

Abstract
Cancer treatment deserves more research efforts despite intensive conventional treatment modalities for many types of malignancies. Metastasis and resistance to chemotherapy and radiotherapy receive a lot of global research efforts. The current advances in cancer biology may improve targeting the critical metabolic differences that distinguish cancer cells from normal cells. Cancer cells are highly glycolytic for energy production, exhibit the Warburg effect, establish aggressive acidic microenvironment, maintain cancer stem cells, exhibit resistance to chemotherapy, have low antioxidant systems but different ΔΨm (delta psi, mitochondrial transmembrane potential), express P-glycoprotein for multidrug resistance, upregulate glucose transporters and monocarboxylate transporters and are under high steady-state reactive oxygen species conditions. Normal cells differ in all these aspects. Lactate produced through the Warburg effect helps cancer metastasis. Targeting glycolysis reactions for energy production in cancer cells seems promising in decreasing the proliferation and metastasis of cancer cells. 3-bromopyruvate makes use of cancer biology in treating cancer cells, cancer stem cells and preventing metastasis in human cancer as discussed in this review. Updated advances are analyzed here, which include research analysis of background, experience, readings in the field of cancer biology, oncology and biochemistry.
Key words: 3-bromopyruvate, cancer biology, lactate, pyruvate, Warburg effect
Abstract
ملخص البحث:ملخص البحث: تناقش هذه المراجعة كيفية التعامل مع الخلايا السرطانية للتمكن من العلاج الانتقائي لها ويعتبر الشفاء من مرض السرطان من التحدي الكبير للأطباء، بالرغم من تواجد طرق العلاج التقليدية. كذلك فان نمو سرطاني ثانوي (Metastasis) ومقاومة الورم للعلاج الكيميائي والشعاعي لا زالت موجودة. لذا فان علاج مرض السرطان لابد أن يستفيد من التطورات الحديثة في بيولوجية السرطان وذلك باستهداف الخلايا السرطانية فقط.
INTRODUCTION
Many forms of cancer are still beyond curative treatment. Diagnostic procedures for various types of tumors have improved dramatically, which has helped in the early detection of cancer. Even with aggressive treatment modalities using advanced surgical techniques combined with chemotherapy and radiotherapy, cancer recurrence is still a big concern with an unfavorable prognosis in some cases. Mortality rates due to cancer are still high. Chemotherapy and adjuvant radiotherapy are known to harm normal cells. Metastasis influences the survival rate of cancer patients.
In this review article, major biochemical differences between the metabolisms in normal cells versus cancer cells with regard to energy pathways are highlighted. Advances in targeting energy pathways of cancer cells are also analyzed.
MITOCHONDRIAL DEFECTS DRIVE CANCER CELLS TO UTILIZE GLYCOLYSIS
The major source of energy in normal cells comes from mitochondrial oxidative phosphorylation reactions. Krebs cycle also occurs in the mitochondria. Glycolysis pathway occurs in the cytoplasm and seems less important with regard to energy production in normal cells. However, the situation is reversed in cancer cells, i.e., the glycolysis pathway becomes the most important, whereas the mitochondrial pathways seem less important based on the presence of defects in mitochondrial respiration.[1,2]
The mitochondrial defect theory versus the somatic mutation theory may determine the cause of carcinogenesis. Therefore, it has to be questioned whether mitochondrial defects cause carcinogenesis or emerge as a direct result of genetic mutations due to the transformation of normal cells to malignant cells under the effect of viruses, chemicals, radiation, or any other causes. Before the discovery of DNA and cloning of the human genome, the mitochondrial defect theory was accepted to a large extent. However, the current prevailing view is that cancer is primarily a genetic disease involving nuclear mutations in oncogenes (gain of function) and tumor suppressor genes (loss of functions), the so-called “driver gene” mutations that regulate the tumorigenic phenotype, including the induction of hypoxia with consequent alterations in the patterns of metabolism. Polymerase chain reaction (PCR), reverse transcription-PCR, DNA microarray, and many other molecular techniques revolutionized cancer research and clinical oncology by guiding researchers toward the presence of upregulated mutant oncogenes and the downregulated mutant tumor suppressor genes. This resulted in an evidence-based targeted therapy in clinical oncology. In cancer cells, survival pathways are activated, for example, Akt survival pathway due to mitochondrial defects leading to the dependency of cancer cells on glycolysis.[1]
The Akt pathway (phosphatidylinositol 3-kinase [PI3K]-Akt pathway) is a signal transduction pathway that utilizes PI3K and Akt or protein kinase B to promote cellular survival and growth in response to extracellular signals.[3] Activated Akt mediates cell survival, growth, proliferation, cell migration and angiogenesis. This occurs through phosphorylating a range of intracellular proteins. Abnormal Akt activation is usually associated with malignancy[4] where Akt gene amplification activates glycolysis enzymes[5] that causes an increase in glucose metabolism via increasing the translocation of glucose transporters 1 and 4 (GLUT-1 and GLUT-4) to the plasma membrane and increasing the hexokinase (HK) expression.[6]
Glycolytic phenotype induces a new environment suitable for cancer cells only (cancer microenvironment) and harmful for normal cells.[7] The cancer microenvironment is characterized by hypoxia and acidosis due to acid extrusion, for example, lactate. Acidification of cancer microenvironment is primarily due to the activity of the Na+/H+ exchanger and the H+/lactate co-transporter. There is a difference between intracellular pH (pHi) and extracellular pH (pHe) in normal cells and that of cancer cells [Figure 1]. The gradient from pHe to pHi is reversed in tumors. The origin of this reversal in tumor pH gradient is mainly due to oncogene activation.[8] Acidic extracellular microenvironment in cancer cells facilitates altered energy metabolism in cancer.[9]
Figure 1.
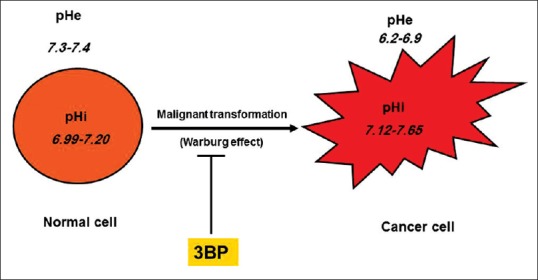
Cancer cells differ from normal cells as regards pH. Intracellular pH is more alkaline in cancer cells. Extracellular pH is strongly acidic in cancer cells and much more acidic than normal cells.
PASTEUR EFFECT, CRABTREE EFFECT, AND WARBURG EFFECT
The major source of energy supply in normal cells is mitochondrial oxidative phosphorylation that can provide normal cells with thirty adenosine triphosphate (ATP) molecules per one glucose molecule. This occurs in an aerobic environment (needs the presence of oxygen). Pasteur noticed that when there is no oxygen, normal cells get a lower amount of energy through glycolysis (two ATP molecules per one glucose molecule) and that glycolysis is inhibited in the presence of oxygen.[10] The Crabtree effect states that in tumor and transformed cells, oxidative phosphorylation (cellular respiration) decreases on exposure to elevated glucose levels.[11]
Unlike normal cells that utilize glucose to produce lactate and two ATP molecules in the absence of oxygen (anerobic glycolysis), Warburg reported that cancer cells use glycolysis as the major energy pathway with the production of enormous amounts of lactate even in the presence of oxygen (aerobic glycolysis).[12] Oncogene activation, for example, Myc results in the upregulation of enzymes of glycolysis, for example, lactate dehydrogenase (LDH-A).[13] Hypoxia together with Myc stabilizes hypoxia-inducible factor resulting in enhanced transcription of genes related to glycolysis, for example, GLUT-1.[14] Hypoxia in tumors decreases the expression of co-stimulatory molecules in monocytes, for example, CD80, which is important for the process of antigen presentation by antigen-presenting cells (APCs) to help the function of T-helper lymphocytes in antigens and this is recognized as an important immunological defense step.[15]
WARBURG EFFECT MAINTAINS HIGH-REACTIVE OXYGEN SPECIES CONDITION IN CANCER CELLS
The production of enormous amounts of lactate characterizes the glycolytic phenotype of cancer cells.[16] Glucose oxidation does not stop at the production of pyruvate, but proceeds to produce lactate via the activity of LDH.[17] Catabolism of glucose in cancer cells ends with the production of lactate even in the presence of oxygen (unlike normal cells where aerobic glycolysis ends with the production of pyruvate). Lactate had no H2O2 scavenging effect either in a cell-free system or in a cell-based system, while pyruvate scavenged H2O2 significantly in a dose-dependent manner, both in a cell-free environment and in a cell-based environment. Lactate did not protect glioma cells against cell death induced by exogenous H2O2. Pyruvate was a protective agent for the experimental glioma cells against cytotoxicity mediated by H2O2.[17] Lactate produced through the Warburg effect is a reflection of the activity of LDH which converts pyruvate to lactate. This consumes pyruvate, abolishes the antioxidant effect gained from pyruvate and causes enhanced steady-state reactive oxygen species (ROS) condition in cancer cells. Interestingly, LDH is a prognostic marker for cancer progression and can predict patient survival.[18] Formation of lactate (Warburg effect) in cancer cells seems to maintain that increased steady-state ROS condition.[17]
Steady-state ROS status is high in cancer cells versus normal cells.[19] Endogenous oxidative stress in cancer cells is high in cultured cancer cells and also in tumor cells.[20,21] Increased endogenous ROS in cancer cells is multifactorial in origin which can be attributed to mitochondrial dysfunction, abnormal metabolism, loss of functional[22,23,24,25] and oncogene activation, for example, c-Myc, Ras, and Bcr-Abl, which were reported to induce ROS generation.[26,27] Increased persistent ROS may cause oxidative damage to DNA, proteins, and lipids in primary cancer cells, which may be associated with a decreased activity of antioxidant enzymes, for example, superoxide dismutase (SOD) and catalase.[28,29] Cancer cells are characterized by a poor antioxidant power compared with normal cells.[30] Antioxidant molecules for example, glutathione peroxidases, SOD and peroxiredoxin may act as tumor suppressors.[31,32] Increased ROS status in cancer cells causes a rise in mitochondrial dysfunction, oncogene activation, aberrant metabolism and antioxidant defects, which may lead to a further elevation in steady-state ROS conditions.[33] As cancer cells evolve, they undergo ROS-mediated selection to select the most aggressive malignant cells to survive at the expense of weak cancer cells.[34,35] In normal cells, the situation is different where pyruvate is the end product of glycolysis in normal cells under aerobic conditions. It scavenges ROS and keeps normal cells at low steady-state conditions. Exogenous lactate preserved the steady-state ROS condition in cancer cells, whereas exogenous pyruvate decreased it significantly.[17]
HEXOKINASE II AS AN IMPORTANT TARGET FOR CANCER THERAPY
Targeting HK II in brain tumors, for example, glioma cells may be a promising therapeutic target in invasive tumors, for example, glioblastoma multiforme (GBM).[36] There are four isoforms of HK enzyme (I–IV).[37] HK II is a therapeutic target that marks malignant human gliomas.[38] In GBM, there is a metabolic shift from oxidative phosphorylation to glycolysis. This causes immortalization and resistance to cancer cell death leading to metabolic adaptation to the malignant status or remodeling in cancer cells, which eventually develop resistance to the current conventional therapeutics.[39]
18Fluorodeoxyglucose-positron emission tomography (18FDG-PET) is a diagnostic technique that was established based on the fact that glycolytic phenotype is a major common characteristic among tumors in general, in which HK II phosphorylates glucose to glucose-6-phosphate (G6P). 18FDG-PET is widely used in the clinical diagnosis and follow-up of cancers to determine the effectiveness of treatment and the presence or absence of active metabolic lesions. 18FDG is an analog of glucose which undergoes no further catabolism in the glycolysis pathway.[40] Reduced glucose metabolism in pediatric GBM correlates with the clinical improvement.[41]
3-BROMOPYRUVATE: A SINGLE SIMPLE DRUG WITH MULTIPLE TARGETS IN CANCER THERAPY
HK II seems to be an important target of 3-bromopyruvate (3BP) [Figure 2]. The product of HK II (G6P) seems critical for cancer cell metabolism, survival, division, and migration. Moreover, HK II has a preventive antioxidant activity.[42] Inhibition of HK II sensitized cancer cells to oxidative stress therapy.[43] Interestingly, the expression of HK II is reported to be very low in normal cells, while it is highly expressed in cancer cells. Therefore, HK II is a potential marker for cancer cells, for example, malignant human gliomas.[38,44]
Figure 2.

Hexokinase II catalyzes a critical metabolic step for cancer cells. ATP: Adenosine triphosphate, NAD+: Oxidized nicotinamide adenine dinucleotide, NADH: Reduced nicotinamide adenine dinucleotide, G6P: Glucose-6-phosphate, R5P: Ribose-5-phosphate, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, GAP: Glyceraldehyde-3-phosphate, HK II: Hexokinase II, NADPH: Reduced nicotinamide adenine dinucleotide phosphate.
3BP targets many points in the glycolysis pathway, for example, at the beginning (HK II step), middle (glyceraldehyde-3-phosphate dehydrogenase [GAPDH] step) and at the end of glycolysis (LDH) steps.[45,46,47] This grants 3BP superiority over many well-known anti-glycolytics which target only one point in glycolysis. HK II may be regarded as an anti-apoptotic protein, that facilitates malignancy.[48] HK II is tightly attached to the mitochondrial voltage-dependent anion channels that links the mitochondrial pathway for ATP production to the cytoplasmic glycolysis pathway which seems critical for phosphorylating glucose to G6P [Figure 3].[49]
Figure 3.

Reported enzyme targets of 3-bromopyruvate (shown in red boxes). 3-bromopyruvate targets Warburg effect at many points of glycolysis pathway. ATP: Adenosine triphosphate, ADP: Adenosine diphosphate, HK II: Hexokinase II, VDAC: Voltage-dependent anion channels, IMM: Inner mitochondrial membrane, OMM: Outer mitochondrial membrane, ANC: Adenine nucleotide carrier, PIC: Inorganic phosphate carrier, GAP: Glyceraldehyde-3-phosphate, GAPDH: Glyceraldehyde-3-phosphate dehydrogenase, PC: Pyruvate carboxylase, PDH: Pyruvate dehydrogenase, 1,3DPG: 1,3 diphosphoglycerate.
3BP disrupts the link between mitochondrial and cytoplasmic power plants for energy production by targeting the entry of pyruvate to Krebs cycle. 3BP inhibits pyruvate dehydrogenase which synthesizes acetyl coenzyme A (CoA) from pyruvate through oxidative decarboxylation, leading to decreased tissue levels of acetyl CoA.[50,51] Acetyl CoA is a critical energy molecule in many energy pathways [Figure 4]. A new mechanism of action was attributed to 3BP which is H2O2 production. Concomitant treatment of glioma cells using 3BP with antioxidants, for example, N-acetyl-L-cysteine, reduced glutathione, or pyruvate was protective to glioma cells against 3BP-induced cancer cell death.[43]
Figure 4.
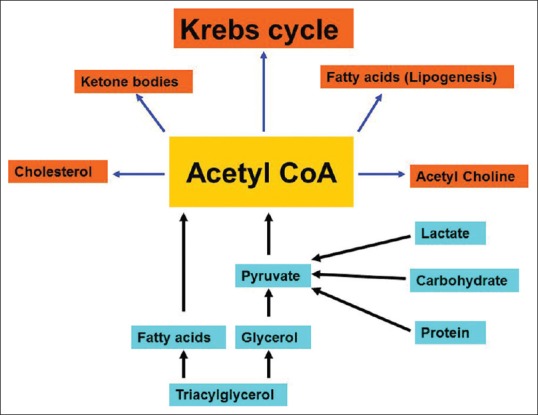
Origin and fate of acetyl coenzyme A. Acetyl coenzyme A is a critical intermediate for energy-generating pathways. Catabolism of proteins, lipids, and carbohydrates gives rise to pyruvate and then acetyl coenzyme A. Acetyl coenzyme A is utilized for different synthetic pathways, among which ketogenesis, Krebs, and lipogenesis are the most important energy pathways.
3BP was reported to cause a significant dose-dependent depletion of energy reserves in glioma cells.[43] It induces two different modes of cell death: Necrosis and apoptosis. This may give 3BP the superiority over chemotherapeutics inducing apoptotic cell death as necrosis is stronger than apoptosis and helps prevent the recurrence of tumors.[43,52]
3-BROMOPYRUVATE IS PROTECTIVE AND LESS TOXIC TO NORMAL CELLS
Compared to cancer cells, normal cells are not significantly harmed during glycolysis inhibition as they have intact mitochondria and may make use of other energy substrates, for example, pyruvate, lipids, and proteins to synthesize ATP.[53] Serial doses of 3BP affected cancer cells selectively, i.e., 3BP was less toxic to normal hepatocytes. On the other hand, 3BP efficiently depleted ATP in hepatocellular carcinoma (HCC) cells[54,55] and decreased the viability of that highly metastatic cell line in vitro[54] and it eradicated xenograft tumors of HCC in all tested animals.[54] Interestingly, 3BP was reported to be nontoxic to neurons.[56] Moreover, serial doses of 3BP protected the neurons of hippocampus against excitotoxicity exerted by kainic acid.[56] Pathologic examination of all tissue samples of mice receiving systemic injection of 3BP confirmed the safety of 3BP in a wide dosage range (5–25 mg/kg). All tissue samples investigated appeared healthy. There were no reported corrosive effects of 3BP.[57]
LACTATE IS A CRITICAL METABOLITE TRANSPORTED THROUGH THE MONOCARBOXYLATE TRANSPORTERS
Lactate represents the beneficial outcome of the Warburg effect on cancer cells [Figures 5 and 6]. High levels of lactate in tumors may support the occurrence of tumor metastasis, resistance to treatment, recurrence and reduced survival rate in patients having different types of cancer.[58] Lactate proved to have an angiogenic stimulatory effect on tumor cells. Influx of tumor-derived lactate through the endothelial cell monocarboxylate transporter 1 (MCT1) supports a nuclear factor-κB/interleukin (IL)-8 pathway that stimulates tumor angiogenesis.[59] Angiogenesis is vital to feed cancer cells in primary and metastatic tumors and to support survival, growth and metastasis of tumors. Tumor angiogenesis can predict patient survival. Taking GBM as an example, an increased survival rate was reported in patients who responded well to anti-angiogenesis treatment.[60]
Figure 5.

Importance of Warburg effect to cancer cells. Lactate is a key metabolic intermediate for cancer cells. VEGF: Vascular endothelial growth factor, HIF-1α: hypoxia-inducible factor 1α
Figure 6.
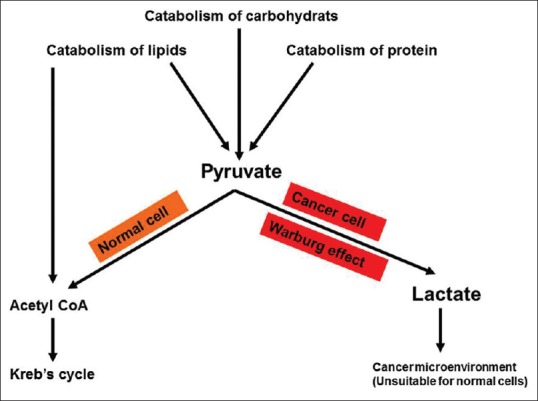
Fate of pyruvate in cancer cells versus normal cells. Pyruvate is the end product of aerobic glycolysis in normal cells and fuels Krebs. Lactate is the end product of glycolysis in cancer cells (Warburg effect) and helps in establishing cancer microenvironment.
Lactate carries a lot of benefits for cancer cells at the expense of normal cells. Lactate is continuously produced in the cytoplasm of cancer cells and is extruded to the outside via the abundant MCT. This helps the establishment of the hostile microenvironment that is suitable for cancer cell survival and progression. Tumor cells use a chemical reduction of the last product of glycolysis (pyruvate) into lactate through LDH activity to oxidize the reducing equivalent reduced nicotinamide adenine dinucleotide to oxidized nicotinamide adenine dinucleotide (NAD+) to keep continuous glycolysis (NAD+ acts as a coenzyme for GAPDH).
These chemical reactions allow tumor cells to survive in this aggressive microenvironment and enhance tumor metastasis to distant anatomical sites.[61,62,63,64] Increased lactate production from glycolysis upregulation participates in microenvironmental acidosis that leads to somatic evolution that selects the most malignant phenotypes which are resistant to acid-induced toxicity.[65] Such evolution occurs at the expense of normal cells and weak cancer cells that cannot afford this acidic microenvironment. Lactate is a proinflammatory mediator that is secreted by tumor cells to facilitate the antigen-dependent secretion of proinflammatory cytokines, for example, IL-17. Lactate activates the IL-23/IL-17 proinflammatory pathway that helps in the establishment of a cancer microenvironment.[66]
Cancer cells can escape from attack by the immune system through many mechanisms, for example, tumor-derived lactate (produced through Warburg effect) decreases cellular immunity performed through the action of T-lymphocytes. Endogenous lactate produced from different tumor cell lines impairs the differentiation of dendritic cells that act as APCs.[67] Lactate significantly reduces the cytotoxic effects exerted by human T cytotoxic lymphocytes (CTLs) by about 50% by suppressing their proliferation and decreasing the production of cytokines. Cytokines produced by CTLs were reduced by about 95%.[68] Lactate produced by melanoma spheroids (three-dimensional) was higher than lactate produced by monolayer culture (two-dimensional) and exerted an inhibitory effect on human CTLs (through the inhibition of recognition of tumor-associated antigens) in spheroid co-cultures. High lactate levels in tumors are correlated with their metastatic potential and they are good prognostic factors to predict metastasis, recurrence of tumors and the short expected patient survival in human cancer.[69,70]
Metabolism in tumors is different from metabolism in normal cells where tumor metabolism is influenced by hypoxia, oncogene activation and tumor suppressor genes inhibition. Upregulated Myc oncogene in human cancers leads to the upregulation of glycolytic enzymes such as LDH-A.[13,71] Moreover, cancer cells are characterized by frequent mutations that lead to genetic changes as a loss of tumor suppressor genes resulting in the decreased utilization of oxygen and enhanced synthesis of lactate.[72]
3-BROMOPYRUVATE: AS A FUNCTIONAL AND STRUCTURAL ANTAGONIST OF BOTH PYRUVIC AND LACTIC ACIDS
3BP is a structural analog of pyruvate and is transported through the same cellular transporters (MCT) as pyruvate.[73] 3BP targets glycolytic enzymes' upstream of pyruvate formation step as it targets HK, GAPDH and LDH steps in glycolysis.[45,46,47,54,74] Antagonizing effects of pyruvate abolish the antioxidant effect of pyruvate in cancer cells, which may enhance the steady-state ROS condition. Interestingly, adding exogenous pyruvate as a treatment to glioma cells scavenged significantly the 3BP-induced H2O2 production (antioxidant effect), while adding exogenous lactate did not.[17] Exogenous pyruvate protected glioma cells against 3BP-induced ATP depletion.[17] Moreover, exogenous pyruvate enhanced the migratory power of glioma cells and exerted a dose-dependent protection against 3BP-induced cell death of glioma cells.[17] All the protective effects of pyruvate against 3BP-induced effects on glioma were overcome on treating glioma cells with gradually increasing doses of 3BP.[17]
Pyruvic acid can be reversibly converted to lactate via LDH. Within the same tumor, cancer cells were reported to take lactate extruded from anaerobic regions to be given to aerobic regions of the same tumor, in which lactate is converted to pyruvate to start the Krebs cycle.[75] Hyperglycolytic tumors that produce enormous amounts of pyruvate to be converted to lactate seem to be more sensitive to the pyruvate's antagonizing effect induced by 3BP.[43] Normal cells, in which pyruvate and energy production have alternative pathways, seem less sensitive to the effects of 3BP.[43]
THERAPEUTIC BENEFITS OF GLYCOLYSIS INHIBITION IN CLINICAL ONCOLOGY
Among the most attractive therapeutic benefits of glycolysis inhibition using 3BP is the inhibition of angiogenesis.[76] Numerous studies reported that 3BP induced the reversal of cancer cells chemoresistance, where 3BP was reported to inhibit the efflux of chemotherapy through the ATP-binding cassette transporters and to antagonize the P-glycoprotein-mediated efflux in MCF-7/ADR drug-resistant breast cancer cells.[53,77,78] Multidrug resistance reversal using 3BP might take place through decreasing ATP content in cancer cells, decreasing HK II activity, inhibiting ATPase activity, and reducing the expression of P-glycoprotein in chemoresistant cancer cells.[77,78,79] Importantly, 3BP kills cancer stem cells, prevents cancer recurrence, and reduces chemoresistance and radioresistance that are commonly encountered in clinical oncology.[79]
3BP was reported to dramatically improve the therapeutic outcome of a patient having fibrolamellar hepatic carcinoma and another patient having metastatic melanoma.[80,81] The dramatic improvements faced in these Stage IV patients strongly suggest the introduction of glycolysis inhibition treatment in the clinical field.
3-BROMOPYRUVATE MAKES USE OF CANCER BIOLOGY TO TARGET CANCER CELLS
The antitumor agent 3BP was reported to have a short half-life under physiological conditions, which will decrease its side effects on normal cells and allow for rapid recovery of normal tissues, for example, liver and kidneys from its pharmacological effects.[82] Affinity of 3BP transport across cancer cells was reported to increase at pH 6.0 (the extracellular milieu of cancer cells) and was better than that at the pH of normal cells (pH 7.4).[83]
MCTs, for example, MCT1 and MCT4 facilitate the efflux of lactate and were reported to be upregulated in cancer cells. MCTs mediate the entry of chemotherapeutic agents, for example, 3BP into the cells that selectively kills cancer cells. Based on this, MCT expression may be used as a molecular marker to predict the response to chemotherapy.[83] Moreover, uptake of 3BP decreased with MCTs inhibitors. There was a higher sensitivity of cancer cells for 3BP upon increasing the level of expression of MCT4 and this may also explain its safety to normal cells.[83]
Compared to normal cells, cancer cells differ in Δψm (termed delta psi and refers to the mitochondrial transmembrane potential). Δψm is used as an indicator for cell death. ΔΨm is lost early during caspase-independent cell death. During the degradation phase of apoptosis, there is a release of catabolic hydrolases and caspase activators from the mitochondria. Those catabolic enzymes together with impairment of the bioenergetic and redox functions of mitochondria finally cause cell death. Upon apoptosis induction, there is a dissipation of ΔΨm, efflux of proteins and eventually cell death.[84]
Recently, gold nanoparticles attached to 3BP were reported to target the mitochondrial membrane potential more selectively and precisely than nontargeted construct or free 3BP, which is expected to enhance their effect on cancer cells rather than normal cells. Therapeutic effects may be better enhanced upon laser irradiation. Interestingly, gold nanoparticles were reported to preferentially kill cancer cells.[85,86]
In endometrial cancer cells, upregulation of the GLUT6 was closely associated with the cancer phenotype while GLUT6 suppression (using small interfering RNA) inhibited both glycolysis and survival of endometrial carcinoma cells that underwent necrosis. GLUT6 promotes glycolysis and survival of endometrial cancer cells in spite of the expression of other GLUTs. The effect of 3BP to decrease the expression level of GLUT6 has not yet been investigated and deserves more research efforts. 3BP was reported to inhibit lipogenesis through pyruvylation of acetyl CoA (precursor of de novo synthesis of fatty acids) and to decrease cancer cellular level of acetyl CoA.[87]
Recently, treatment with 3BP was effective for treating breast cancer. P-glycoprotein mediates the efflux of chemotherapeutics as one of the main mechanisms for multidrug resistance in many cancer types where multidrug-resistant breast cancer cells can efflux the chemotherapeutics used in treatment. 3BP was reported to increase the sensitivity of resistant breast cancer cells to doxorubicin (283-fold), paclitaxel (85-fold), daunorubicin (201-fold) and epirubicin. The main mechanisms reported to achieve this chemosensitization effect are the ability of 3BP to reverse P-glycoprotein-mediated efflux in multidrug-resistant breast cancer cells. Moreover, 3BP decreased the ATPase activity, P-glycoprotein expression and the intracellular level of ATP and HK II.[69]
CLINICAL IMPLICATIONS
Being antagonized by reduced glutathione and N-acetyl cysteine, 3BP can be considered as a safe chemotherapeutic agent. When signs of overdosage, toxicity, or undesirable side effects develop after 3BP administration, reduced glutathione or N-acetyl cysteine can be given immediately as a rescue therapy.
CONCLUSION
Energy metabolism is altered in cancer cells and differs from normal cells.[9] 3BP showed a potent action against cancer cells. It interferes with many of their biological pathways which make it an attractive candidate for the management of cancer patients. 3BP was reported to be efficient, safe, less toxic and well tolerated compared to many anticancer agents and further clinical trials are needed to explore its full potential as a clinical anticancer agent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006;175:913–23. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amoêdo ND, Valencia JP, Rodrigues MF, Galina A, Rumjanek FD. How does the metabolism of tumour cells differ from that of normal cells. Biosci Rep. 2013;33 doi: 10.1042/BSR20130066. pii: E00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis. 2004;9:667–76. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 4.Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J Cell Physiol. 2012;227:3178–84. doi: 10.1002/jcp.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jang M, Kim SS, Lee J. Cancer cell metabolism: Implications for therapeutic targets. Exp Mol Med. 2013;4:45–e45. doi: 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 7.Stubbs M, McSheehy PM, Griffiths JR, Bashford CL. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today. 2000;6:15–9. doi: 10.1016/s1357-4310(99)01615-9. [DOI] [PubMed] [Google Scholar]
- 8.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na/H exchanger in metastasis. Nat Rev Cancer. 2005;5:786–95. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Yang JM. Altered energy metabolism in cancer: A unique opportunity for therapeutic intervention. Cancer Biol Ther. 2013;14:81–9. doi: 10.4161/cbt.22958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh AK, Sloviter HA. Glycolysis and pasteur effect in rat reticulocytes. J Biol Chem. 1973;248:3035–40. [PubMed] [Google Scholar]
- 11.Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. 1929;23:536–45. doi: 10.1042/bj0230536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 13.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–63. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 15.Lahat N, Rahat MA, Ballan M, Weiss-Cerem L, Engelmayer M, Bitterman H. Hypoxia reduces CD80 expression on monocytes but enhances their LPS-stimulated TNF-alpha secretion. J Leukoc Biol. 2003;74:197–205. doi: 10.1189/jlb.0303105. [DOI] [PubMed] [Google Scholar]
- 16.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 17.El Sayed SM, El-Magd RM, Shishido Y, Chung SP, Diem TH, Sakai T, et al. 3-Bromopyruvate antagonizes effects of lactate and pyruvate, synergizes with citrate and exerts novel anti-glioma effects. J Bioenerg Biomembr. 2012;44:61–79. doi: 10.1007/s10863-012-9409-4. [DOI] [PubMed] [Google Scholar]
- 18.Suh SY, Ahn HY. Lactate dehydrogenase as a prognostic factor for survival time of terminally ill cancer patients: A preliminary study. Eur J Cancer. 2007;43:1051–9. doi: 10.1016/j.ejca.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 19.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- 21.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365–72. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 22.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–52. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 23.Rodrigues MS, Reddy MM, Sattler M. Cell cycle regulation by oncogenic tyrosine kinases in myeloid neoplasias: From molecular redox mechanisms to health implications. Antioxid Redox Signal. 2008;10:1813–48. doi: 10.1089/ars.2008.2071. [DOI] [PubMed] [Google Scholar]
- 24.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–62. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 25.Horn HF, Vousden KH. Coping with stress: Multiple ways to activate p53. Oncogene. 2007;26:1306–16. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 26.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31(Pt 6):1441–4. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 27.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–44. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 28.Lu W, Ogasawara MA, Huang P. Models of reactive oxygen species in cancer. Drug Discov Today Dis Models. 2007;4:67–73. doi: 10.1016/j.ddmod.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979;39:1141–9. [PubMed] [Google Scholar]
- 30.Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61:290–302. doi: 10.1016/j.addr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–80. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 32.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, et al. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24:8038–50. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 33.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 34.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002;3:420–5. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desagher S, Glowinski J, Prémont J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci. 1997;17:9060–7. doi: 10.1523/JNEUROSCI.17-23-09060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grobben B, De Deyn PP, Slegers H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 2002;310:257–70. doi: 10.1007/s00441-002-0651-7. [DOI] [PubMed] [Google Scholar]
- 37.Wilson JE. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J Exp Biol. 2003;206(Pt 12):2049–57. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 38.Paggi MG, Fanciulli M, Del Carlo C, Citro G, Carapella CM, Floridi A. The membrane-bound hexokinase as a potential marker for malignancy in human gliomas. J Neurosurg Sci. 1990;34:209–13. [PubMed] [Google Scholar]
- 39.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 40.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2:683–93. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 41.Mineura K, Yasuda T, Kowada M, Sakamoto T, Ogawa T, Shishido F, et al. Positron emission tomographic evaluations in the diagnosis and therapy of multifocal glioblastoma. Report of a pediatric case. Pediatr Neurosci. 1985;12:208–12. doi: 10.1159/000120253. [DOI] [PubMed] [Google Scholar]
- 42.da-Silva WS, Gomez-Puyou A, de Gomez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem. 2004;279:39846–55. doi: 10.1074/jbc.M403835200. [DOI] [PubMed] [Google Scholar]
- 43.El Sayed SM, Abou El-Magd RM, Shishido Y, Chung SP, Sakai T, Watanabe H, et al. D-amino acid oxidase gene therapy sensitizes glioma cells to the antiglycolytic effect of 3-bromopyruvate. Cancer Gene Ther. 2012;19:1–18. doi: 10.1038/cgt.2011.59. [DOI] [PubMed] [Google Scholar]
- 44.Smith TA. Mammalian hexokinases and their abnormal expression in cancer. Br J Biomed Sci. 2000;57:170–8. [PubMed] [Google Scholar]
- 45.Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001;173:83–91. doi: 10.1016/s0304-3835(01)00667-x. [DOI] [PubMed] [Google Scholar]
- 46.Ganapathy-Kanniappan S, Geschwind JF, Kunjithapatham R, Buijs M, Vossen JA, Tchernyshyov I, et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 2009;29:4909–18. [PMC free article] [PubMed] [Google Scholar]
- 47.Mulet C, Lederer F. Bromopyruvate as an affinity label for baker's yeast flavocytochrome b2. Kinetic study of the inactivation reaction. Eur J Biochem. 1977;73:443–7. doi: 10.1111/j.1432-1033.1977.tb11336.x. [DOI] [PubMed] [Google Scholar]
- 48.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: Cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–86. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedersen PL. Voltage dependent anion channels (VDACs): A brief introduction with a focus on the outer mitochondrial compartment's roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr. 2008;40:123–6. doi: 10.1007/s10863-008-9165-7. [DOI] [PubMed] [Google Scholar]
- 50.Murray RK, Granner DK, Mayes PA, Rodwell VW. Columbus, OHIO: hLange Medical Books/McGraw-Hill Medical Publishing Division; 2003. Harper's illustrated biochemistry.(26th edition), Overview of Metabolism; pp. 122–7. [Google Scholar]
- 51.Rícný J, Tucek S. Acetyl coenzyme A and acetylcholine in slices of rat caudate nuclei incubated in the presence of metabolic inhibitors. J Biol Chem. 1981;256:4919–23. [PubMed] [Google Scholar]
- 52.Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: A specific form of programmed cell death? Exp Cell Res. 2003;283:1–16. doi: 10.1016/s0014-4827(02)00027-7. [DOI] [PubMed] [Google Scholar]
- 53.Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One. 2011;6:e27222. doi: 10.1371/journal.pone.0027222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, et al. Advanced cancers: Eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun. 2004;324:269–75. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 55.Pereira da Silva AP, El-Bacha T, Kyaw N, dos Santos RS, da-Silva WS, Almeida FC, et al. Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem J. 2009;417:717–26. doi: 10.1042/BJ20080805. [DOI] [PubMed] [Google Scholar]
- 56.Lee KH, Park JH, Won R, Lee H, Nam TS, Lee BH. Inhibition of hexokinase leads to neuroprotection against excitotoxicity in organotypic hippocampal slice culture. J Neurosci Res. 2011;89:96–107. doi: 10.1002/jnr.22525. [DOI] [PubMed] [Google Scholar]
- 57.Buijs M, Vossen JA, Geschwind JF, Ishimori T, Engles JM, Acha-Ngwodo O, et al. Specificity of the anti-glycolytic activity of 3-bromopyruvate confirmed by FDG uptake in a rat model of breast cancer. Invest New Drugs. 2009;27:120–3. doi: 10.1007/s10637-008-9145-0. [DOI] [PubMed] [Google Scholar]
- 58.Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60:916–21. [PubMed] [Google Scholar]
- 59.Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–60. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- 60.Sorensen AG, Emblem KE, Polaskova P, Jennings D, Kim H, Ancukiewicz M, et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012;72:402–7. doi: 10.1158/0008-5472.CAN-11-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gatenby RA, Gawlinski ET. The glycolytic phenotype in carcinogenesis and tumor invasion: Insights through mathematical models. Cancer Res. 2003;63:3847–54. [PubMed] [Google Scholar]
- 62.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006;66:5216–23. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- 63.Jang M, Kim SS, Lee J. Cancer cell metabolism: Implications for therapeutic targets. Exp Mol Med. 2013;45:e45. doi: 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–34. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 65.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 66.Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, et al. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol. 2008;180:7175–83. doi: 10.4049/jimmunol.180.11.7175. [DOI] [PubMed] [Google Scholar]
- 67.Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107:2013–21. doi: 10.1182/blood-2005-05-1795. [DOI] [PubMed] [Google Scholar]
- 68.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 69.Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, et al. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2001;51:349–53. doi: 10.1016/s0360-3016(01)01630-3. [DOI] [PubMed] [Google Scholar]
- 70.Walenta S, Schroeder T, Mueller-Klieser W. Lactate in solid malignant tumors: Potential basis of a metabolic classification in clinical oncology. Curr Med Chem. 2004;11:2195–204. doi: 10.2174/0929867043364711. [DOI] [PubMed] [Google Scholar]
- 71.Husain Z, Seth P, Sukhatme VP. Tumor-derived lactate and myeloid-derived suppressor cells: Linking metabolism to cancer immunology. Oncoimmunology. 2013;2:e26383. doi: 10.4161/onci.26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–3. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 73.Zorzano A, Fandos C, Palacín M. Role of plasma membrane transporters in muscle metabolism. Biochem J. 2000;349(Pt 3):667–88. doi: 10.1042/bj3490667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shoshan MC. 3-Bromopyruvate: Targets and outcomes. J Bioenerg Biomembr. 2012;44:7–15. doi: 10.1007/s10863-012-9419-2. [DOI] [PubMed] [Google Scholar]
- 75.Semenza GL. Tumor metabolism: Cancer cells give and take lactate. J Clin Invest. 2008;118:3835–7. doi: 10.1172/JCI37373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.El Sayed SM, El-Magd RM, Shishido Y, Yorita K, Chung SP, Tran DH, et al. D-Amino acid oxidase-induced oxidative stress, 3-bromopyruvate and citrate inhibit angiogenesis, exhibiting potent anticancer effects. J Bioenerg Biomembr. 2012;44:513–23. doi: 10.1007/s10863-012-9455-y. [DOI] [PubMed] [Google Scholar]
- 77.Yu SJ, Yoon JH, Yang JI, Cho EJ, Kwak MS, Jang ES, et al. Enhancement of hexokinase II inhibitor-induced apoptosis in hepatocellular carcinoma cells via augmenting ER stress and anti-angiogenesis by protein disulfide isomerase inhibition. J Bioenerg Biomembr. 2012;44:101–15. doi: 10.1007/s10863-012-9416-5. [DOI] [PubMed] [Google Scholar]
- 78.Wu L, Xu J, Yuan W, Wu B, Wang H, Liu G, et al. The reversal effects of 3-bromopyruvate on multidrug resistance in vitro and in vivo derived from human breast MCF-7/ADR cells. PLoS One. 2014;9:e112132. doi: 10.1371/journal.pone.0112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Isayev O, Rausch V, Bauer N, Liu L, Fan P, Zhang Y, et al. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget. 2014;5:5177–89. doi: 10.18632/oncotarget.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J Bioenerg Biomembr. 2012;44:163–70. doi: 10.1007/s10863-012-9417-4. [DOI] [PubMed] [Google Scholar]
- 81.El Sayed SM, Mohamed WG, Seddik MA, Ahmed AS, Mahmoud AG, Amer WH, et al. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: A concise literature review and case study. Chin J Cancer. 2014;33:356–64. doi: 10.5732/cjc.013.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Glick M, Biddle P, Jantzi J, Weaver S, Schirch D. The antitumor agent 3-bromopyruvate has a short half-life at physiological conditions. Biochem Biophys Res Commun. 2014;452:170–3. doi: 10.1016/j.bbrc.2014.08.066. [DOI] [PubMed] [Google Scholar]
- 83.Azevedo-Silva J, Queirós O, Ribeiro A, Baltazar F, Young KH, Pedersen PL, et al. The cytotoxicity of 3-bromopyruvate in breast cancer cells depends on extracellular pH. Biochem J. 2015;467:247–58. doi: 10.1042/BJ20140921. [DOI] [PubMed] [Google Scholar]
- 84.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 85.Baltazar F, Pinheiro C, Morais-Santos F, Azevedo-Silva J, Queirós O, Preto A, et al. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol Histopathol. 2014;29:1511–24. doi: 10.14670/HH-29.1511. [DOI] [PubMed] [Google Scholar]
- 86.Marrache S, Dhar S. The energy blocker inside the power house: Mitochondria targeted delivery of 3-bromopyruvate. Chem Sci. 2015;6:1832–45. doi: 10.1039/c4sc01963f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Byrne FL, Poon IK, Modesitt SC, Tomsig JL, Chow JD, Healy ME, et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014;74:5832–45. doi: 10.1158/0008-5472.CAN-14-0254. [DOI] [PubMed] [Google Scholar]