

Abstract
Thrombospondin-1 (TSP-1) is primarily expressed by platelets and endothelial cells (ECs) and rapidly released upon their activation. It functions in haemostasis as a bridging molecule in platelet aggregation, by promoting platelet adhesion to collagen and by protecting von Willebrand factor strings from degradation. In blood of patients undergoing surgery and in co-cultures of neutrophils with platelets or ECs, we observed proteolysis of the 185 kDa full-length TSP-1 to a 160-kDa isoform. We hypothesized that TSP-1 processing may alter its haemostatic properties. Selective enzyme inhibitors in co-cultures revealed that neutrophil proteases elastase and cathepsin G mediate TSP-1 processing. The cut site of cathepsin G was mapped to TSP-1 amino acids R237/T238 by Edman sequencing. Formation of neutrophil extracellular traps protected TSP-1 from complete degradation and promoted controlled processing to the 160-kDa isoform. Haemostatic properties were tested by platelet aggregation, adhesion, coagulation and string formation under flow. Platelets from TSP-1 deficient mice did not differ from wild-type in platelet aggregation but showed severe impairment of platelet adhesion to collagen and string formation under flow. Reconstitution experiments revealed that the 160-kDa TSP-1 isoform was markedly more potent than the 185-kDa full-length molecule in restoring function. Thus, TSP-1 processing by neutrophil proteases yields a 160-kDa isoform which shows enhanced potency to promote platelet adhesion and string formation. This finding reveals a novel mechanism of neutrophil-mediated thrombus formation and provides first evidence for the impact of TSP-1 proteolysis on its haemostatic properties.
Keywords: thrombospondin-1, neutrophil elastase, cathepsin G, proteolysis, platelet adhesion, platelet string formation
Introduction
Thrombospondin-1 (TSP-1) is a multi-domain glycoprotein formed by three identical 185 kDa sub-units that are connected via disulphide bonds. 1 The protein comprises a heparin-binding domain (HBD) of globular structure which is N-terminal of the connecting region, followed by a pro-collagen-homology domain, three properidin-like TSP type 1 modules, three epidermal growth factor (GF)-like TSP type 2 elements that provide structural stability, seven calcium-binding TSP type 3 repeats and a unique lectin-like C-terminal globular domain. 1 Each domain enables TSP-1 to fulfil distinct functions in various biological processes and to interact with a variety of binding partners such as fibrinogen, von Willebrand factor (vWF), the scavenger receptor CD36 or the cell surface receptor CD47. 2 3 The main producers of TSP-1 are platelets and endothelial cells (ECs) where it is stored in α-granules or Weibel–Palade bodies. 4 5 TSP-1 is constitutively expressed and protein levels between 20 and 40 ng/mL are found in human plasma under physiological conditions. 6 7 However, a rapid increase of TSP-1 release can be observed after activation of platelets and ECs. 4 5
Among its various biological functions, the role of TSP-1 in angiogenesis has been extensively studied over the past years. 8 Both pro- and anti-angiogenic properties are attributed to TSP-1. 9 10 11 While the N-terminus mediates adhesion and motility of ECs, the remaining molecular core inhibits angiogenesis by antagonizing survival pathways while also activating apoptotic pathways. 8 12 In particular, TSP-1 binding to CD47 or CD36 surface receptors was found to inhibit nitric oxide (NO) signalling. 13 14 Lee et al reported in 2006 that the matrix metalloprotease a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS)-1 is able to cut TSP-1 leading to a matrix-bound trimer of the 36-kDa N-terminal domain and a soluble, monomeric 110 to 125 kDa C-terminal fragment acting as a potent angiogenesis inhibitor. 15 16 Thus, by removal of the N-terminal HBD the anti-angiogenic effect of TSP-1, mainly attributed to the type 1 repeats and C-terminal globular domain, is promoted.
In addition to its prominent functions in angiogenesis, TSP-1 is also known to play a role in haemostasis. It enhances platelet aggregation by forming a bridge between fibrinogen molecules bound to platelet integrin αIIbβ3. 17 Furthermore, TSP-1 promotes platelet aggregation through binding to CD36 and CD47 receptors which results in the activation of platelets and intra-cellular signalling. 18 Another essential function of TSP-1 is the ability to stabilize platelet aggregates under shear stress. While the plasma protease ADAMTS-13 cuts multimeric vWF to resolve platelet strings, TSP-1 is able to bind to and stabilize vWF thereby protecting it from ADAMTS-13-mediated degradation. 19
In the context of haemostasis, it is still unclear whether proteolytic processing of TSP-1 and removal of the N-terminus promotes a functional change of the protein as observed in angiogenesis. We previously found that two different TSP-1 isoforms of 185 and 160 kDa are constitutively present in human plasma. 6 A substantial increase of the smaller TSP-1 isoform was detected after surgical intervention when blood coagulation and post-operative wound healing were triggered. However, the source and mechanism generating the 160-kDa TSP-1 variant have not been revealed to date and the functional consequences on haemostasis have not been addressed.
During the past years, several plasma or leukocyte-derived proteases were found to have the ability to cut TSP-1 resulting in fragments of variable size ranging from 25 to 160 kDa, but with unknown functional or in vivo relevance. In addition to plasmin, TSP-1 is reportedly susceptible to cleavage by the neutrophil serine proteases cathepsin G and elastase which may remove the N-terminal domain. 20 21 Since it is known that neutrophils are crucially involved in platelet aggregation and in the formation and degradation of fibrin during thrombus deposition, 22 we hypothesized that their activation and protease release might also affect TSP-1 function in haemostasis or thrombosis where the so-called neutrophil extracellular traps (NETs) are implicated.
NETs are a network of extracellular deoxyribonucleic acid (DNA) associated with histones and neutrophil-derived proteins such as elastase and have recently been discovered to contribute to thrombus formation. 23 24 During the process of NETosis DNA is decondensed by modification of histones, for example, via citrullination by peptidylarginine deiminase 4 (PAD4) and subsequently released to the extracellular space. 25 In addition to their proposed role in pathogen trapping and killing, 26 NETs were shown to be associated with various pathological conditions including thrombosis. 27 28
To address the question whether proteolytic processing of TSP-1 may alter its haemostatic properties and hence affect thrombus formation, we (1) identified the proteases mediating TSP-1 fragmentation in co-cultures of neutrophils with platelets or ECs. We (2) characterized the generated TSP-1 isoform at the molecular level and (3) compared the original full-length protein to the smaller isoform regarding their potency to promote platelet adhesion and string formation.
Materials and Methods
Cells and Cell Culture
Human microvascular ECs were isolated from foreskin samples and purified as previously described. 29 ECs were grown in EGM-2 MV medium (Lonza/Clonetics, Walkersville, Maryland, United States) supplemented with 5% foetal calf serum (FCS), hydrocortisone, ascorbic acid, basic human fibroblast GF, insulin-like GF 1, epidermal GF and gentamicin/amphotericin GA-1000 (concentrations not specified). Attachment to cell culture dishes was promoted by fibronectin at 1 µg/mL (EMD Millipore Corporation, Temecula, California, United States). For all experiments, medium was switched to medium without serum to avoid contamination with bovine TSP-1.
Human neutrophils and peripheral blood mononuclear cells (PBMCs) were isolated from ethylenediaminetetraacetic acid (EDTA) whole blood using Histopaque density gradient separation (#10771, #11191, Sigma-Aldrich, St. Louise, Missouri, United States). After centrifugation, the neutrophil layer was aspirated and washed with Dulbecco's phosphate-buffered saline Ca 2+ /Mg 2+ -free (PBSdef) which was followed by lysis of red blood cells and re-establishment of isotonicity with 3% sodium chloride. Neutrophils were then re-suspended in cell culture medium and only isolates with more than 90% purity as determined by the Sysmex XN-350 haemocytometer (Sysmex, Kobe, Japan) were used for experiments. Platelets were isolated via size exclusion with a Sepharose 2B (Sigma-Aldrich) column from whole blood collected in citrate, theophylline, adenosine and dipyridamole tubes. Platelets were washed with PBS and re-suspended in culture medium (containing 1 mM CaCl 2 , but without serum or GFs) for co-culture and stimulation by A23187 (Sigma-Aldrich) or thrombin receptor activator peptide 6 (TRAP-6, BACHEM, Basel, Switzerland). To inhibit platelet activation, prostaglandin E1 (Santa Cruz, Dallas, Texas, United States) was added.
Co-Culture of Endothelial and Blood Cell Populations
ECs were seeded in 2 mL EGM-2 MV to reach confluence in 6 wells (5 × 10 5 cells/well) within 1 day. Cells were then washed twice with 2 mL PBSdef and 1 mL EGM-2 MV w/o GFs and FCS was added. The cells were allowed to release TSP-1 into the supernatant for 48 hours and subsequently a co-culture was performed. Per well, 20 × 10 6 platelets, 2 × 10 6 PBMCs or 2 × 10 6 neutrophils were applied. A total of 500 µL of the conditioned medium were removed from the 6-well and replaced with platelets, PBMCs or neutrophils suspended in 500 µL EGM-2 MV w/o GFs and FCS for co-culture. Moreover, 500 µL of these cell isolates were added to the retrieved 500 µL conditioned medium or to 500 µL EGM-2 MV w/o GFs and FCS for control. Samples were either left untreated or were supplied with activating stimuli. Platelets were activated with 10 µM TRAP-6, PBMCs with 1 µg/mL lipopolysaccharide (LPS, Sigma-Aldrich) and neutrophils with 100 ng/mL phorbol myristate acetate (PMA, Sigma-Aldrich). Alternatively, platelets (4 × 10 6 ) were co-cultured with neutrophils (4 × 10 5 ) without additional stimulus. The supernatant was collected after 0.5, 1, 2 and 4 hours for immunoblotting.
Furthermore, co-cultures were supplemented with inhibitors for the neutrophil proteases cathepsin G and/or elastase (#219372, #324744, Merck KGaA, Darmstadt, Germany). Note that 1 × 10 5 ECs or 4 × 10 6 platelets were combined with 4 × 10 5 neutrophils. Elastase inhibitor II (2.1 mM) and cathepsin G inhibitor I (0.1 mM) were added either separately or combined. The supernatant was harvested after 30 minutes of incubation.
TSP-1 Processing with Purified Neutrophil Proteases
Serum-free, conditioned EC supernatant containing 185 kDa TSP-1 was supplied with purified, human neutrophil elastase at 10 to 50 mU/mL (SERVA Electrophoresis GmbH, Heidelberg, Germany) or cathepsin G at 2 to 50 mU/mL (Merck KGaA) for 30 minutes to 4 hours at 37°C.
Immunoblotting
For sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis, samples (culture supernatants without further concentration or dilution) were mixed with loading buffer containing 11.5% β-mercaptoethanol, 0.4 M dithiothreitol for reducing conditions, but lacking β-mercaptoethanol and dithiothreitol under non-reducing conditions. Proteins were subsequently transferred to a polyvinylidene fluoride membrane (Merck KGaA) overnight at 20 or 35 V. Membranes were blocked and then incubated overnight at 4°C with primary antibodies: mouse anti-TSP-1 Ab11 (D4.6 + A6.1 + MBC200.1, Thermo Fisher Scientific, Waltham, Massachusetts, United States), C-terminus specific mouse anti-TSP-1 Ab4 (clone A6.1, Thermo Scientific) and N-terminus specific goat anti-TSP-1 N20 (Santa Cruz) in a 1:300 dilution or rabbit anti-histone H3 (citrulline R2 + R8 + R17) (Abcam, Cambridge, United Kingdom) at 1:1,000 dilution. Thereafter, membranes were incubated for 1 hour at 1:50,000 dilution of species-specific horseradish peroxidase-conjugated secondary antibody (Thermo Fisher Scientific). After addition of the chemiluminescent reagent (Lumigen, Inc., Southfield, Michigan, United States), membranes were exposed to X-ray film and images were processed with Adobe Photoshop CS6.
NET Formation
NET experiments were based on a co-culture of neutrophils (1 × 10 5 ) and platelets (1 × 10 7 ). Cells were activated by the calcium ionophore A23187 (4 µM) in the absence or presence of the NETosis inhibitor GSK484 at 2 mM (Cayman Chemicals, Ann Arbor, Michigan, United States). Where indicated, 50 µM cathepsin G inhibitor and 1 mM elastase inhibitor were added (concomitantly with GSK484), then incubated for 30 minutes before stimulation with A23187. NETs generated by activated neutrophils were digested with 500 mU/mL micrococcal nuclease (Sigma-Aldrich) for 10 minutes and culture supernatant was retrieved by centrifugation at 5,000 × g for 90 seconds for immunoblotting of TSP-1 and citrullinated histone H3 (citH3) or for elastase activity assays (BioVision, Milpitas, California, United States) conducted according to the manufacturer's instructions.
DNA Release Assay
Neutrophils and platelets were seeded into black 96-well tissue culture plates. Prior to activation with A23187, the cells were pre-incubated for 30 minutes with the PAD4 inhibitor GSK484 at 2 mM. After addition of 4 µM A23187 and 5 µM Sytox Green (Thermo Fisher Scientific), the released, cell-free DNA was measured at 405 nm in a plate reader (Varioskan Flash, Thermo Fisher Scientific) at intervals of 15 minutes over a period of 6 hours.
In Vitro Platelet Aggregation and Adhesion Under Flow
Parallel flow chambers (µ-slide VI 0.4 Luer) of the ibidi pump system ( ibidi , Munich, Germany) were coated with collagen (moeLab GmbH, Langenfeld, Germany) overnight. Before the experiment, the flow chambers and the perfusion set were pre-perfused for 1 minute with mouse plasma 1:10 diluted with Tyrode's Hepes (TH) buffer supplemented with 1 mM CaCl 2 , 0.5% bovine serum albumin and 10 mg/mL fibrinogen. Heparinized blood was drawn from the vena cava of 8- to 12-week-old mice. A total of 500 µL of blood were either left untreated or were supplied with 2 or 0.5 µg purified 160 or 185 kDa TSP-1 for 10 minutes. Platelets were labelled by the addition of fluorescent anti-mouse GPIbβ antibody (emfret Analytics, Eibelstadt, Germany) at 1:300 prior to 1:3 dilution of the whole sample with TH buffer. Mouse blood was perfused through parallel flow chambers at a shear rate of 7 dyne/cm 2 for 7 minutes. Platelets were visualized with an IX83 microscope (Olympus, Tokyo, Japan) and 10 images per sample were taken within 2 minutes with an Orca Flash 4.0 camera (Hamamatsu, Bridgewate, New Jersey, United States) using the CellSens Dimensions software (Olympus). Images were analysed for the number of adherent platelets, the total area of platelet aggregates in 0.1 mm 2 and for the number and mean length of platelet strings (after manual labelling of the strings in red) using the Fiji -version of the ImageJ software: Composite colour images were split into red, green and blue channels and the red channel was subjected to automated thresholding using the MaxEntropy algorithm followed by the ‘analyse particles’ routine using a minimum size of 50 pixels. The analysis of platelet strings is further illustrated in Supplementary Fig. S7 , available in the online version.
Mice deficient in TSP-1 and of C57Bl/6J background (B6.129S2-Thbs1 tm1Hyn/J) were obtained from Charles River Laboratories, Sulzfeld, Germany. Wild-type (WT) mice (C57BL/6J) were used for comparison.
Results
Platelets and ECs Release Full-Length 185 kDa TSP-1 but do not Generate the 160 kDa Isoform
When human plasma samples are analysed for the presence of TSP-1 isoforms immediately prior to and 1 day after surgery (resection of liver metastases), two distinct TSP-1 protein variants are detected ( Fig. 1A and Supplementary Fig. S1A , available in the online version) as we have previously reported. 6 While both the 185-kDa full-length protein and a smaller 160-kDa TSP-1 isoform are present before surgical intervention, a predominance of the 160-kDa TSP-1 protein is observed in the post-operative period involving haemostasis and the initiation of wound healing.
Fig. 1.
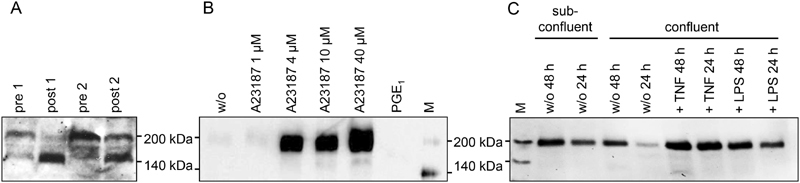
Thrombospondin-1 (TSP-1) isoforms circulating in human plasma as compared with TSP-1 proteins secreted by isolated platelets and endothelial cells (ECs). ( A ) Blood samples of two colorectal cancer patients were retrieved immediately before (pre) and 1 day after (post) resection of liver metastases and plasma was processed as previously described. 6 ( B ) Human platelets were isolated from whole blood of healthy volunteers by size exclusion chromatography in the presence of 100 nM prostaglandin E1 (PGE 1 ). They were then either left untreated (w/o), further inhibited with 100 nM PGE 1 or activated with different concentrations A23187 (1–40 µM) for 30 minutes at 37°C before collection of the supernatant. ( C ) TSP-1 released into the culture medium of human dermal microvascular endothelial cells after 24 or 48 hours was analysed for confluent and untreated, tumor necrosis factor-α (TNFα) (100 ng/mL) or lipopolysaccharide (LPS) (1 µg/mL) stimulated cultures and compared with untreated sub-confluent (proliferating) ECs. Shown are representative immunoblots (Ab11) with reduced protein samples of culture supernatants (without further concentration or dilution). Please refer to Supplementary Fig. S1 (available in the online version) for quantitation of immunoblots. Experiments were repeated 3 to 4 times. M, biotinylated protein marker.
To identify the source of the smaller 160 kDa TSP-1 variant, isoform generation and secretion of TSP-1 was investigated in platelets and ECs which constitute the main producers of TSP-1 in blood. Isolated human platelets showed a dose-dependent release of 185 kDa TSP-1 ( Fig. 1B and Supplementary Fig. S1B , available in the online version) when stimulated with increasing concentrations of calcium ionophore A23187. Platelets also consistently secreted the full-length 185 kDa TSP-1 protein in response to other agonists such as adenosine diphosphate or TRAP-6 (data not shown).
With respect to human microvascular ECs, a similar observation was made. Confluent and sub-confluent cultures were compared, and confluent cells were activated with tumor necrosis factor-α or LPS or were left untreated. Comparable to platelets, ECs selectively released full-length 185 kDa TSP-1 into the supernatant which was increased after pro-inflammatory stimulation of cells ( Fig. 1C and Supplementary Fig. S1C , available in the online version). Moreover, proliferating (sub-confluent) cells also produced 185 kDa TSP-1 molecules. The generation of a 160-kDa TSP-1 isoform was not observed under any of the experimental conditions.
The 160 kDa TSP-1 Isoform is Generated in Co-Cultures of Neutrophils with ECs or Platelets
We continued to investigate whether TSP-1 might be processed to the 160-kDa isoform upon co-culture with other blood cell types. Addition of freshly isolated human platelets or PBMCs to ECs and incubation for up to 4 hours consistently yielded the 185-kDa TSP-1 protein, with increasing intensity throughout the incubation period ( Fig. 2A and B and Supplementary Fig. S2A and S2B , available in the online version). In contrast, when neutrophils were combined with ECs or platelets a 160-kDa TSP-1 molecule was generated over time while the amount of the full-length 185 kDa protein decreased ( Fig. 2C and D and Supplementary Fig. S2C and S2D , available in the online version). Addition of selected stimuli to activate blood cells in the various co-cultures did not alter the type of TSP-1 isoform generated. However, TSP-1 processing from 185 to 160 kDa was enhanced, when EC–neutrophil co-cultures were stimulated with PMA, while platelet TSP-1 was instantly processed to 160 kDa when neutrophils were added (presumably due to neutrophil activation in platelet co-culture) and no further neutrophil stimulus was required. Incubation of the various blood cell types with conditioned EC medium (supernatant of ECs grown for 48 hours) as opposed to direct cell contact, yielded comparable results (data not shown). Concomitant control cultures of isolated leukocytes for up to 4 hours revealed that PBMCs and neutrophils (with or without cell activation) did not release substantial amounts of the TSP-1 glycoprotein during the incubation period ( Supplementary Fig. S4A and S4B , available in the online version).
Fig. 2.
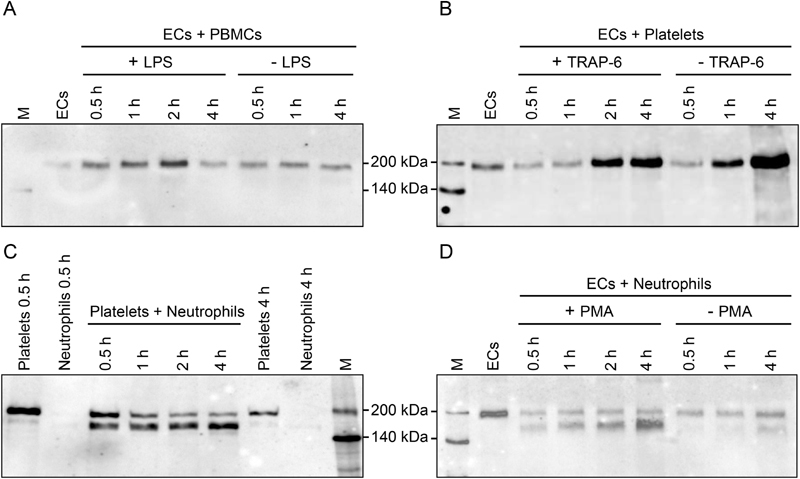
Thrombospondin-1 (TSP-1) isoforms generated in co-cultures of vascular cell populations. Confluent endothelial cell (EC) cultures grown in serum-free medium for 48 hours were either left untreated (ECs) or were supplied with freshly isolated human ( A ) peripheral blood mononuclear cells (PBMCs), ( B ) platelets or ( D ) neutrophils. Where indicated (+) a stimulus was added to activate PBMCs (lipopolysaccharide [LPS] at 1 µg/mL), platelets (thrombin receptor activator peptide 6 [TRAP-6] at 10 µM) or neutrophils (phorbol myristate acetate [PMA] at 100 ng/ml). ( C ) Platelets were also combined with neutrophils or kept separately for control. Supernatant was collected after 0.5, 1, 2 and 4 hours of culture. Reduced protein samples were analysed by immunoblots with Ab11. Each experiment was repeated four times with blood drawn from different donors. Please refer to Supplementary Fig. S2 (available in the online version) for quantitation of immunoblots. M, biotinylated protein marker.
The 160 kDa TSP-1 Fragment is Proteolytically Generated by Neutrophil-Derived Elastase and Cathepsin G and Corresponds in Size to the Shorter Plasma Isoform
To test whether neutrophil-derived proteases are mediating TSP-1 processing to the smaller isoform, co-cultures of neutrophils with ECs or platelets were performed in the presence of inhibitors for the neutrophil proteases elastase and cathepsin G ( Fig. 3A and B and Supplementary Fig. S3A and S3B , available in the online version). When the inhibitors were given separately, processing was partially inhibited leading to the concurrent appearance of both TSP-1 variants (185, 160 kDa). Concomitant treatment with both inhibitors had a co-operative inhibitory effect on proteolysis of the 185-kDa protein to the 160-kDa molecule.
Fig. 3.
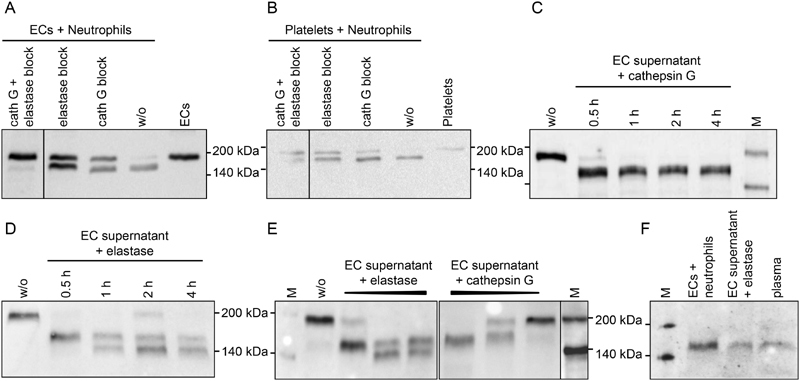
Thrombospondin-1 (TSP-1) processing by neutrophil-derived proteases. Freshly isolated human neutrophils were added to ( A ) confluent, serum-free cultures of endothelial cells or ( B ) platelets isolated from the same donor and incubated for 30 minutes at 37°C in the absence or presence of elastase inhibitor II (2.1 mM) and/or cathepsin G inhibitor I (0.1 mM). ( C–E ) Serum-free endothelial cell (EC) supernatant containing 185 kDa TSP-1 (w/o) was incubated with the purified proteases ( C ) cathepsin G at 10 mU/mL or ( D ) elastase at 20 mU/mL for 30 minutes to 4 hours, or ( E ) was exposed to increasing concentrations of elastase (10–50 mU/mL) and cathepsin G (2–50 mU/mL) for 30 minutes at 37°C. ( F ) The 160-kDa TSP-1 proteins generated either by co-culture of ECs with neutrophils or by 30-minute elastase digest of EC supernatant were compared with the smaller TSP-1 isoform prevalent in human plasma post-surgery. TSP-1 protein was detected in supernatants by immunoblotting with Ab11. Experiments were performed at least three times with blood from different donors. Please refer to Supplementary Fig. S3 (available in the online version) for quantitation of immunoblots. M, biotinylated protein marker.
In the next step, processing of TSP-1 by neutrophil-derived proteases was confirmed with purified enzymes. EC supernatant containing full-length TSP-1 was incubated with cathepsin G or elastase revealing fragmentation of 185 to 160 kDa TSP-1 by the neutrophil proteases in a time-dependent ( Fig. 3C and D and Supplementary Fig. S3C and S3D , available in the online version) and dose-dependent ( Fig. 3E and Supplementary Fig. S3E , available in the online version) manner. While cathepsin G consistently yielded the 160-kDa TSP-1 isoform, elastase additionally generated 140 kDa TSP-1 fragments after prolonged incubation or at high enzyme concentrations. Of note, when 160 kDa fragments generated by protease digest or produced in co-culture of ECs with neutrophils were compared with a post-surgical human plasma sample, proteins were found to match in molecular weight ( Fig. 3F and Supplementary Fig. S3F , available in the online version).
Neutrophil-Mediated Proteolysis of TSP-1 Removes the N-Terminus and Yields a Trimeric Core Fragment of 160 kDa Chains
To elucidate which domains of TSP-1 were removed by neutrophil-derived proteases, immunoblotting was performed with antibodies specific to N-terminal or C-terminal epitopes of TSP-1. While the antibody specific for the C-terminus detected both the 185- and 160-kDa TSP-1 isoform, the N-terminus specific antibody revealed the full-length TSP-1 and a smaller 25 kDa N-terminal fragment which was removed after processing of TSP-1 with elastase or cathepsin G ( Fig. 4A and Supplementary Fig. S4C , available in the online version). Furthermore, TSP-1 isoforms were analysed with intact or reduced disulphide bonds ( Fig. 4B ). In reduced samples, the original 185 kDa protein was confirmed for EC supernatant which was processed to 160 kDa when cathepsin G was added. Under non-reducing conditions, both the untreated and cathepsin G-treated samples showed high molecular weight molecules migrating at the upper edge of the resolving gel with an estimated molecular weight of > 400 to 450 kDa, thus pointing to trimeric complexes. Based on these analyses, we concluded that the 160-kDa TSP-1 fragments resulting from cleavage by neutrophil proteases were lacking the N-terminus and likely to form a trimeric core molecule encompassing the inter-chain disulphide bonds.
Fig. 4.
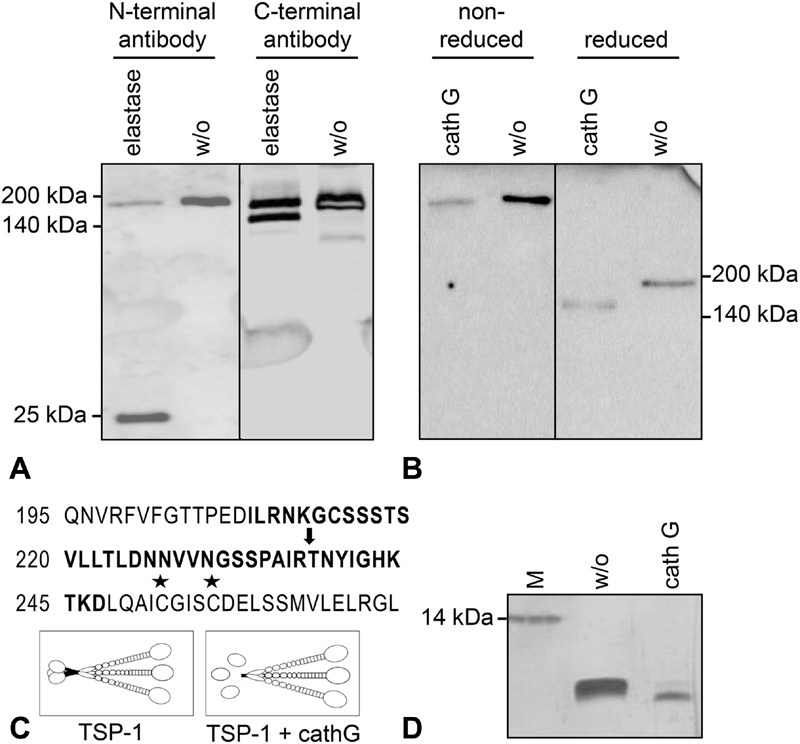
Molecular characterization of the 160-kDa thrombospondin-1 (TSP-1) isoform. Serum-free endothelial cell (EC) supernatant was either left untreated (w/o) or was digested with elastase (20 mU/mL) or cathepsin G (10 mU/mL) for 30 minutes at 37°C. ( A ) TSP-1 was detected in reduced protein samples by immunoblotting with two distinct antibodies, specific for the N-terminal or the C-terminal domain of TSP-1. ( B ) Reduced and non-reduced protein samples were compared on a gradient gel (4–20%) and TSP-1 was immunostained with Ab11, a combination of three monoclonal antibodies covering N- and C-terminal epitopes. ( C ) Illustration of the cathepsin G cut site (arrow) in the TSP-1 protein sequence as determined by mass spectrometry and Edman sequencing refers to amino acid numbering of secreted TSP-1 (not including the signal peptide); asterisks mark the cysteines involved in inter-chain disulphide bonds for trimerization. TSP-1 cleavage by cathepsin G results in the release of the monomeric N-terminal HBD (25 kDa) and a trimeric C-terminal core fragment of 160 kDa chains. ( D ) Silver staining of a synthetic peptide (comprising TSP-1 amino acids 208–247, bold letters in panel C) without or with digest by cathepsin G (50 mU/mL for 45 minutes).
To identify the exact cut-site of cathepsin G in TSP-1, the 160-kDa TSP-1 fragment generated after protease digest was analysed by mass spectrometry and by Edman protein sequencing. Mass spectrometry was able to narrow the potential cut site of cathepsin G to the area of amino acids 213 to 244 (of the secreted TSP-1 molecule, not including the signal peptide sequence) which is N-terminal of the connecting region. Edman sequencing revealed the specific cut site to be located between amino acid R237 and T238 ( Fig. 4C ). Moreover, the cut site was confirmed by cathepsin G cleavage of a synthetically generated 40 amino acid peptide covering the TSP-1 region of interest ( Fig. 4D ).
NET Formation Promotes the Controlled Processing of TSP-1 by Neutrophil-Derived Proteases
Since NET formation has recently been implicated in thrombosis, we further assessed whether the release of NETs by activated neutrophils may have an impact on the proteolytic processing of TSP-1. Thus, co-cultures of platelets and neutrophils were either left untreated or were activated with the calcium ionophore A23187, a potent trigger of NET formation. Furthermore, the NETosis inhibitor GSK484 (interfering with histone citrullination by PAD4) was added to the co-cultures prior to addition of the stimulus to allow for comparable neutrophil activation in the absence of NET formation. Release of neutrophil DNA was assessed in a time course by Sytox Green incorporation, showing a strong increase of free DNA in the supernatant after co-culture treatment with A23187 ( Fig. 5A ). Addition of GSK484 was able to block DNA release to a level close to untreated co-cultures. To test whether the NETosis inhibitor GSK484 still allows for the activation of neutrophils and thus the release of neutrophil proteases, elastase was measured in culture supernatant by activity assay ( Fig. 5B ). While A23187 triggered substantial elastase release, an even higher amount of elastase activity was detected in the presence of the NETosis inhibitor thus reflecting potent neutrophil activation and degranulation with release of soluble substances into the supernatant.
Fig. 5.
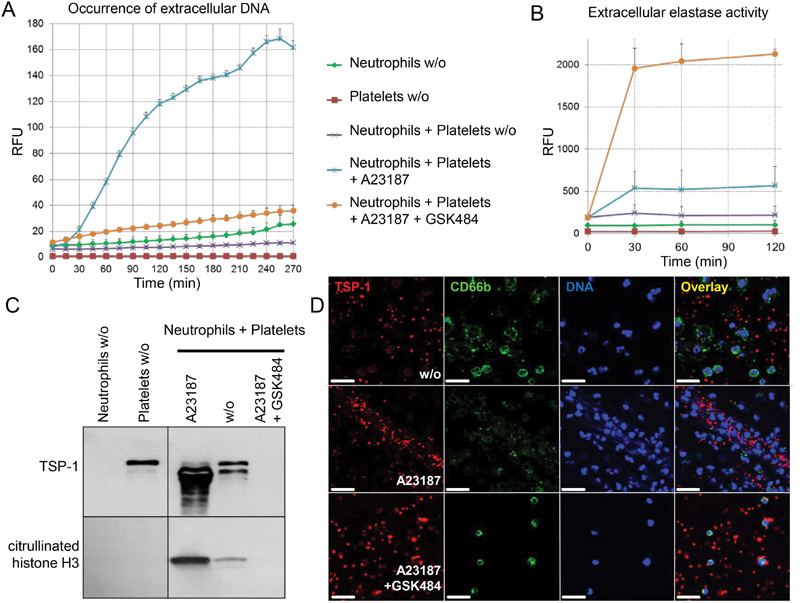
Impact of neutrophil extracellular trap (NET) formation on neutrophil-mediated proteolysis of thrombospondin-1 (TSP-1). Co-cultures of neutrophils and platelets were left untreated or stimulated with 4 µM A23187 in the absence or presence of the NETosis inhibitor GSK484 at 2 mM. ( A ) Release of neutrophil deoxyribonucleic acid (DNA) was assessed by incorporation of Sytox Green dye and measurement of relative fluorescence units (RFUs) over 5 hours. ( B ) Elastase was evaluated by activity assay. Supernatant was retrieved after 0, 30, 60 and 120 minutes. ( C ) Occurrence of TSP-1 isoforms (upper panel) and citrullination of histone H3 (lower panel) was determined at 120 minutes by immunoblotting. ( D ) Co-cultures seeded on cover slips were left untreated (w/o), stimulated by 4 µM A23187 for NET formation or exposed to A23187 in the presence of 2 mM GSK484 to block NETosis. After fixation and permeabilization, cultures were stained for TSP-1 (red), the granulocyte marker CD66b (green) and DNA (blue). Scale bar: 20 µm. All experiments were repeated at least three times with cells isolated from different human donors. Immunoblots and fluorescence images depict one representative experiment, while DNA release and elastase activity data are given as mean and standard deviation of three independent experiments.
The occurrence of TSP-1 isoforms and citH3, as a marker of NETosis, was determined in the supernatant of co-cultures by immunoblotting ( Fig. 5C ). Citrullination of histone H3 was not detectable for untreated neutrophils or platelets that were cultured separately. Neutrophils in co-culture with platelets produced low amounts of citH3 which was effectively increased by A23187 stimulation. This effect was blocked through the addition of the NETosis inhibitor GSK484. Immunoblot results were further confirmed ( Supplementary Fig. S5A , available in the online version) with a citH3 enzyme-linked immunosorbent assay. 30
Regarding TSP-1 isoforms, platelets secreted the full-length 185 kDa molecule, which was processed to 160 kDa in the presence of neutrophils as we had previously observed. Activation by the calcium ionophore A23187 and thus NET formation was able to promote TSP-1 release and processing resulting in increased levels of the 160 kDa TSP-1 isoform. Of interest, inhibition of NETosis by GSK484 led to the total loss of TSP-1 over 2 hours ( Fig. 5C ), presumably due to degradation by the massive release of proteases from activated neutrophils in the absence of NETosis ( Fig. 5B ). This notion was confirmed when we limited GSK484 exposure to 60 minutes and observed increased TSP-1 proteolysis which was efficiently blocked by inhibitors against cathepsin G and elastase ( Supplementary Fig. S5B , available in the online version). To address whether these effects would also hold true for other, more physiological stimuli of NETosis and for other TSP-1 sources, we investigated the fate of TSP-1 processing when NETs were comparably induced in platelet–neutrophil co-cultures by LPS ( Supplementary Fig. S6A and S6B , available in the online version) or in endothelial–neutrophil co-cultures by A23187 ( Supplementary Fig. S6C and S6D , available in the online version). We confirmed that NETs were induced (DNA was released) which was associated with the enhanced secretion and controlled processing of TSP-1 to the 160-kDa isoform, while NET blockade resulted in TSP-1 loss.
To investigate the localization of TSP-1 during NET formation, co-cultured platelets and neutrophils were immunostained for the neutrophil marker CD66b, the platelet marker CD41 and for TSP-1 to be analysed by confocal fluorescence microscopy ( Fig. 5D and Supplementary Fig. S5C , available in the online version). DNA in neutrophil nuclei and NET structures was visualized by Hoechst 33342 dye. Unstimulated neutrophils showed intact lobulated nuclei and CD66b cell surface staining. TSP-1 was mostly found to co-localize with CD41 positive platelets which were randomly distributed and clearly separated from unstimulated neutrophils. Neutrophils activated by calcium ionophore flattened out showing less lobulated, enlarged nuclei and decondensed chromatin as well as formation of NETs with extracellular DNA. A23187 treatment triggered local enrichment of TSP-1 in NET structures. When the inhibitor GSK484 was added prior to stimulation of co-cultures with A23187, neutrophil nuclei appeared intact and no NET structures were apparent. In this setting, TSP-1 was not associated with neutrophils but found in/on remaining platelets. Whether TSP-1 was partly degraded could not be deduced from the analysis, since the applied antibody was raised against a short TSP-1 peptide sequence.
The Proteolytically Processed 160 kDa TSP-1 Isoform has Enhanced Potency to Promote Platelet Adhesion and String Formation Under Flow
Ultimately, it was pertinent to assess possible functional consequences of neutrophil-mediated TSP-1 processing from the full-length 185 kDa molecule to the 160 kDa isoform regarding the haemostatic properties of TSP-1. Whole blood retrieved from TSP-1 knockout (KO) mice (as compared with WT mice) was analysed with respect to platelet adhesion, aggregation and string formation under in vitro flow conditions ( Fig. 6A , Supplementary Fig. S7 and Supplementary Videos A–D , available in the online version). In line with the previously reported TSP-1 functions in promoting platelet attachment to collagen and protecting vWF strings from degradation, the number of adherent platelet aggregates and the mean length of formed platelet strings were significantly higher for WT than for TSP-1 KO mice ( Fig. 6B and C ). To be able to compare the two TSP-1 isoforms in function, commercially obtained, platelet-purified 185 kDa TSP-1 was treated with cathepsin G to yield the 160-kDa fragment, and the reaction was subsequently stopped by adding cathepsin G inhibitor. Conversely, when the inhibitor was given prior to the addition of cathepsin G, TSP-1 remained intact. Substitution of TSP-1 KO blood with the two purified TSP-1 isoforms at high concentration (4 µg/mL) could restore platelet adhesion and string formation to the WT situation. However, at lower concentrations (1 µg/mL) the processed 160 kDa isoform was significantly more potent than the original 185 kDa molecule, showing a higher number of adherent platelet aggregates and longer platelet strings. These results were confirmed with recombinantly generated TSP-1 isoforms ( Supplementary Fig. S8 , available in the online version).
Fig. 6.
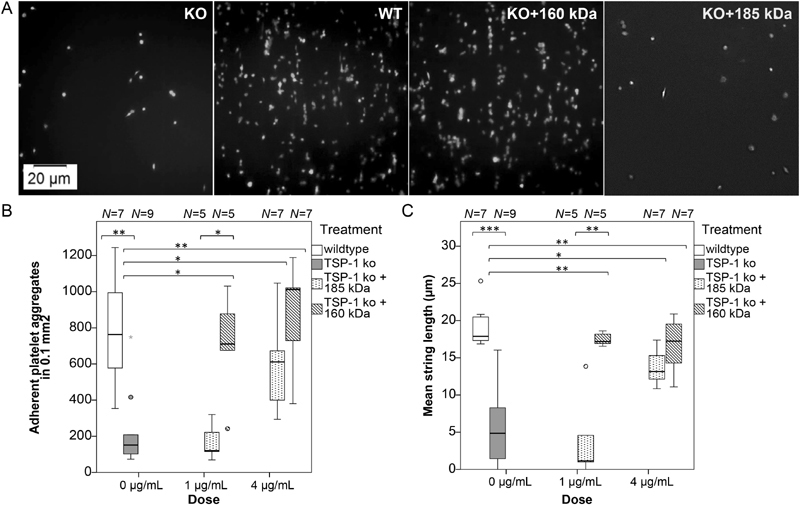
Comparison of thrombospondin-1 (TSP-1) isoforms in promoting platelet adhesion and string formation on collagen under flow. Blood retrieved from wild-type (WT) or TSP-1 knockout (KO) mice was supplied with anti-GPIbβ antibody to fluorescently label platelets and was perfused over collagen-coated slides in an ibidi flow chamber for 7 minutes at 7 dyne/cm 2 . TSP-1 KO blood was either left untreated or was substituted with 1 or 4 µg/mL of purified 160 or 185 kDa TSP-1 protein. ( A ) Representative microscopic images of the different treatment groups were taken after 7 minutes (under continuing flow). ( B ) The number of adherent platelet aggregates and ( C ) the mean length of formed platelet strings were analysed with Fiji software as outlined in Supplementary Fig. S7 ( available in the online version). Boxplots illustrate the data distribution of 5 to 9 independent experiments (representing blood from individual mice); statistically significant differences between groups were assessed by Mann–Whitney U test (SPSS 23.0).
Supplementary Videos
Comparison of thrombospondin-1 (TSP-1) isoforms in promoting platelet adhesion and string formation on collagen under flow. Blood retrieved from wild-type ( A ) or TSP-1 knockout (KO) mice was supplied with anti-GPIbβ antibody to fluorescently label platelets and was perfused over collagen-coated slides in an ibidi flow chamber for 7 minutes at 7 dyne/cm 2 . TSP-1 KO blood was either ( B ) left untreated or was substituted with 1 µg/mL of purified ( C ) 160 kDa or ( D ) 185 kDa TSP-1 protein. Time series were recorded with an Olympus IX83 inverted microscope. Images were captured every 10 seconds over 5 minutes in total, using a 40× objective and the Hamamatsu Orca Flash 4 sCMOS camera. Online content including video sequences viewable at: https://www.thieme-connect.de/products/ejournals/html/10.1055/s-0038-1675229
Supplementary Video B
Supplementary Video C
Supplementary Video D
Discussion
The extracellular matrix protein TSP-1 is susceptible to processing by various proteases generating fragments of different length and function. For example, the matrix metalloproteinase ADAMTS-1 was found to cleave TSP-1 between E293 and L294 (amino acid numbering without signal peptide) releasing a trimeric N-terminus of 36 kDa and monomeric C-terminal fragments of 110 to 125 kDa with enhanced anti-angiogenic properties. 16 Bonnefoy and Legrand reported in 2000 that TSP-1 secreted by human umbilical vein ECs is processed upon the addition of plasmin, cathepsin G and elastase to fragments of comparable size. 21 In this study, we were able to extend the observations by Bonnefoy and Legrand identifying cathepsin G and elastase as being responsible for TSP-1 processing from a full-length 185 kDa protein to 160 kDa fragments in a co-culture setting of ECs or platelets with neutrophils. Moreover, processing was blocked by inhibitors for the neutrophil proteases. The processed 160 kDa TSP-1 fragments were found to be trimeric, lacking the 25-kDa N-terminal HBD, and matched in size with TSP-1 molecules found in post-operative human plasma. The exact cut site of cathepsin G was identified to reside between R237 and T238.
Of interest, TSP-1 has previously been shown to act as a competitive inhibitor of plasmin, elastase and cathepsin G, blocking their proteolytic activity upon their binding to the TSP-1 type 3 domain. 31 While these investigations elucidated the effects of TSP-1 on protease activity, our study focused on the impact of proteolytic processing on TSP-1 function, in particular in haemostasis. TSP-1 is known to contribute to platelet aggregation by forming a bridge between platelet-bound fibrinogen and integrin and by protecting vWF from cleavage by ADAMTS-13, thus promoting thrombus formation. 17 19 More recently, Kuijpers et al showed that TSP-1 binding to CD36 supports platelet adhesion and thrombus stability on collagen. 32 While these studies did not evaluate TSP-1 fragmentation during haemostasis, earlier investigations by Rabhi-Sabile et al reported that smaller (165 kDa) TSP-1 molecules lacking the N-terminus remained platelet-bound after cathepsin G induced platelet aggregation. 20 Based on the application of blocking antibodies, they concluded that these TSP-1 fragments retained function in platelet aggregation, but they did not address or reveal a functional gain of the shorter isoform. We have now compared the full-length TSP-1 molecule and the processed 160 kDa protein for their potency to promote platelet adhesion and string formation. The experimental design was generally based on whole blood or purified platelets from WT versus TSP-1 KO mice, to be reconstituted with purified TSP-1 isoforms for functional comparison. However, in commonly applied assays such as rotational thromboelastometry, a cone and plate analyser (Impact-R) or light transmission aggregometry (PAP8, moeLab GmbH, Supplementary Fig. S5 , available in the online version), platelet aggregation did not differ significantly between blood samples from WT versus TSP-1 KO mice and hence could not be rescued by the addition of purified TSP-1 isoforms. These data are in accordance with Lawler et al who showed that platelets of TSP-1 KO mice had normal aggregation. 33 However, Isenberg et al reported in 2008 that TSP-1 acts as an antagonist of NO signalling to promote platelet aggregation which necessitates the addition of NO donors during in vitro investigations. 34 Thus, we added the NO donor diethylamine NONOate in light transmission aggregometry experiments and could confirm the delaying effect of NO on the thrombin-induced aggregation of WT as well as TSP-1 KO platelets ( Supplementary Fig. S9 , available in the online version). Of note, there was no difference between WT and KO, and the addition of purified TSP-1 had no impact.
We then switched from static to flow conditions and evaluated adhesion, aggregation and string formation of platelets in an in vitro flow chamber. In this setting, platelets from TSP-1 KO mice showed severely impaired function pointing to a predominant role of TSP-1 in promoting platelet adhesion on collagen and protecting vWF strings under flow. Moreover, we could restore these functions by the addition of purified TSP-1 to blood samples of TSP-1 KO mice. Importantly, the 160-kDa fragment was significantly more potent than the full-length 185 kDa protein at limiting TSP-1 concentrations of 1 µg/mL, whereas both molecules restored platelet adhesion and string formation at higher protein levels (4 µg/mL). While constitutive TSP-1 plasma levels are comparably low and range at 20 to 40 ng/mL, the local concentration may be expected to increase substantially after platelet activation, as indicated by TSP-1 serum values of 1 to 10 µg/mL after complete in vitro platelet activation. 7 Thus, neutrophil-mediated TSP-1 proteolysis may serve to enhance the haemostatic properties of platelet- and EC-released TSP-1 under early or limiting conditions during thrombus formation.
Since both the number of adherent platelet aggregates on collagen and the average length of formed platelet strings differed significantly between blood samples reconstituted by 160 versus 185 kDa TSP-1 protein, the processing of TSP-1 may affect CD36-mediated platelet binding to collagen as well as vWF protection from degradation. Of note, both functions are attributed to the type I repeats and might be facilitated by more accessible domain interactions after HBD removal. Alternatively, the loss of the N-terminal domain rather than the molecular change of the remaining core molecule may account for the functional difference between isoforms, since the TSP-1 HBD is known to provide a variety of interaction sites for platelet surface molecules. 35 Thus, TSP-1 may be more stable after removal of the N-terminus, which has previously been implicated in protein internalization and degradation of TSP-1 by binding to the low-density lipoprotein receptor-related protein. 36
In addition to fostering vWF strings, TSP-1 is known to have a propensity for self-polymerization and cross-linking to other proteins by thiol-disulphide exchange. 31 37 Since this might constitute another mechanism for enhanced platelet adhesion and string formation, we triggered TSP-1 polymerization by calcium depletion as previously described 38 and then compared TSP-1 multimers by SDS-agarose discontinuous gel electrophoresis ( Supplementary Fig. S10 , available in the online version). Interestingly, TSP-1 polymerization could be induced for the platelet-purified 185 kDa TSP-1 protein, but not for the cathepsin G-generated 160 kDa isoform. Comparably, recombinantly produced 160 or 185 kDa TSP-1 did not undergo polymerization upon EDTA treatment, eliminating this TSP-1 feature as the potential mechanism accounting for the enhanced haemostatic properties of the 160-kDa isoform.
We further extended our analysis to the role of NETs in TSP-1 proteolysis, since NET formation was discovered to affect haemostasis and play a particular role in thrombosis. 27 When we induced NET formation in co-cultures of platelets and neutrophils, TSP-1 was highly released and entirely processed to 160 kDa. In contrast, inhibition of NETosis resulted in rapid proteolysis and complete degradation of TSP-1 which is likely due to the excessive release of neutrophil-derived proteases, as it was blocked by inhibitors of cathepsin G and elastase. These data suggest that NET formation promotes processing of TSP-1 to 160 kDa in a controlled manner, protecting TSP-1 from further degradation by neutrophil proteases in the NET environment which might be of specific relevance in pathophysiological settings. Of note, NET-guided TSP-1 proteolysis also occurred in the presence of human plasma (data not shown) which is a central regulatory element of protease activity under physiological conditions.
In summary, the contact or concomitant activation of neutrophils and platelets/ECs results in the release of TSP-1 molecules which are processed by neutrophil proteases to a trimeric 160 kDa molecule lacking the N-terminal domain. NET formation supports the generation of the smaller TSP-1 isoform and protects the protein from further degradation. Importantly, the 160-kDa TSP-1 fragment shows enhanced potency to promote platelet adhesion on collagen and string formation under flow. This finding reveals a novel mechanism by which neutrophils may support thrombus formation at the site of vessel injury and provides first evidence for the impact of TSP-1 proteolysis on its haemostatic properties. Furthermore, neutrophil-mediated processing of TSP-1 might also have clinical implications, in particular with respect to thrombosis. A gene polymorphism (N700S) of TSP-1 has previously been identified to be associated with an increased risk for myocardial infarction 39 and the S700 TSP-1 variant was found to be more susceptible to in vitro digest by the protease trypsin and to exhibit enhanced platelet aggregation properties when compared with the N700 protein. 40 Of interest, the proposed binding site of TSP-1 for cathepsin G and elastase 41 is in proximity to TSP-1 amino acid 700 which led us to compare the N700 and S700 TSP-1 variants for their susceptibility to proteolytic cleavage by neutrophil proteases and the functional impact on platelet string formation. The recombinant, full-length 185 kDa TSP-1 molecule engineered to carry serine in position 700 was markedly more susceptible to cathepsin G digest and also more potent than the N700 variant in promoting platelet adhesion and string formation in whole blood, comparable to the processed 160 kDa isoform ( Supplementary Fig. S8 , available in the online version). This may indicate that excessive neutrophil-mediated proteolysis of TSP-1 might favour pathological processes such as thrombosis and warrants further investigation.
Limitations of the Study
While the applied cathepsin G inhibitor (Merck #219372) is highly selective and only weakly inhibits other tested proteases such as plasmin, elastase or proteinase 3 (as reflected in 1,000-fold higher IC 50 values), the elastase inhibitor (Merck #324744) has a broader spectrum of affected enzymes. It has poor reactivity with cathepsin G but may inhibit neutrophil proteinase 3. 42 43 Hence, we cannot entirely exclude an additional contribution by proteinase 3 to TSP-1 processing, but would like to emphasize that purified cathepsin G or elastase were sufficient to generate the 160-kDa isoform.
Regarding measurement of platelet adhesion and string formation under flow, we gave preference to heparin over citrate or EDTA for anticoagulation of mouse blood, because the structure and function of TSP-1 are known to be dependent on calcium. 44 Of note, the 185- and 160-kDa TSP-1 isoforms exhibited comparable affinity for heparin in vitro (data not shown) which may possibly relate to complex formation between TSP-1, cathepsin G and heparin or may be conferred by the heparin binding sites within the type I repeats 45 which are retained in both the 160- and 185-kDa TSP-1 isoform and constitute the TSP-1 region mediating platelet adhesion to collagen as well as vWF protection. However, we cannot exclude the possibility that the presence of heparin in mouse blood selectively limits the function of full-length 185 kDa TSP-1 in platelet adhesion and string formation by binding to the N-terminal domain (not present in the shorter 160 kDa isoform).
Acknowledgements
We would like to thank Anna Zommer for her support in isolating ECs, Luca Martelanz and Marie-Therese Grasl for their help in retrieving blood samples from mice and Marion Mussbacher for her assistance in platelet aggregation experiments.
Funding Statement
Funding This work was supported by the Austrian Science Fund (project SFB-54 P09).
Footnotes
Conflict of Interest None.
What is known about this topic?
The full-length 185 kDa TSP-1 protein is released by activated platelets and ECs as a homotrimer.
It promotes platelet aggregation as well as platelet adhesion to collagen and protects von Willebrand factor strings from degradation.
What does this paper add?
TSP-1 is rapidly processed to a shorter 160 kDa isoform by neutrophil-derived proteases which is significantly more potent than the original 185 kDa TSP-1 protein in promoting platelet adhesion to collagen and formation of platelet strings.
This is the first study showing an impact of controlled TSP-1 proteolysis on its haemostatic properties and reveals a novel mechanism by which neutrophils promote primary haemostasis.
Supplementary Material
References
- 1.Carlson C B, Lawler J, Mosher D F. Structures of thrombospondins. Cell Mol Life Sci. 2008;65(05):672–686. doi: 10.1007/s00018-007-7484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klenotic P A, Page R C, Li W, Amick J, Misra S, Silverstein R L. Molecular basis of antiangiogenic thrombospondin-1 type 1 repeat domain interactions with CD36. Arterioscler Thromb Vasc Biol. 2013;33(07):1655–1662. doi: 10.1161/ATVBAHA.113.301523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao A G, Lindberg F P, Finn M B, Blystone S D, Brown E J, Frazier W A. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J Biol Chem. 1996;271(01):21–24. doi: 10.1074/jbc.271.1.21. [DOI] [PubMed] [Google Scholar]
- 4.Baenziger N L, Brodie G N, Majerus P W. Isolation and properties of a thrombin-sensitive protein of human platelets. J Biol Chem. 1972;247(09):2723–2731. [PubMed] [Google Scholar]
- 5.Mosher D F, Doyle M J, Jaffe E A. Synthesis and secretion of thrombospondin by cultured human endothelial cells. J Cell Biol. 1982;93(02):343–348. doi: 10.1083/jcb.93.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starlinger P, Moll H P, Assinger A et al. Thrombospondin-1: a unique marker to identify in vitro platelet activation when monitoring in vivo processes. J Thromb Haemost. 2010;8(08):1809–1819. doi: 10.1111/j.1538-7836.2010.03908.x. [DOI] [PubMed] [Google Scholar]
- 7.Starlinger P, Alidzanovic L, Schauer D et al. Platelet-stored angiogenesis factors: clinical monitoring is prone to artifacts. Dis Markers. 2011;31(02):55–65. doi: 10.3233/DMA-2011-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawler P R, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2(05):a006627. doi: 10.1101/cshperspect.a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staniszewska I, Zaveri S, Del Valle L et al. Interaction of alpha9beta1 integrin with thrombospondin-1 promotes angiogenesis. Circ Res. 2007;100(09):1308–1316. doi: 10.1161/01.RES.0000266662.98355.66. [DOI] [PubMed] [Google Scholar]
- 10.Jiménez B, Volpert O V, Crawford S E, Febbraio M, Silverstein R L, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6(01):41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 11.Iruela-Arispe M L, Lombardo M, Krutzsch H C, Lawler J, Roberts D D. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100(13):1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 12.Taraboletti G, Morbidelli L, Donnini S et al. The heparin binding 25 kDa fragment of thrombospondin-1 promotes angiogenesis and modulates gelatinase and TIMP-2 production in endothelial cells. FASEB J. 2000;14(12):1674–1676. doi: 10.1096/fj.99-0931fje. [DOI] [PubMed] [Google Scholar]
- 13.Isenberg J S, Ridnour L A, Dimitry J, Frazier W A, Wink D A, Roberts D D. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. 2006;281(36):26069–26080. doi: 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 14.Isenberg J S, Jia Y, Fukuyama J, Switzer C H, Wink D A, Roberts D D. Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J Biol Chem. 2007;282(21):15404–15415. doi: 10.1074/jbc.M701638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iruela-Arispe M L. Regulation of thrombospondin1 by extracellular proteases. Curr Drug Targets. 2008;9(10):863–868. doi: 10.2174/138945008785909365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee N V, Sato M, Annis D S et al. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25(22):5270–5283. doi: 10.1038/sj.emboj.7601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonnefoy A, Hantgan R, Legrand C, Frojmovic M M. A model of platelet aggregation involving multiple interactions of thrombospondin-1, fibrinogen, and GPIIbIIIa receptor. J Biol Chem. 2001;276(08):5605–5612. doi: 10.1074/jbc.M010091200. [DOI] [PubMed] [Google Scholar]
- 18.Dorahy D J, Thorne R F, Fecondo J V, Burns G F. Stimulation of platelet activation and aggregation by a carboxyl-terminal peptide from thrombospondin binding to the integrin-associated protein receptor. J Biol Chem. 1997;272(02):1323–1330. doi: 10.1074/jbc.272.2.1323. [DOI] [PubMed] [Google Scholar]
- 19.Bonnefoy A, Daenens K, Feys H B et al. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. 2006;107(03):955–964. doi: 10.1182/blood-2004-12-4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabhi-Sabile S, Pidard D, Lawler J, Renesto P, Chignard M, Legrand C. Proteolysis of thrombospondin during cathepsin-G-induced platelet aggregation: functional role of the 165-kDa carboxy-terminal fragment. FEBS Lett. 1996;386(01):82–86. doi: 10.1016/0014-5793(96)00408-5. [DOI] [PubMed] [Google Scholar]
- 21.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000;98(04):323–332. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 22.Francis C W, Marder V J. Degradation of cross-linked fibrin by human leukocyte proteases. J Lab Clin Med. 1986;107(04):342–352. [PubMed] [Google Scholar]
- 23.Maugeri N, Campana L, Gavina M et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074–2088. doi: 10.1111/jth.12710. [DOI] [PubMed] [Google Scholar]
- 24.Semeraro F, Ammollo C T, Morrissey J H et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(07):1952–1961. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:38. doi: 10.3389/fimmu.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brinkmann V, Reichard U, Goosmann Cet al. Neutrophil extracellular traps kill bacteria Science 2004303(5663):1532–1535. [DOI] [PubMed] [Google Scholar]
- 27.Fuchs T A, Brill A, Duerschmied D et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfeiler S, Stark K, Massberg S, Engelmann B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica. 2017;102(02):206–213. doi: 10.3324/haematol.2016.142471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchberger E, El Harchi M, Payrhuber D et al. Overexpression of the transcriptional repressor complex BCL-6/BCoR leads to nuclear aggregates distinct from classical aggresomes. PLoS One. 2013;8(10):e76845. doi: 10.1371/journal.pone.0076845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thålin C, Daleskog M, Göransson S P et al. Validation of an enzyme-linked immunosorbent assay for the quantification of citrullinated histone H3 as a marker for neutrophil extracellular traps in human plasma. Immunol Res. 2017;65(03):706–712. doi: 10.1007/s12026-017-8905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogg P J. Thrombospondin 1 as an enzyme inhibitor. Thromb Haemost. 1994;72(06):787–792. [PubMed] [Google Scholar]
- 32.Kuijpers M J, de Witt S, Nergiz-Unal R et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler Thromb Vasc Biol. 2014;34(06):1187–1192. doi: 10.1161/ATVBAHA.113.302917. [DOI] [PubMed] [Google Scholar]
- 33.Lawler J, Sunday M, Thibert V et al. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest. 1998;101(05):982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isenberg J S, Romeo M J, Yu C et al. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood. 2008;111(02):613–623. doi: 10.1182/blood-2007-06-098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elzie C A, Murphy-Ullrich J E. The N-terminus of thrombospondin: the domain stands apart. Int J Biochem Cell Biol. 2004;36(06):1090–1101. doi: 10.1016/j.biocel.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Mikhailenko I, Krylov D, Argraves K M, Roberts D D, Liau G, Strickland D K. Cellular internalization and degradation of thrombospondin-1 is mediated by the amino-terminal heparin binding domain (HBD). High affinity interaction of dimeric HBD with the low density lipoprotein receptor-related protein. J Biol Chem. 1997;272(10):6784–6791. doi: 10.1074/jbc.272.10.6784. [DOI] [PubMed] [Google Scholar]
- 37.Detwiler T C, Turk J L, Browne P C. Thiol-disulfide exchange by thrombospondin. Semin Thromb Hemost. 1987;13(03):276–280. doi: 10.1055/s-2007-1003502. [DOI] [PubMed] [Google Scholar]
- 38.Turk J L, Detwiler T C. Thiol-disulfide exchange by thrombospondin: evidence for a thiol and a disulfide bond protected by calcium. Arch Biochem Biophys. 1986;245(02):446–454. doi: 10.1016/0003-9861(86)90236-5. [DOI] [PubMed] [Google Scholar]
- 39.Topol E J, McCarthy J, Gabriel S et al. Single nucleotide polymorphisms in multiple novel thrombospondin genes may be associated with familial premature myocardial infarction. Circulation. 2001;104(22):2641–2644. doi: 10.1161/hc4701.100910. [DOI] [PubMed] [Google Scholar]
- 40.Narizhneva N V, Byers-Ward V J, Quinn M J et al. Molecular and functional differences induced in thrombospondin-1 by the single nucleotide polymorphism associated with the risk of premature, familial myocardial infarction. J Biol Chem. 2004;279(20):21651–21657. doi: 10.1074/jbc.M311090200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hogg P J, Jiménez B M, Chesterman C N. Identification of possible inhibitory reactive centers in thrombospondin 1 that may bind cathepsin G and neutrophil elastase. Biochemistry. 1994;33(21):6531–6537. doi: 10.1021/bi00187a021. [DOI] [PubMed] [Google Scholar]
- 42.Attucci S, Korkmaz B, Juliano Let al. Measurement of free and membrane-bound cathepsin G in human neutrophils using new sensitive fluorogenic substrates Biochem J 2002366(Pt 3):965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korkmaz B, Attucci S, Hazouard E et al. Discriminating between the activities of human neutrophil elastase and proteinase 3 using serpin-derived fluorogenic substrates. J Biol Chem. 2002;277(42):39074–39081. doi: 10.1074/jbc.M202918200. [DOI] [PubMed] [Google Scholar]
- 44.Lawler J, Chao F C, Cohen C M. Evidence for calcium-sensitive structure in platelet thrombospondin. Isolation and partial characterization of thrombospondin in the presence of calcium. J Biol Chem. 1982;257(20):12257–12265. [PubMed] [Google Scholar]
- 45.Guo N H, Krutzsch H C, Nègre E, Zabrenetzky V S, Roberts D D. Heparin-binding peptides from the type I repeats of thrombospondin. Structural requirements for heparin binding and promotion of melanoma cell adhesion and chemotaxis. J Biol Chem. 1992;267(27):19349–19355. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.