

Abstract
Severe thrombocytopenia, characterized by dysplastic megakaryocytes and intracranial bleeding, was diagnosed in six individuals from a consanguineous kindred. Three of the individuals were successfully treated by bone marrow transplant. Whole-exome sequencing and homozygosity mapping of multiple family members, coupled with whole-genome sequencing to reveal shared non-coding variants, revealed one potentially functional variant segregating with thrombocytopenia under a recessive model: GALE p.R51W (c.C151T, NM_001127621). The mutation is extremely rare (allele frequency = 2.5 × 10−05), and the likelihood of the observed co-segregation occurring by chance is 1.2 × 10−06. GALE encodes UDP-galactose-4-epimerase, an enzyme of galactose metabolism and glycosylation responsible for two reversible reactions: interconversion of UDP-galactose with UDP-glucose and interconversion of UDP-N-acetylgalactosamine with UDP-N-acetylglucosamine. The mutation alters an amino acid residue that is conserved from yeast to humans. The variant protein has both significantly lower enzymatic activity for both interconversion reactions and highly significant thermal instability. Proper glycosylation is critical to normal hematopoiesis, in particular to megakaryocyte and platelet development, as reflected in the presence of thrombocytopenia in the context of congenital disorders of glycosylation. Mutations in GALE have not previously been associated with thrombocytopenia. Our results suggest that GALE p.R51W is inadequate for normal glycosylation and thereby may impair megakaryocyte and platelet development. If other mutations in GALE are shown to have similar consequences, this gene may be proven to play a critical role in hematopoiesis.
Introduction
Inherited thrombocytopenia is a rare hematological condition with a wide range of clinical presentation. Patients can present with or without congenital anomalies and with small, normal or large platelets. Inherited thrombocytopenia has been historically classified by clinical phenotype (absence/presence of megakaryocytes; platelet size) and more recently by underlying genetic lesion. Genes that harbor mutations responsible for inherited thrombocytopenia (1–3) function in megakaryopoiesis (ANKRD26, ETV6, FLI1, FYB, GATA1, GFI1B, HOXA11, MPL, NBEAL2, RBM8A, RUNX1, THPO); platelet production (ACTN1, CYCS, GP1BA, GP1BB, GP9, ITGA2B, ITGB3, MKL1, MYH9, PRKACG, TUBB1, WAS); or platelet clearance (ABCG6, ABCG8, ADAMTS13, GNE, SLFN14, STIM1, vWF). Patients with some forms of congenital disorders of glycosylation (CDGs) also present with thrombocytopenia (4–7).
From two generations of an extended, consanguineous family, six individuals presented at young ages with severe thrombocytopenia, characterized by dysplastic. We investigated the genetic cause of the condition in the family and identified a homozygous mutation in a gene important for galactose metabolism and glycosylation, but not previously associated with thrombocytopenia.
Results
Clinical features
Six individuals from two generations of kindred AH presented with severe thrombocytopenia, and in some individuals, mild anemia and febrile neutropenia. Multiple marriages in the kindred were consanguineous (Fig. 1A). Proband IV.1 presented at age 13 months with thrombocytopenia (platelets 44 × 109/L) and neutropenia (absolute neutrophil counts, 1.00 × 109/L). Her other blood tests and physical exam were normal. Peripheral blood histology showed big and pale platelets. Bone marrow aspiration revealed normal cellularity but markedly dysplastic with hypolobated nuclei and hypogranularity (Fig. 1B). The proband had two affected aunts (III.1 and III.3) and three affected cousins (III.11, III.16, III.18). All affected relatives presented as children with increased bleeding tendency in the context of thrombocytopenia (Supplementary Material, Table S1), leading in three patients to intracranial hemorrhages. These three patients underwent successful bone marrow transplant. The clinical features of the family were originally described in 2003 (8), before the birth of the proband. At that time, four genes associated with congenital thrombocytopenia [NFE2, FLI1, FOG1 and GFI1B1 (9–13)] were sequenced, but all sequences were normal.
Figure 1.
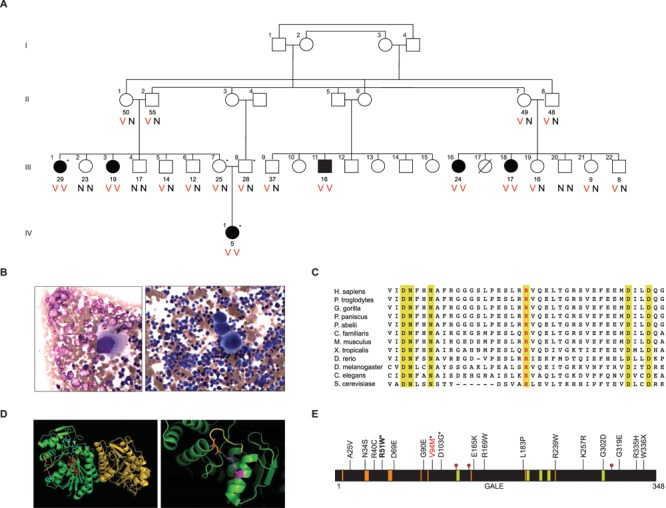
Inherited thrombocytopenia in family AH; GALE conservation, structure and mutations. (A) Six individuals in four-generation family AH presented with severe thrombocytopenia (black symbols). Current ages in years of individuals are listed below symbols. V indicates variant allele GALE p.R51W (c.C151T); N indicates reference allele. Asterisks indicate individuals evaluated by whole-exome sequencing. (B) Bone marrow aspirate treated with Wright-Giemsa stain from affected family member IV.1. Arrow indicates a megakaryocyte with hypogranular cytoplasm and hypolobated nuclei. (C) Protein sequence alignment from multiple species of the GALE region including Arg51 (red text) showing evolutionary conservation. Gold indicates complete conservation through S. cerevisiae. (D) GALE crystal structure (PDB ID:1HZJ), viewed in PyMOL (72). Left: GALE homodimer (green and gold), highlighting Arg51 (magenta), Asn340 (orange), UDP-N-acetylgalactosamine (blue), NAD+ cofactor (red), C-terminal end of GALE (yellow). Right: close-up view of Arg51 near the C-terminal end of GALE. Dotted yellow line indicates the 3-Å distance between Arg51 and Asn340. (E) Galactosemia-associated mutations of GALE reported in the literature. GALE domains are the substrate-binding site (gold), NAD+-binding site (orange) and catalytic sites (red dots at residues 132, 157, 307). Asterisks indicate mutations reported as homozygotes; others were reported as compound heterozygotes. GALE p.V94M (red text) is associated with severe generalized galactosemia. GALE p.R51W is shown in bold.
Gene discovery
To identify the genetic basis of thrombocytopenia in family AH, whole-exome sequencing was performed on the affected proband IV.1, her unaffected parents III.7 and III.8 and affected aunt III.1. All available family members were then genotyped for all candidate variants. The only potentially damaging coding sequence mutation consistent with recessive inheritance of the phenotype was GALE p.R51W (c.C151T, NM_001127621) at chr1:24,125,191 (hg19) (Fig. 1A). The mutation was homozygous in all affected family members, heterozygous in the proband’s parents and either heterozygous or absent from healthy relatives. GALE p.R51W has been identified in three heterozygotes in the ExAC database and is absent from dbSNP150, the 1000 Genomes Project and our in-house database of 2800 exomes, including many individuals from the Middle East. All in silico analyses predicted GALE p.R51W to be damaging: PolyPhen score 1.00, SIFT P = 0.00 and PROVEAN score −7.78. Arg51 is completely conserved through yeast (Saccharomyces cerevisiae) (Fig. 1C). However, structurally, Arg51 does not appear to interfere with binding pockets of the substrate or with the NAD+ cofactor (Fig. 1D).
In order to identify non-coding variants that co-segregated with the illness, we carried out homozygosity mapping with the 6 affected individuals and 14 of their unaffected relatives (Supplementary Material, Figs S1 and S2). The only genomic region homozygous in all affected relatives and not homozygous in any unaffected relatives was chr1:23,907,702-27,666,378 (hg19), a region of 3.76MB that includes GALE. The 3.76MB shared homozygous region is very gene-rich, including three genes implicated in hematopoiesis: RPL11, RUNX3 and LIN28A (14–17). Mutations in RPL11 are associated with Diamond–Blackfan anemia, an inherited bone marrow failure syndrome resulting in anemia and physical abnormalities (15,18–20). Partial loss of function of RUNX3 in human hematopoietic progenitor cells leads to defects in megakaryopoiesis and erythropoiesis (21). RUNX3 is also very closely related by sequence to RUNX1, mutations in which lead to a familial platelet disorder with leukemia predisposition. Double deletion of RUNX1 and RUNX3 in mice leads to a bone marrow failure phenotype (17). LIN28A is expressed in stem cells early in development and confers pluripotency. Partial loss of expression of LIN28A leads to skewing of hematopoietic lineages, with increased numbers of myeloid cells and fewer B-cells (14).
In order to evaluate whether a non-coding genomic variant in the homozygous region might alter expression of these genes, we integrated RNA analysis and whole-genome sequence of informative family members and unrelated controls. RNA-Seq was undertaken using as template RNA isolated from lymphoblasts of the proband, her parents and controls, with cells curated in the same way for all samples. Neither expression of RPL11 nor expression of RUNX3 differed between the proband and her parents, or between the proband and unrelated controls, or between the parents and unrelated controls (Supplementary Material, Table S2). No expression of LIN38A could be detected in lymphoblast-derived RNA from any of the samples, not surprising given that LIN38A is expressed specifically during embryonic development. Whole-genome sequencing revealed 79 variants with allele frequency < 0.005 in the shared homozygous region. Sixteen of these 79 variants either altered a moderately conserved site (GERP > 2.0) or were in proximity to a gene implicated in hematopoiesis, regardless of conservation at the site (Supplementary Material, Table S3). The only rare variant within 100kb of RPL11 was rs18987607 in RPL11 intron 2, with a GERP score of −1.3 and the reference allele G and variant allele A about equally frequent in mammalian species. The most promising variant at RUNX3 was rs565347299, upstream of the transcription start site, with a GERP score of +4.3 and embedded in predicted transcription factor binding sites for CUX1 and MEF2A (Supplementary Material, Fig. S3) (22). However, the absence of any difference in RUNX3 expression between the proband and her parents or controls suggested that homozygosity for variant alleles in this region did not alter RUNX3 expression. The only rare variant near LIN28A was rs536013503, with GERP score −2.4 and the variant allele T acting as the reference site in most other mammalian species. While these approaches cannot formally exclude the possibility that a non-coding variant in this region is responsible for the phenotype of family AH, these observations were sufficiently negative to encourage us to carry out further studies of GALE p.R51W.
Impaired enzyme activities of GALE p.R51W
UDP-galactose-4-epimerase, the enzyme encoded by GALE, is critical to galactose metabolism and to the production of glycosylation substrates. The enzyme catalyzes two reversible reactions: conversion between UDP-galactose (UDP-gal) and UDP-glucose (UDP-glc) and conversion between UDP-N-acetylgalactosamine (UDP-galNAc) and UDP-N-acetylglucosamine (UDP-glcNAc). Defective conversion of UDP-galactose to UDP-glucose leads to generalized features of galactosemia, including vomiting, hypotonia, seizures, jaundice, galactosuria and hepatomegaly (23,24). Less severe defects in GALE enzyme activity lead to milder clinical signs of galactosemia (25).
To understand the relationship of GALE genotype to clinical phenotype, we compared GALE p.R51W to two previously reported GALE mutations: p.V94M and p.D103G (Fig. 1E) (23,26). Homozygosity for GALE p.V94M causes the most severe symptoms of galactosemia, attributed to nearly absent enzyme activity for the conversion of UDP-galactose to UDP-glucose (27). In contrast, homozygosity for GALE p.D103G does not lead to any known disease. To compare the activities of these enzymes, we analyzed recombinant human GALE proteins purified from Escherichia coli, and their substrate and product sugars, by ultraperformance liquid chromatography (UPLC) mass spectrometry (Fig. 2A and B). Compared to wild-type protein, GALE p.R51W has lower activity both for conversion of UDP-galactose to UDP-glucose, with 40% activity relative to normal, and for conversion of UDP-N-acetylgalactosamine to UDP-N-acetylglucosamine, with 39% activity relative to normal (Fig. 2B, Supplementary Material, Table S4). GALE p.V94M and GALE p.D103G activities were consistent with previous reports. That is, GALE p.V94M activity was only 1% of normal for conversion of UDP-galactose to UDP-glucose and 5% of normal for conversion of UDP-N-acetylgalactosamine to UDP-N-acetylglucosamine. GALE p.D103G activity was 51% of normal for conversion of UDP-galactose to UDP-glucose, slightly higher than this activity for GALE p.R51W. GALE p.D103G activity was 48% of normal for conversion of UDP-N-acetylgalactosamine to UDP-N-acetylglucosamine, significantly higher than this activity for GALE p.R51W (P = 0.01).
Figure 2.
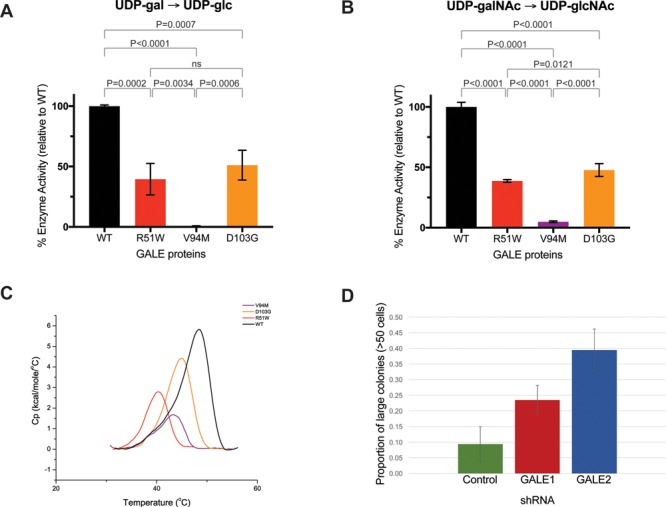
Enzyme activity and differential scanning calorimetry. Enzyme assays in figures (A) and (B) were performed with 10-ng protein. Substrates were (A) UDP-galactose or (B) UDP-N-acetylgalactosamine. Enzyme activities were measured as fraction product (Supplementary Material, Table S2) and were normalized to wildtype protein measurements. The mean values of normalized enzyme activities were plotted + standard deviation for each allele. Experiments were performed in at least triplicate (23,28,29). (C) Representative endotherm plots for each GALE allele. Endotherm data was normalized for protein concentration, with intrascan baseline correction. Black indicates wildtype protein (normal), red indicates GALE p.R51W, purple indicates GALE p.V94M and orange indicates GALE p.D103G. (D) Size distributions of megakaryocyte colonies after treatment of CD34+ hematopoietic progenitor cells with control or anti-GALE shRNA probes. Cells were plated in MegaCult collagen-based media. Slides were fixed, stained and classified by colony size. Data represents two independent experiments, each with plating and counting in duplicate.
NAD+ is a critical co-factor of GALE, and impaired binding to NAD+ is a possible mechanism for the effects of some GALE variants (28). We measured binding to NAD+ for wild-type GALE protein, and for GALE p.R51W, GALE p.V94M and GALE p.D103G (Supplementary Material, Fig. S4). Compared to wild-type GALE protein, NAD+ binding by GALE p.R51W was reduced by 41% (P = 0.004), intermediate between reduction of NAD+ binding by GALE p.V94M of 63% (P = 0.0001) and by GALE p.D103G of 33% (P = 0.60). NAD+ binding by GALE p.R51W appears to be somewhat compromised but not eliminated.
We next tested whether GALE p.R51W rescues yeast growth upon galactose challenge given knockout of gal10, the yeast gene homologous to GALE (23,29). In yeast, Gal10p catalyzes the interconversion of UDP-galactose and UDP-glucose, but not the interconversion of UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine. GALE p.R51W does rescue gal10-/- yeast upon galactose challenge (Supplementary Material, Fig. S5). Given this result, it is not surprising that homozygotes for GALE p.R51W do not have clinical signs of galactosemia.
Thermal instability of GALE p.R51W protein
Residue Arg51 of GALE is in close proximity to the C-terminus of the protein (Fig. 1D), raising the possibility that substitution of tryptophan for arginine at this site might destabilize the protein. To test this hypothesis, we measured the melting points (Tm) of normal and several variant GALE proteins using differential scanning calorimetry (DSC) (Fig. 2C, Table 1). The melting point of GALE p.R51W, 40.5°+0.16°C, is significantly lower than any other GALE protein that we measured, including GALE p.V94M, which is associated with severe galactosemia. The GALE p.R51W melting point is also lower than melting points of multiple other GALE variant proteins previously reported in the literature (23,30,31). The 8.4°C difference in melting points of GALE p.R51W versus normal GALE protein strongly suggests that GALE p.R51W is thermally unstable relative to other GALE proteins.
Table 1.
Melting temperatures of GALE proteins measured by scanning calorimetry
| WT | R51W | V94M | D103G | |
|---|---|---|---|---|
| Tm, mean (s.d.) | 48.93 (0.13) | 40.48 (0.32) | 44.78 (0.87) | 44.50 (0.57) |
| Tm difference from WT (°C) | 8.45 | 4.15 | 4.43 | |
| P-value for difference from WT | 4.8 × 10−9 | 7.8 × 10−5 | 5.0 × 10−6 |
Effects of reduced expression of GALE on CD34+ cell proliferation and megakaryocyte differentiation
Results of the thermal stability analysis suggest that GALE p.R51W is unstable and hence deficient in cells. To evaluate the consequence of GALE deficiency on hematopoiesis, we treated cord blood-derived CD34+ hematopoietic progenitor cells with shRNA probes designed to knock down function of GALE (Supplementary Material, Fig. S6). Knockdown of GALE slowed the proliferation of megakaryocyte precursors in liquid culture (Supplementary Material, Fig. S7; Table S5). Furthermore, megakaryocyte colonies from CD34+ cells treated with anti-GALE shRNA probes were significantly larger than colonies treated with control shRNA (Fig. 2D; Supplementary Material, Table S6). Colonies with abnormally large numbers of cells may reflect the presence of immature and possible defects in megakaryocyte differentiation (32,33). These results indicate that perturbation of GALE expression in vitro leads to cellular hematologic abnormalities.
Discussion
Epimerase deficiency galactosemia, due to deficiency of GALE, has a wide range of clinical severity (23,26,30,34–40), ranging from peripheral benign effects detectable only in blood cells to generalized disease with acute features including hypotonia, poor feeding, vomiting, jaundice, hepatomegaly/liver dysfunction, aminoaciduria and cataracts (34,41). The most severe effects are due to homozygosity for GALE p.V94M, which is defective in conversion of UDP-galactose and UDP-N-acetylgalactosamine to their glucose counterparts, leading to build-up of galactose metabolites and hence to severe galactosemia (14,17). The biochemical defects of GALE p.R51W are different. Enzyme activities of GALE p.R51W are intermediate between normal and GALE p.V94M for both interconversion reactions, but the GALE p.R51W enzyme is significantly less thermally stable than is GALE p.V94M. We speculate that these features of GALE p.R51W would not lead to build up of galactose, but would lead to less cellular enzyme.
The fundamental question is why patients homozygous for GALE p.R51W have severe thrombocytopenia, given that no hematopoietic defects have been reported for persons with other GALE mutations. That is, why would deficiency of GALE enzyme due to GALE p.R51W lead specifically to severe hematopoietic defects? Several lines of evidence are suggestive.
First, based on genetic criteria, homozygosity for GALE p.R51W is an excellent candidate for the cause of the phenotype of family AH. GALE p.R51W is the only coding sequence change that perfectly co-segregates with the phenotype in the family, and no non-coding variant that co-segregates with the phenotype has a convincing functional effect. GALE residue 51 is completely conserved as arginine in all species; the mutation is predicted to be highly damaging by in silico tools; and no homozygotes for GALE p.R51W appear in any database.
Second, defects in other genes regulating sugar metabolism and glycosylation are associated with bone marrow failure. Mutations in ALG8, encoding alpha-1,3-glucosyltransferase, cause CDG type 1K, features of which include anemia and thrombocytopenia (5,7). Mutations in SLC37A4, encoding a glucose-6-phosphate transporter, cause glycogen storage disease type 1B, which includes neutropenia (42). Mutations in G6PC3, encoding glucose-6-phosphatase, cause severe congenital neutropenia type 4 (43). Mutations in SLC35A1 (Fig. 3), encoding the CMP-sialic acid transporter, cause CDG type 2F, which can lead to thrombocytopenia and the presence of giant platelets (6).
Figure 3.
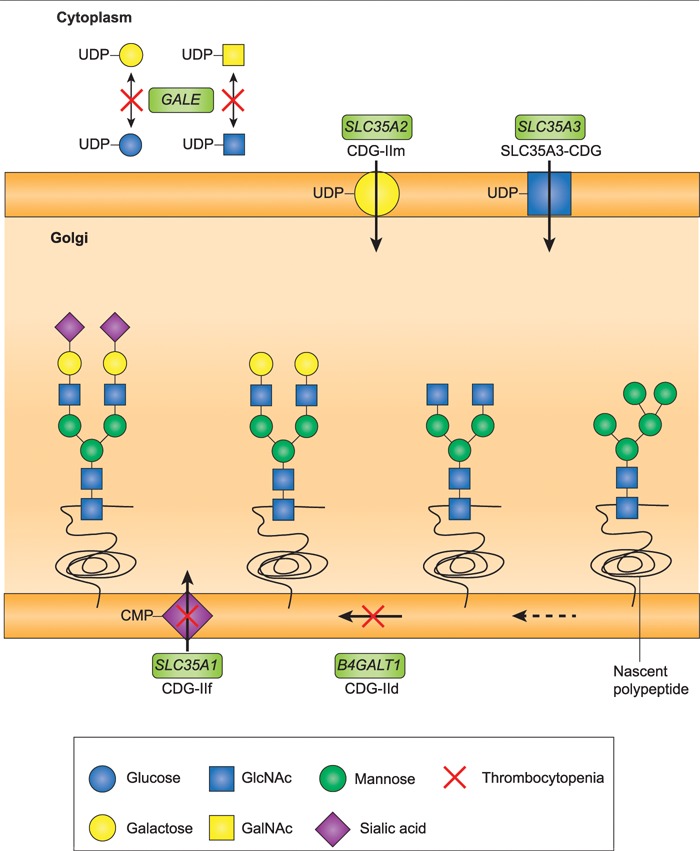
N-linked glycosylation pathway and thrombocytopenia. Mutations in critical steps of the N-linked glycan biosynthesis pathway are associated with thrombocytopenia. Glycan biosynthesis begins in the cytoplasm, with subsequent modifications in the endoplastic reticulum and the Golgi. Membrane transporters including SLC35A1, SLC35A2 and SLC35A3 allow building-block sugars to enter the Golgi. Enzymes in the pathway, including B4GALT1, control the levels of these sugars, and add or subtract sugars from the glycan chain. GALE is responsible for interconversion of these sugars in the cytoplasm, in preparation for their entry into the Golgi and integration into glycan chains. Mutations in this pathway cause CDGs. Two forms of CDG include thrombocytopenia among presenting symptoms: CDG-IId, caused by mutation of B4GALT1, and CDG-IIf, caused by mutation of SLC35A1. Genes responsible for steps in the pathway are indicated in green boxes, with the CDG disorder under the gene name. Steps in the pathway with mutations associated with thrombocytopenia are indicated by red Xs. Figure is adapted from Freeze 2006 (73).
Third, models of GALE deficiency in Caenorhabditis elegans, Drosophila melanogaster and Chinese hamster ovary (CHO) cells indicate that GALE is essential for normal development of multiple organ systems (44–46). In C. elegans, complete knockout of gale-1, the single homolog of human GALE, is embryonic lethal (46). Partial loss-of-function leads to abnormal levels of UDP-galactose and UDP-galactosamine. Mutant worms develop more slowly than wildtype, with misshapen gonads and activated unfolded protein response. In D. melanogaster, complete loss of dGALE, the homolog of human GALE, is embryonic lethal (45). Complementation with E. coli GALE (eGALE), which only interconverts UDP-galactose and UDP-glucose, along with Plesiomonas shigelloides GALE (wbgU), which only interconverts UDP-galactosamine and UDP-glucosamine, rescues GALE function and restores viability. However, single complementation with either eGALE or wbgU leads to reduced fecundity. In CHO cells, deficiency of GALE leads to impaired N-linked and O-linked glycosylation, and to synthesis of abnormal low-density-lipoprotein (LDL) receptors (44). These observations suggest that complete loss-of-function of GALE in humans is likely to be lethal, and the consequences of glycosylation defects may be broad and yet to be fully appreciated.
Fourth, a patient reported more than 20 years ago suggests that family AH may not be the sole representative of GALE deficiency and thrombocytopenia. A 4-year-old female patient of European–American ancestry was diagnosed with clinical features very similar to the affected individuals in family AH: severe thrombocytopenia with dysplastic, occasional anemia and febrile neutropenia (47). She also had mild peripheral galactosemia, detected by the absence of GALE enzyme activity from her red blood cells. Like the affected relatives of family AH, this patient did not present with clinical signs of galactosemia. Her GALE genotype was not determined and unfortunately she was lost to follow-up.
Finally, deficiency of glycosylation due to inadequate GALE may have a major effect on hematopoiesis. The effects of GALE knockdown on growth and differentiation of hematopoietic progenitor cells suggest that GALE is important for megakaryocyte development. Emerging evidence has also begun to shed light on the importance of glycosylation for homing of hematopoietic cells (48,49) and for platelet biogenesisx (50–52). In particular, glycosylation products with sialylated N-acetyllactosamine (LacNAc) help regulate platelet lifespan and synthesis of hepatic thrombopoietin. These products are found in abundance on β1-integrin, a membrane protein enriched on platelets that is important for homing and for interactions with the extracellular matrix (53–56). N-acetyllactosamine is a dimer of galactose and N-acetylglucosamine. Synthesis of N-acetyllactosamine is regulated by β1,4-galactosyltransferase 1 (β4GALT1). N-acetylglucosamine is a product of one of the conversion reactions catalyzed by GALE. In mice, deletion of β4GALT1 results in severe thrombocytopenia, with defective megakaryocyte localization to bone marrow (57), and ablation of both GALE and β4GALT1 leads to reduced engraftment of hematopoietic progenitor cells (58). These observations suggest that GALE may play an important role in glycosylation of molecules involved in cell adhesion and homing. Defects in GALE enzyme function may lead to errors in glycosylation of β1-integrin or another critical cell surface molecule and therefore to impairment of homing of megakaryocyte and platelet progenitors or to their normal interaction with the extracellular matrix.
In conclusion, we have identified GALE p.R51W, a homozygous mutation in an extended kindred including multiple individuals presenting with severe thrombocytopenia and intracranial bleeding. GALE p.R51W has reduced enzymatic activity and is thermally unstable. The unique features of this mutation may explain why the phenotype of family AH is severe thrombocytopenia and not acute galactosemia. Another family with mutation in GALE and a similar phenotype would support the link of GALE to hematopoiesis. Confirmation of GALE as a gene responsible for thrombocytopenia would further elucidate the role of glycosylation in platelet biogenesis and hematopoiesis.
Materials and Methods
Study subjects
The study was approved by the institutional review boards of the University of Washington, Seattle Children’s Hospital, and Rabin Medical Center. Informed consent was obtained from all study subjects.
Genomics
Genomic DNA was extracted from peripheral blood of patients. Whole exomes were captured using VCRome v2.1 (NimbleGen) and then sequenced on a HiSeq 250 (Illumina) with mean depth of coverage of targeted regions of 120×. More than 96.5% of targeted basepairs had >8 high-quality reads. Sequencing reads were aligned to hg19 with BWA. Variant calling was performed using the Genome Analysis Toolkit (59). Variant annotation and interpretation were performed as previously described (60–62). Candidate variants were validated with Sanger sequencing.
For homozygosity mapping, 1 million polymorphisms were genotyped used the HumanOmniExpress-24 v1.1 BeadChip (Illumina). Identity-by-descent analysis and homozygosity mapping were performed using PLINK (63).
Whole-genome sequencing of 1-μg DNA was carried out to an average depth of 32× on a Illumina HiSeq X instrument with 2 × 150 bp paired-end reads. Sequence reads were aligned using iSAAC aligner v.01.15.02.08 (64). Single-nucleotide variants and small indels were called using iSAAC variant caller v1.0.6 (64). Large genomic insertions and deletions were called using Control-FREEC v6.46 (65). Structural variants were evaluated by Manta Structural Variant Caller (66) in iSAAC-aligned BAM files and by Lumpy v0.2.11 (67) in BAM files aligned by BWA 0.7.12-r1039 (68).
For RNA sequencing, RNA was extracted from patient and control cells using TRIzol reagent (Thermo) as previously described (62). RNA-sequencing libraries were prepared in duplicate for each sample using the TruSeq stranded mRNA library kit (Illumina) and sequenced on a HiSeq2500 (Illumina). Sequencing reads were mapped using TopHat v2.1.1 (69). Gene expression values in fragments per kilobase exon per million mapped reads were calculated using Cufflinks v2.2.1 (69). Differentially expressed genes between samples were identified by Cuffdiff program of the Cufflinks suite.
Biochemistry
The human GALE cDNA sequence was PCR-amplified with flanking NdeI and XhoI restriction sites and cloned into the pET-31 expression vector (Novagen; kind gift of Hazel Holden, University of Wisconsin, Madison). Variant alleles p.R51W, p.V94M and p.D103G were created with the Q5 site-directed mutagenesis kit (New England Biolabs). Expression vectors were transformed into Rosetta2(DE3) E. coli (Millipore Sigma). Single clones were grown in LB media at 37°C until OD600 reached 0.6. Bacterial cultures were then induced with 500-μM IPTG and then grown at 18°C overnight. Bacterial cultures were then pelleted and lysed with xTractor Cell Lysis Buffer (Clontech). Recombinant hGALE proteins were then purified with column chromatography using nickel agarose columns (Clontech) and the following buffers: equilibration buffer 25-mm Tris, pH 8.0, 250-mm NaCl, 10% glycerol (TBSG), 20-mm imidazole; wash buffer TBSG, 40-mm imidazole; elution buffer TBSG, 300-mm imidazole. Elution fractions were dialyzed with TBSG to remove imidazole.
Enzyme assays were adapted from previous procedures (23,28,29). Unless otherwise specified, 10-ng enzyme was combined with either 0.8-mm UDP-gal or UDP-galNAc and 0.5-mm NAD+ in 40-mm glycine, pH 8.7. Reactions were incubated for 30 min at 37°C and quenched at a 1:1 ratio with acetonitrile (ACN). Quenched reactions were centrifuged at 3800 rpm for 15 min using the SH-3000 rotor (Sorvall), and supernatant was further diluted with ACN to achieve a 80:20 ACN:H2O ratio. To analyze the amounts of sugar substrate and product, 5-μL post-quenched sample was analyzed on the CORTECS UPLC HILIC column, 90 Å, 1.6 um, 2.1 mm × 150 mm (Waters) using the Acquity UPLC/Xevo TQ MS/MS system (Waters). Detailed parameters for liquid chromatography and mass spectrometry procedures are provided in Supplementary Materials.
For DSC, protein samples were freshly dialyzed overnight, then diluted to 10 μM prior to loading onto the Microcal VP-Capillary DSC system (Malvern). Twelve scans were performed per run, with starting temperature of 25°C and final temperature of 65°C, at a scan rate of 60°C/h. Calorimetry scans were analyzed using the Microcal VP-DSC software (Malvern). Scans were normalized to concentration and intrascan baseline prior to determining the melting points (Tm).
To measure NAD+ binding, wild-type and variant GALE proteins were diluted to 15 μM in TBSG, further diluted to 2.5 μM in 10-mm HEPES, pH 8.8, and 200-μl samples were aliquoted in triplicate in black plates. Samples were excited at 350 nm and 400–500 nm emission spectra were collected using a Biotek Synergy H1 plate reader with a gain setting of 100.
UPLC-tandem mass spectrometry was carried out with the Waters Acquity LC, Waters Xevo TQ MS/MS system. The UPLC column was CORTECS UPLC HILIC Column, 90 Å, 1.6 um, 2.1 mm × 100 mm (15-cm column) (Waters). Column temperature was 30°C. Chromatographic pump conditions and gradients are indicated in Supplementary Material, Table S7; MS tuning parameters are indicated in Supplementary Material, Table S8; and sugar retention times in Supplementary Material, Table S9. Peak areas were integrated with TargetLynx software (Waters).
Yeast growth assays
Yeast growth assays were performed as previously described (23,37,39). The yeast strain used was JFy3835 a gal10-, gal80- haploid strain derived from W303 (MATa ade2-1 his3-11,15 leu 2-3,112 ura3-1 trp1-1 can1-100 RAD5+), a kind gift of J. Fridovitch-Keil. Low copy number (CEN) plasmids (pMM33) containing GALE cDNA sequence were transformed into JFy3835 and grown with and without 0.05% galactose. pMM33-hGALE-R51W was created via Q5 site-directed mutagenesis (New England Biolabs) using pMM33-hGALE-WT (also donated by J. Fridovich-Keil). The experiment was performed in the Synergy H1 microplate reader (Biotek) over 48 h, with measurements taken every 30 min.
Hematopoiesis experiments
For lentivirus preparation, shRNA for GALE (TRCN0000049461) was purchased from Sigma in pLKO.1 lentiviral vector. pLKO-GFP-luciferase was prepared as previously described (70,71). Lentiviral particles were produced by transfecting HEK-293T cells with the lentiviral plasmids and third-generation packaging plasmids. Virus was harvested 48 h after transfection and concentrated with ultracentrifugation. Viruses were titered with serial dilution on HEK-293T cells.
For quantitative PCR, RNA was extracted using Trizol reagent (Thermo). Reverse transcription was performed using Superscript II (Thermo). Quantitative PCR was carried out in triplicate with TaqMan Gene Expression Assay reagents (Applied Biosystems). TaqMan probes for GALE (Hs00166181_m1) and control GAPDH were used. Assays were performed on the ABI 7900HT Real-Time PCR System. Knockdown efficiency is shown in Supplementary Material, Fig. S6.
CD34+ cells from human cord blood were purchased from AllCells. Thawed cells were expanded for 4 h at 37°C in tissue culture incubator with X-VIVO10 media (Lonza) supplemented with 1% BSA (Thermo), 2-mm L-glutamine (Thermo), penicillin/streptomycin (Thermo), SCF (100 ng/mL), FLT3 (100 ng/mL), TPO (50 ng/mL) and IL-6 (20 ng/mL). All cytokines were purchased from Peprotech. Cells were seeded on RetroNectin-coated (Takara; 10 μg/cm2) non-TC-treated 96-well plates at a density of 10–100 000 cells per well. The multiplicity of infection was 50 for all viruses. Viruses were concentrated onto cells via centrifugation at 2500 × rpm for 30 min. Viruses were removed with washing 24 h after infection. GFP+ cells were sorted on day 3 with the BD FACS Aria cytometer.
For collagen-based megakaryocyte colony formation assays, 50 000 cells were added to 1.2-mL MegaCult-C media containing cytokines (04901; StemCell Technologies). Cells in media were mixed with collagen and plated on to dual chamber slides and maintained in a humidified chamber for 10 days. Slides were dehydrated, fixed and stained according to manufacturer’s instructions (StemCell Technologies).
For liquid culture expansion of cells in megakaryocyte growth conditions, CD34+ cells were placed into the following media. Megakaryocyte stage 1: SFEM supplemented with penicillin/streptomycin (Thermo), LDL (20 μg/mL; StemCell Technologies), TPO (50 ng/mL), SCF (50 ng/mL), IL-6 (10 ng/mL) and IL-9 (15 ng/mL). Megakaryocyte stage 2: same as megakaryocyte Stage 1 except [SCF] is 1 ng/mL. All cytokines were purchased from Peprotech.
Supplementary Material
Acknowledgements
We thank family AH for participating in the study. We thank J. Fridovich-Keil, H. Holden, J. Thoden and P. Bradley for reagents and technical advice and E. Ahler, J. Fan, C.N. Kamischke, H.D. Kulasekara, M.K. Lee, K.R. Loeb, M. Sadilek, M.L. Sharon, J. Sumida, X. Yang and the University of Washington Molecular Analysis Facility for technical assistance. This work was supported by grants from the National Institutes of Health [R24DK099808 to A.Sh. and M.C.K.; R35CA197458 to M.C.K.; and F30DK103462 and T32HD007183 to A. Seo]; the ARCS Foundation [to A. Seo]; the Israel Cancer Association [to H.T.]; and the American Cancer Society [to M.C.K.].
Conflict of Interest statement. None declared.
References
- 1. Shimamura A. and Alter B.P. (2010) Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev., 24, 101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang M.Y., Keel S.B., Walsh T., Lee M.K., Gulsuner S., Watts A.C., Pritchard C.C., Salipante S.J., Jeng M.R., Hofmann I. et al. (2015) Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica, 100, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson B., Fletcher S.J. and Morgan N.V. (2016) Inherited thrombocytopenia: novel insights into megakaryocyte maturation, proplatelet formation and platelet lifespan. Platelets, 27, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Izumi R., Niihori T., Suzuki N., Sasahara Y., Rikiishi T., Nishiyama A., Nishiyama S., Endo K., Kato M., Warita H. et al. (2014) GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromuscul. Disord., 24, 1068–1072. [DOI] [PubMed] [Google Scholar]
- 5. Hock M., Wegleiter K., Ralser E., Kiechl-Kohlendorfer U., Scholl-Burgi S., Fauth C., Steichen E., Pichler K., Lefeber D.J., Matthjis G. et al. (2015) ALG8-CDG: novel patients and review of the literature. Orphanet J. Rare Dis., 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones C., Denecke J., Strater R., Stolting T., Schunicht Y., Zeuschner D., Klumperman J., Lefeber D.J., Spelten O., Zarbock A. et al. (2011) A novel type of macrothrombocytopenia associated with a defect in alpha2,3-sialylation. Am. J. Pathol., 179, 1969–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schollen E. (2004) Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency). J. Med. Genet., 41, 550–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tamary H., Yaniv I., Stein J., Dgany O., Shalev Z., Shechter T., Resnitzky P., Shaft D., Zoldan M., Kornreich L. et al. (2003) A clinical and molecular study of a Bedouin family with dysmegakaryopoiesis, mild anemia, and neutropenia cured by bone marrow transplantation. Eur. J. Haematol., 71, 196–203. [DOI] [PubMed] [Google Scholar]
- 9. Levin J., Peng J.P., Baker G.R., Villeval J.L., Lecine P., Burstein S.A. and Shivdasani R.A. (1999) Pathophysiology of thrombocytopenia and anemia in mice lacking transcription factor NF-E2. Blood, 94, 3037–3047. [PubMed] [Google Scholar]
- 10. Nichols K.E., Crispino J.D., Poncz M., White J.G., Orkin S.H., Maris J.M. and Weiss M.J. (2000) Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet., 24, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saleque S., Cameron S. and Orkin S.H. (2002) The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev., 16, 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shivdasani R.A. (2001) Molecular and transcriptional regulation of megakaryocyte differentiation. Stem Cells, 19, 397–407. [DOI] [PubMed] [Google Scholar]
- 13. Spyropoulos D.D., Pharr P.N., Lavenburg K.R., Jackers P., Papas T.S., Ogawa M. and Watson D.K. (2000) Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol. Cell. Biol., 20, 5643–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaudhuri A.A., So A.Y., Mehta A., Minisandram A., Sinha N., Jonsson V.D., Rao D.S., O'Connell R.M. and Baltimore D. (2012) Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc. Natl. Acad. Sci. U.S.A., 109, 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quarello P., Garelli E., Carando A., Brusco A., Calabrese R., Dufour C., Longoni D., Misuraca A., Vinti L., Aspesi A. et al. (2010) Diamond-Blackfan anemia: genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica, 95, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robledo S., Idol R.A., Crimmins D.L., Ladenson J.H., Mason P.J. and Bessler M. (2008) The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA, 14, 1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C.Q., Krishnan V., Tay L.S., Chin D.W., Koh C.P., Chooi J.Y., Nah G.S., Du L., Jacob B., Yamashita N. et al. (2014) Disruption of Runx1 and Runx3 leads to bone marrow failure and leukemia predisposition due to transcriptional and DNA repair defects. Cell Rep., 8, 767–782. [DOI] [PubMed] [Google Scholar]
- 18. Lipton J.M. and Ellis S.R. (2009) Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol. Oncol. Clin. North Am., 23, 261–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cmejla R., Cmejlova J., Handrkova H., Petrak J., Petrtylova K., Mihal V., Stary J., Cerna Z., Jabali Y. and Pospisilova D. (2009) Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum. Mutat., 30, 321–327. [DOI] [PubMed] [Google Scholar]
- 20. Zhang Z., Jia H., Zhang Q., Wan Y., Zhou Y., Jia Q., Zhang W., Yuan W., Cheng T., Zhu X. et al. (2013) Assessment of hematopoietic failure due to Rpl11 deficiency in a zebrafish model of Diamond-Blackfan anemia by deep sequencing. BMC Genomics, 14, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balogh P. and Goldfarb A. (2017) RUNX3 is required for megakaryocyte-erythroid specification in primary human progenitors. Exp. Hematol., 53, S130. [Google Scholar]
- 22. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., Lee R., Bessy A., Cheneby J., Kulkarni S.R., Tan G. et al. (2018) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res., 46, D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wohlers T.M., Christacos N.C., Harreman M.T. and Fridovich-Keil J.L. (1999) Identification and characterization of a mutation, in the human UDP-galactose-4-epimerase gene, associated with generalized epimerase-deficiency galactosemia. Am. J. Hum. Genet., 64, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thoden J.B., Wohlers T.M., Fridovich-Keil J.L. and Holden H.M. (2001) Molecular basis for severe epimerase deficiency galactosemia. X-ray structure of the human V94m-substituted UDP-galactose 4-epimerase. J. Biol. Chem., 276, 20617–20623. [DOI] [PubMed] [Google Scholar]
- 25. Timson D.J. (2016) The molecular basis of galactosemia—past, present and future. Gene, 589, 133–141. [DOI] [PubMed] [Google Scholar]
- 26. Maceratesi P., Daude N., Dallapiccola B., Novelli G., Allen R., Okano Y. and Reichardt J. (1998) Human UDP-galactose 4' epimerase (GALE) gene and identification of five missense mutations in patients with epimerase-deficiency galactosemia. Mol. Genet. Metab., 63, 26–30. [DOI] [PubMed] [Google Scholar]
- 27. Wohlers T.M. and Fridovich-Keil J.L. (2000) Studies of the V94M-substituted human UDPgalactose-4-epimerase enzyme associated with generalized epimerase-deficiency galactosaemia. J. Inherit. Metab. Dis., 23, 713–729. [DOI] [PubMed] [Google Scholar]
- 28. McCorvie T.J., Liu Y., Frazer A., Gleason T.J., Fridovich-Keil J.L. and Timson D.J. (2012) Altered cofactor binding affects stability and activity of human UDP-galactose 4'-epimerase: implications for type III galactosemia. Biochim. Biophys. Acta, 1822, 1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCorvie T.J., Wasilenko J., Liu Y., Fridovich-Keil J.L. and Timson D.J. (2011) In vivo and in vitro function of human UDP-galactose 4'-epimerase variants. Biochimie, 93, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Timson D.J. (2005) Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase. FEBS J., 272, 6170–6177. [DOI] [PubMed] [Google Scholar]
- 31. Pey A.L., Padin-Gonzalez E., Mesa-Torres N. and Timson D.J. (2014) The metastability of human UDP-galactose 4'-epimerase (GALE) is increased by variants associated with type III galactosemia but decreased by substrate and cofactor binding. Arch. Biochem. Biophys., 562, 103–114. [DOI] [PubMed] [Google Scholar]
- 32. Lefebvre P., Lin J., Linzer D.I. and Cohen I. (2001) Murine prolactin-like protein E synergizes with human thrombopoietin to stimulate expansion of human megakaryocytes and their precursors. Exp. Hematol., 29, 51–58. [DOI] [PubMed] [Google Scholar]
- 33. Yang H., Miller W.M. and Papoutsakis E.T. (2002) Higher pH promotes megakaryocytic maturation and apoptosis. Stem Cells, 20, 320–328. [DOI] [PubMed] [Google Scholar]
- 34. Alano A., Almashanu S., Chinsky J.M., Costeas P., Blitzer M.G., Wulfsberg E.A. and Cowan T.M. (1998) Molecular characterization of a unique patient with epimerase-deficiency galactosaemia. J. Inherit. Metab. Dis., 21, 341–350. [DOI] [PubMed] [Google Scholar]
- 35. Chhay J.S., Vargas C.A., McCorvie T.J., Fridovich-Keil J.L. and Timson D.J. (2008) Analysis of UDP-galactose 4'-epimerase mutations associated with the intermediate form of type III galactosaemia. J. Inherit. Metab. Dis., 31, 108–116. [DOI] [PubMed] [Google Scholar]
- 36. Park H.D., Park K.U., Kim J.Q., Shin C.H., Yang S.W., Lee D.H., Song Y.H. and Song J. (2005) The molecular basis of UDP-galactose-4-epimerase (GALE) deficiency galactosemia in Korean patients. Genet. Med., 7, 646–649. [DOI] [PubMed] [Google Scholar]
- 37. Quimby B.B., Alano A., Almashanu S., DeSandro A.M., Cowan T.M. and Fridovich-Keil J.L. (1997) Characterization of two mutations associated with epimerase-deficiency galactosemia, by use of a yeast expression system for human UDP-galactose-4-epimerase. Am. J. Hum. Genet., 61, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schulz J.M., Watson A.L., Sanders R., Ross K.L., Thoden J.B., Holden H.M. and Fridovich-Keil J.L. (2004) Determinants of function and substrate specificity in human UDP-galactose 4'-epimerase. J. Biol. Chem., 279, 32796–32803. [DOI] [PubMed] [Google Scholar]
- 39. Wasilenko J., Lucas M.E., Thoden J.B., Holden H.M. and Fridovich-Keil J.L. (2005) Functional characterization of the K257R and G319E-hGALE alleles found in patients with ostensibly peripheral epimerase deficiency galactosemia. Mol. Genet. Metab., 84, 32–38. [DOI] [PubMed] [Google Scholar]
- 40. Openo K.K., Schulz J.M., Vargas C.A., Orton C.S., Epstein M.P., Schnur R.E., Scaglia F., Berry G.T., Gottesman G.S., Ficicioglu C. et al. (2006) Epimerase-deficiency galactosemia is not a binary condition. Am. J. Hum. Genet., 78, 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fridovich-Keil J.L. (2006) Galactosemia: the good, the bad, and the unknown. J. Cell. Physiol., 209, 701–705. [DOI] [PubMed] [Google Scholar]
- 42. Klein C. (2009) Congenital neutropenia. Hematology Am. Soc. Hematol. Educ. Program, 344–350. [DOI] [PubMed] [Google Scholar]
- 43. Boztug K., Appaswamy G., Ashikov A., Schaffer A.A., Salzer U., Diestelhorst J., Germeshausen M., Brandes G., Lee-Gossler J., Noyan F. et al. (2009) A syndrome with congenital neutropenia and mutations in G6PC3. N. Engl. J. Med., 360, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kingsley D.M., Kozarsky K.F., Hobbie L. and Krieger M. (1986) Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase deficient mutant. Cell, 44, 749–759. [DOI] [PubMed] [Google Scholar]
- 45. Daenzer J.M., Sanders R.D., Hang D. and Fridovich-Keil J.L. (2012) UDP-galactose 4'-epimerase activities toward UDP-Gal and UDP-GalNAc play different roles in the development of Drosophila melanogaster. PLoS Genet., 8, e1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brokate-Llanos A.M., Monje J.M., Murdoch Pdel S. and Munoz M.J. (2014) Developmental defects in a Caenorhabditis elegans model for type III galactosemia. Genetics, 198, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosoff P.M. (1995) Myelodysplasia and deficiency of uridine diphosphate-galactose 4-epimerase. J. Pediatr., 127, 605–608. [DOI] [PubMed] [Google Scholar]
- 48. Dimitroff C.J., Lee J.Y., Rafii S., Fuhlbrigge R.C. and Sackstein R. (2001) Cd44 is a major E-selectin ligand on human hematopoietic progenitor cells. J. Cell Biol., 153, 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sackstein R., Merzaban J.S., Cain D.W., Dagia N.M., Spencer J.A., Lin C.P. and Wohlgemuth R. (2008) Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med., 14, 181–187. [DOI] [PubMed] [Google Scholar]
- 50. Wang Y., Jobe S.M., Ding X., Choo H., Archer D.R., Mi R., Ju T. and Cummings R.D. (2012) Platelet biogenesis and functions require correct protein O-glycosylation. Proc. Natl. Acad. Sci. U.S.A., 109, 16143–16148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ju T. and Cummings R.D. (2005) Protein glycosylation: chaperone mutation in Tn syndrome. Nature, 437, 1252. [DOI] [PubMed] [Google Scholar]
- 52. Wandall H.H., Rumjantseva V., Sorensen A.L., Patel-Hett S., Josefsson E.C., Bennett E.P., Italiano J.E. Jr., Clausen H., Hartwig J.H. and Hoffmeister K.M. (2012) The origin and function of platelet glycosyltransferases. Blood, 120, 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hemmoranta H., Satomaa T., Blomqvist M., Heiskanen A., Aitio O., Saarinen J., Natunen J., Partanen J., Laine J. and Jaatinen T. (2007) N-glycan structures and associated gene expression reflect the characteristic N-glycosylation pattern of human hematopoietic stem and progenitor cells. Exp. Hematol., 35, 1279–1292. [DOI] [PubMed] [Google Scholar]
- 54. Nasirikenari M., Veillon L., Collins C.C., Azadi P. and Lau J.T. (2014) Remodeling of marrow hematopoietic stem and progenitor cells by non-self ST6Gal-1 sialyltransferase. J. Biol. Chem., 289, 7178–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grozovsky R., Giannini S., Falet H. and Hoffmeister K.M. (2015) Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr. Opin. Hematol., 22, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grozovsky R., Giannini S., Falet H. and Hoffmeister K.M. (2015) Regulating billions of blood platelets: glycans and beyond. Blood, 126, 1877–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giannini S., Lee-Sundlov M.M., Adelman M., Begonja A.J., Lau J.T., Falet H. and Hoffmeister K.M. (2017) Blood, 130, S1018. [Google Scholar]
- 58. Mercier F., Shi J., Sykes D.B., Miller E., Oki T., Man C.-H., Yan X.-J., Kfoury Y., Lee D., Doench J.G. et al. (2017) Blood, 130, S2493. [Google Scholar]
- 59. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gulsuner S., Walsh T., Watts A.C., Lee M.K., Thornton A.M., Casadei S., Rippey C., Shahin H., Consortium on the Genetics of Schizophrenia (COGS), PAARTNERS Study Group et al. (2013) Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell, 154, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seo A., Walsh T., Lee M.K., Ho P.A., Hsu E.K., Sidbury R., King M.C. and Shimamura A. (2016) FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas, 45, 858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang M.Y., Churpek J.E., Keel S.B., Walsh T., Lee M.K., Loeb K.R., Gulsuner S., Pritchard C.C., Sanchez-Bonilla M., Delrow J.J. et al. (2015) Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet., 47, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., Bakker P.I., Daly M.J. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raczy C., Petrovski R., Saunders C.T., Chorny I., Kruglyak S., Margulies E.H., Chuang H.Y., Källberg M., Kumar S.A., Liao A. et al. (2013) Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics, 29, 2041–2043. [DOI] [PubMed] [Google Scholar]
- 65. Boeva V., Popova T., Bleakley K., Chiche P., Cappo J., Schleiermacher G., Janoueix-Lerosey I., Delattre O. and Barillot E. (2012) Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 28, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S. and Saunders C.T. (2016) Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics, 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- 67. Layer R.M., Chiang C., Quinlan A.R. and Hall I.M. (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol., 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v1301 [qbio.GN].
- 69. Trapnell C., Pachter L. and Salzberg S.L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Doulatov S., Vo L.T., Chou S.S., Kim P.G., Arora N., Li H., Hadland B.K., Bernstein I.D., Collins J.J., Zon L.I. et al. (2013) Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell, 13, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Doulatov S., Vo L.T., Macari E.R., Wahlster L., Kinney M.A., Taylor A.M., Barragan J., Gupta M., McGrath K., Lee H.Y. et al. (2017) Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci. Transl. Med., 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.The PyMOL Molecular Graphics System, Version 1.8 Schrödinger LLC. [Google Scholar]
- 73. Freeze H.H. (2006) Genetic defects in the human glycome. Nat. Rev. Genet., 7, 537–551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.