues to how this penetration may be working physically (Fig. S3A), and together with organoid quantification can serve as the foundation for future computational analyses. Moreover, the organoid-TPN delivery platform could serve as a tool for testing the effects of knocking down different genetic targets across organoids that represent the diversity of patient tumors, while incorporating the challenges of receptor-specific penetration and delivery that traditional 2D culture screens lack.
While the relative expression levels and distributions of relevant αvβ3/5 integrins and neuropilin-1 vary between individual tumors and their derived cell lines – as with virtually all tumor markers - we found that the vast majority of PDACs in a human tissue microarray, along with the cell lines we tested, were positive for both (Fig. 1). Staining for neuropilin-1 in orthotopic allograft tumors and the adjacent normal pancreatic tissue highlights the differences in expression patterns that set the stage for tumor-specific penetration (Fig. S6A). Encouragingly, iRGD-mediated TPN uptake was observed in these orthotopic tumor models, commensurate with the level of NRP-1 seen in the tumors (Figs. S5 & S6B). Moreover, the presence of iRGD-recognized integrins on angiogenic blood vessels and tumor cells (43), as well as neuropilin-1 in stromal elements (e.g. myofibroblasts) (44), is well-documented, as are their roles in potentially abetting tumor growth. As such, we expect that iRGD-based approaches would be applicable to a large proportion of PDACs.
From the therapeutic angle, we focused on targeting KRAS mRNA due to the high rate of KRAS mutations in human tumors. While artificial inducible models of KRAS withdrawal or anti-KRAS shRNA expression have shown robust responses, studies using small molecule drugs to target this pathway in vivo have not, to date, been able to achieve clinical therapeutic impact (26). Indeed, in contrast to other oncogenes like EGFR and ERBB2, KRAS does not have a clinically-approved small molecule or antibody inhibitor, although research on this topic is ongoing (45). While KRAS siRNA sequences used in this paper are not specific for mutated KRAS mRNA compared to wild-type, strategies for the development of mutation-specific siRNAs have been described in the literature (46) and may add some degree of greater specificity for tumor cells. Therapeutic siKRAS delivery has been achieved in lung cancer models (28), but systemic treatment of pancreatic cancer, which does not benefit from a first-pass effect and lies in an organ with low overall bloodflow, has not yet been reported. We are thus encouraged by our in vivo results showing knockdown in a ‘gold standard’ PDAC platform (immune-competent KPC mice), and evidence that systemic administration of our modified, targeted siKras-bearing TPNs slowed tumor growth in an aggressive allograft model of pancreatic cancer (Fig. 4). The tumor-slowing effect was most pronounced after the first week of treatment, suggesting particular efficacy in more advanced tumors, and the overall tumor doubling time was increased by 32% compared to untreated tumors. Improvements in imaging techniques may enable more sophisticated preclinical therapeutic trials in the future. Unfortunately, KRAS knockdown is unlikely to suffice as a monotherapy given the strong possibility of resistance due to compensatory mutations and altered expression profiles. Thus, it will be necessary to redouble efforts to identify and credential new targets that will enhance or synergize with KRAS pathway blockade. shRNA and CRISPR/Cas9-powered screens are generating unprecedented lists of genetic targets that have the potential to become new RNAi therapies. A recently defined cancer dependency map has unveiled the importance of gene expression in addition to mutations for tumor survival (47). These findings suggest that in addition to efforts focused on mutated oncogenes, the majority of hits derived from genetic screens remain to be tested for efficacy as therapeutic targets. In this work, we have demonstrated that TPNs are well-poised to serve as a tool to establish a target validation platform for single and multiple siRNA knockdown candidates (Fig. S2), as well as a delivery vehicle for credentialed combinations of siRNA interventions. Furthermore, with iRGD being successfully employed to deliver chemotherapeutics to pancreatic cancer (22), it may be possible to combine gene-targeted therapies with traditional cytotoxic drugs to more comprehensively combat this disease.
In summary, we have engineered peptide-based nanocomplexes specifically designed to address the constraints and challenges of systemically treating pancreatic cancer. In particular, these tumor-penetrating nanocomplexes can deliver siRNA addressing a key driving genetic mutation in PDAC, utilizing embedded mechanisms for penetrating through the tumor environment using iRGD, whose receptors are widely expressed in human pancreatic cancers. With validation of both the penetrating properties and therapeutic efficacy of these particles in various in vitro and in vivo models of PDAC, we believe the approach can easily be adapted to enable translation of our growing genetic understanding of PDAC.
Supplementary Material
5. Acknowledgements
The authors would like to thank Dr. Heather Fleming (MIT) for critical reading of the manuscript. They would also like to thank Lauren Brais, Dorisanne Ragon, Ewa Sicinska, and the clinical research coordinator and pathology teams at the Dana-Farber Cancer Institute and Brigham and Women’s Hospital for their assistance with consenting patients and obtaining tissue for organoid culture. The authors acknowledge the Koch Institute core facilities in the Swanson Biotechnology Center (funded by the Koch Institute Support Grant P30-CA14051 from the NCI), particularly the Nanotechnology Materials Core for expertise in TEM imaging and advanced instrumentation, the Flow Cytometry Core, the Hope Babette Tang Histology Facility, and the Microscopy Core (especially Jeffrey Wyckoff for intravital imaging); as well as the MIT Division of Comparative Medicine and Committee on Animal Care. This work was funded in part by a grant from the Lustgarten Foundation, a Core Center Grant (P30-ES002109) from the National Institute of Environmental Health Sciences, a Starr Cancer Consortium grant from the Starr Foundation, the Marie-D. & Pierre Casimir-Lambert Fund, the MIT-Harvard Center of Cancer Nanotechnology Excellence (NIH U54CA151884), the Marble Center for Cancer Nanomedicine and NCI U01 CA176058 (to W.C.H.). The content of the information within this document does not necessarily reflect the position or the policy of the Government. J.H.L. gratefully acknowledges funding from the NIH/NIGMS (MSTP T32GM007753) and from the Ludwig Fellowship for metastasis research. L.H. acknowledges funding from Koch Institute Quinquennial Cancer Research Fellowship. S.R. was supported by the Dana-Farber Leadership Council, NIH T32 CA009172, American Society of Clinical Oncology/Conquer Cancer Foundation Young Investigator Award, Hope Funds for Cancer Research Postdoctoral Fellowship, the Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research, Perry S. Levy Endowed Fellowship, and the Harvard Catalyst and Harvard Clinical and Translational Science Center (UL1 TR001102). A.J.A. was supported by the Pancreatic Cancer Action Network Samuel Stroum Fellowship, Hope Funds for Cancer Research Postdoctoral Fellowship, American Society of Clinical Oncology Young Investigator Award, Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research, Perry S. Levy Endowed Fellowship, and the Harvard Catalyst and Harvard Clinical and Translational Science Center (UL1 TR001102). S.N.B. and T.J. are HHMI Investigators.
Financial support (also listed in the acknowledgments):
Support for the project as a whole: Marble Center for Cancer Nanomedicine; Lustgarten Foundation grant; Starr Cancer Consortium grant (Starr Foundation); Marie-D. & Pierre Casimir-Lambert Fund; NCI P30-CA14051; NCI CCNE U54CA151884
J.H.L.: NIH/NIGMS (MSTP T32GM007753); Ludwig Fellowship for metastasis research
L.H.: Koch Institute Quinquennial Cancer Research Fellowship
S.R.: Dana-Farber Leadership Council, NIH T32 CA009172, American Society of Clinical Oncology/Conquer Cancer Foundation Young Investigator Award, Hope Funds for Cancer Research Postdoctoral Fellowship, Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research, Perry S. Levy Endowed Fellowship, and the Harvard Catalyst and Harvard Clinical and Translational Science Center (UL1 TR001102)
A.J.A.: Pancreatic Cancer Action Network Samuel Stroum Fellowship, Hope Funds for Cancer Research Postdoctoral Fellowship, American Society of Clinical Oncology Young Investigator Award, Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research, Perry S. Levy Endowed Fellowship, and the Harvard Catalyst and Harvard Clinical and Translational Science Center (UL1 TR001102)
W.C.H.: NCI U01 CA176058
B.M.W.: Lustgarten Foundation, Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research
S.N.B. and T.J.: HHMI Investigators
Footnotes
Disclosure of Potential Conflicts of Interests:
The authors do not have any conflicts of interests relevant to the work presented in this manuscript
6 References
- 1.National Cancer Institute. Fast Stats: An interactive tool for access to SEER cancer statistics. http://seer.cancer.gov/faststats (last accessed 9-27-2017).
- 2.American Cancer Society, Cancer Facts & Figures 2013. American Cancer Society: Atlanta, GA, 2013. [Google Scholar]
- 3.Ryan DP, Hong TS, Bardeesy N, Pancreatic Adenocarcinoma. New England Journal of Medicine 2014, 371 (11), 1039–1049. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. , Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine 2013, 369 (18), 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. , FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. New England Journal of Medicine 2011, 364 (19), 1817–1825. [DOI] [PubMed] [Google Scholar]
- 6.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. , Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol 2010, 28 (22), 3617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rougier P, Riess H, Manges R, Karasek P, Humblet Y, Barone C, et al. , Randomised, placebo-controlled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. European Journal of Cancer 2013, 49 (12), 2633–42. [DOI] [PubMed] [Google Scholar]
- 8.Ioka T, Okusaka T, Ohkawa S, Boku N, Sawaki A, Fujii Y, et al. , Efficacy and safety of axitinib in combination with gemcitabine in advanced pancreatic cancer: subgroup analyses by region, including Japan, from the global randomized Phase III trial. Japanese Journal of Clinical Oncology 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Philip PA, Benedetti J, Corless CL, Wong R, O’Reilly EM, Flynn PJ, et al. , Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol 2010, 28 (22), 3605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. , Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of Clinical Oncology 2007, 25 (15), 1960–1966. [DOI] [PubMed] [Google Scholar]
- 11.Manji GA, Olive KP, Saenger YM, Oberstein P, Current and emerging therapies in metastatic pancreatic cancer. Clinical Cancer Research 2017, 23 (7), 1670–1678. [DOI] [PubMed] [Google Scholar]
- 12.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR, Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21 (3), 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. , Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324 (5933), 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. , Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62 (1), 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, et al. , Phase IB study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clinical Cancer Research 2016, 22 (12), 2848–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lou K-J, Stromal uncertainties in pancreatic cancer. Science-Business eXchange 2014, 7 (23). [Google Scholar]
- 17.Rhim Andrew D, Oberstein Paul E, Thomas Dafydd H, Mirek Emily T, Palermo Carmine F, Sastra Stephen A, et al. , Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25 (6), 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E, C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A 2009, 106 (38), 16157–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. , Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16 (6), 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang D, Wang H, Sun C, Zhao H, Hu K, Qin W, et al. , Development of a high quantum yield dye for tumour imaging. Chemical Science 2017, 8 (9), 6322–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Li X, Sha H, Zhang L, Bian X, Han X, et al. , sTRAIL-iRGD is a promising therapeutic agent for gastric cancer treatment. Scientific Reports 2017, 7 (1), 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Lin P, Perrett I, Lin J, Liao YP, Chang CH, et al. , Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer. J Clin Invest 2017, 127 (5), 2007–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu X, Wu J, Liu Y, Yu M, Zhao L, Zhu X, et al. , Ultra-pH-responsive and tumor-penetrating nanoplatform for targeted siRNA delivery with robust anti-cancer efficacy. Angewandte Chemie 2016, 55 (25), 7091–7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hidalgo M, Pancreatic cancer. N Engl J Med 2010, 362 (17), 1605–1617. [DOI] [PubMed] [Google Scholar]
- 25.di Magliano MP, Logsdon CD, Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144 (6), 1220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Downward J, Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003, 3 (1), 11–22. [DOI] [PubMed] [Google Scholar]
- 27.Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, et al. , A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. Journal of Thoracic Oncology 2011, 6 (8), 1435–7. [DOI] [PubMed] [Google Scholar]
- 28.Xue W, Dahlman JE, Tammela T, Khan OF, Sood S, Dave A, et al. , Small RNA combination therapy for lung cancer. Proc Natl Acad Sci U S A 2014, 111 (34), E3553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, et al. , Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther 2014, 13 (12), 2876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, et al. , Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol Ther 2010, 18 (7), 1357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dahlman JE, Barnes C, Khan O, Thiriot A, Jhunjunwala S, Shaw TE, et al. , In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, et al. , Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464 (7291), 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda S, Keiser K, Nair JK, Charisse K, Manoharan RM, Kretschmer P, et al. , siRNA conjugates carrying sequentially assembled trivalent N-Acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chemical Biology 2015. [DOI] [PubMed] [Google Scholar]
- 34.Zorde Khvalevsky E, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. , Mutant KRAS is a druggable target for pancreatic cancer. Proceedings of the National Academy of Sciences 2013, 110 (51), 20723–20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren Y, Cheung HW, von Maltzhan G, Agrawal A, Cowley GS, Weir BA, et al. , Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Sci Transl Med 2012, 4 (147), 147ra112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren Y, Hauert S, Lo JH, Bhatia SN, Identification and characterization of receptor-specific peptides for siRNA delivery. ACS Nano 2012, 6 (10), 8620–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo JH, Kwon EJ, Zhang AQ, Singhal P, Bhatia SN, Comparison of modular PEG incorporation strategies for stabilization of peptide-siRNA nanocomplexes. Bioconjug Chem 2016, 27 (10), 2323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. , Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7 (5), 469–483. [DOI] [PubMed] [Google Scholar]
- 39.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE, Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protocols 2009, 4 (11), 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu H, Liang ZY, Ren XY, Liu TH, Small interfering RNAs targeting mutant K-ras inhibit human pancreatic carcinoma cells growth in vitro and in vivo. Cancer Biology & Therapy 2006, 5 (12), 1693–8. [DOI] [PubMed] [Google Scholar]
- 41.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. , Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160 (1–2), 324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. , Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med 2015, 21 (11), 1364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desgrosellier JS, Cheresh DA, Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010, 10 (1), 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, et al. , Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res 2012, 72 (16), 4047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostrem JM, Shokat KM, Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 2016, 15 (11), 771–785. [DOI] [PubMed] [Google Scholar]
- 46.Schwarz DS, Ding H, Kennington L, Moore JT, Schelter J, Burchard J, et al. , Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet 2006, 2 (9), e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. , Defining a cancer dependency map. Cell 2017, 170 (3), 564–576 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.