

Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive disorder, characterized by a progressive degeneration of anterior horn cells of the spinal cord resulting in hypotonia, skeletal muscle atrophy and weakness. We report the case of a 33-year-old female with SMA type IV (SMA4) who presented with symptoms of spinal cord lesion that was initially missed. Further evaluation resulted in the diagnosis of ependymoma. To the best of our knowledge, this is the first time that the coexistence of SMA4 and ependymoma has been reported.
Keywords: Ependymoma, muscle weakness, spinal muscular atrophy type IV
Abstract
ملخص البحث: يعتبر ضمور العضلات الشوكي من الأمراض الوراثية المتنحية التي تتميز بضمور تدريجي لخلايا القرن الأمامي للحبل الشوكي والتي ينتج عنها ضمور عضلات الهيكل العظمي وضعفها. يعرض الباحثون حالة لسيدة في الثالثة والثلاثين من العمر تعاني من ضمور العضلات الشوكي من النوع الرابع والتي حضرت إلى المستشفى بأعراض مرض الحبل الشوكي لم يتم اكتشافه. وبمزيد من الفحوصات تم تشخيص هذا المرض بأنه الورم البطاني العصبي (ependymoma). يعتقد الباحثون أن هذه أول حالة يرتبط فيها ضمور العضلات الشوكي بالورم البطاني العصبي.
INTRODUCTION
Spinal muscular atrophy type IV (SMA4) is an adult onset, mild form of SMA. It is a rare condition with a prevalence of 1/300,000. SMA4 usually manifests in the second or third decade of life. It is a motor neuron disease, resulting in proximal muscle weakness with lower motor neuron features.[1,2,3] However, the presence of sensory involvement, sphincter abnormalities and upper motor neuron signs should raise the suspicion of other coexisting conditions.[1,2]
Spinal cord ependymomas are the most common intramedullary tumors. They are considered to be benign tumors, but their slow evolution causes a delay in diagnosis, and there is an important functional risk of extensive disabilities.[4,5]
This report presents the case of a 33-year-old female patient with SMA4 and ependymoma. The aim of this case report is to emphasize the importance of careful history taking, keen neurological examination and awareness of red flags that could be indicative of a neurological disease.
CASE REPORT
A 33-year-old right-handed Saudi homemaker with two children had a history of slowly progressive proximal muscle weakness of 15-year duration. She reported that although her weakness was mild, it affected her daily activities. However, she was completely independent. The patient reported that her younger sister had similar symptoms, which raised suspicion that the cause was an underlying genetic condition. Electromyography (EMG) was carried out, which was suggestive of a motor neuron disease. Based on the EMG features and positive family history, SMA was suspected. Genetic examination was undertaken, which showed a signal increase of 50% of the survival motor neuron 2 (SMN2), suggesting the presence of three copies of SMN2 and a signal reduction of nearly 50% of the neuronal apoptosis inhibitory protein (NAIP) gene. The patient was diagnosed with SMA4. Her condition was stable and she was followed regularly in the neurology clinic for approximately 2 years.
However, on subsequent visits, she reported that she had suffered from intermittent episodes of neck pain over a previous couple of months. This pain radiated to both arms and was associated with bilateral hand numbness. Her symptoms were asymmetrical; they were worse in the right arm with sensory disturbance along the medial side of forearm and little and middle fingers. She also noticed that the weakness in her legs weakness was getting worse and in the past month, she had difficulty walking with frequent falls, both of which significantly affected her daily activities. There was no sphincter dysfunction. The patient sought medical attention in the Emergency Department, and it was thought this was part of her underlying neurological disease because of which she was discharged home with close follow-up.
The general physical examination was within normal ranges, stable vital signs and no dysmorphic features. However, she was obese (weight 85 kg and height 163 cm).
The neurological examination was normal and revealed high cortical functions and cranial nerves. She had wasting of proximal muscles with obvious fasciculations. She had hypotonia and hyporeflexia in the upper extremities, with a muscle power score of 4/5 using the medical research council scale. The lower extremities were spastic with muscle power scored at 3/5 using medical research council and hyperactive muscle stretch reflexes, sustained clonus and extensor plantar response. There was a sensory level at T4 dermatome. She had a waddling gait, but her coordination was normal.
On investigations, including magnetic resonance imaging (MRI) of the spinal cord showed an intramedullary mixed solid and cystic lesion that spanned approximately nine vertebral segments and caused cord expansion. The solid component demonstrated low signal intensity on T1 and high signal intensity on T2 with gadolinium enhancement. Radiological diagnosis was suggestive of ependymoma or astrocytoma [Figures 1 and 2]. The MRI of the brain showed no abnormalities, decreasing the possibility of metastasis.
Figure 1.
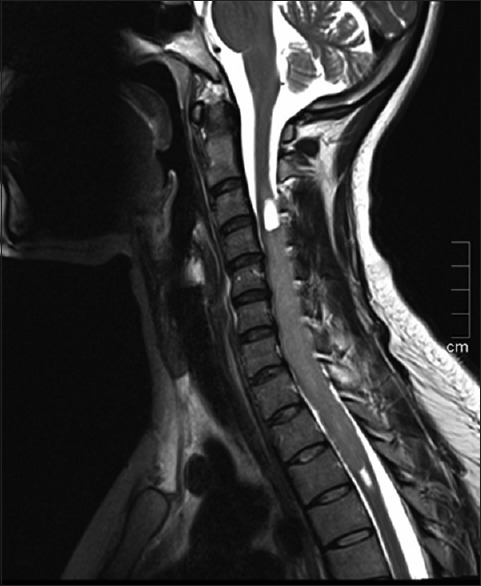
MRI of spinal cord showing intramedullary mixed solid and cystic lesion extending from C3-T4 and causing cord expansion. The solid component demonstrates high signal intensity on T2
Figure 2.
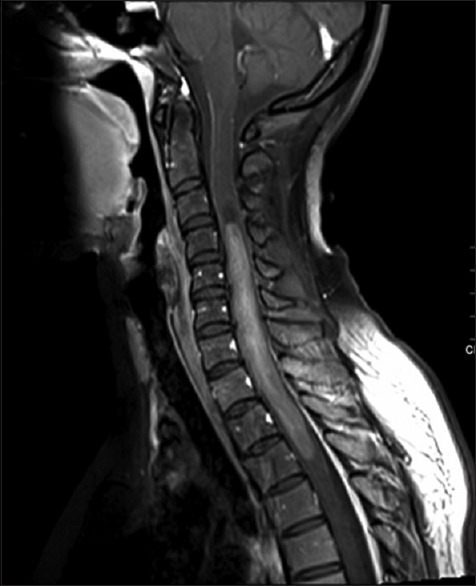
MRI of spinal cord showing intramedullary mixed solid and cystic lesion extending from C3-T4 and causing cord expansion. The solid component with gadolinium enhancement
The patient was admitted immediately and she then started to have sphincter abnormality in the form of urine retention and constipation. She received a course of dexamethasone and her symptoms improved slightly. Under general anesthesia with intraoperative neuromonitor, the patient was placed in the prone position and underwent a posterior spinal approach, namely French door laminoplasty through C3-T4. Under the microscope, a midline dural opening was done and then midline myelotomy. The tumor was exposed and a biopsy taken. After half of the tumor had been removed, there was a sudden drop in blood pressure maximal expiratory pressure by more than 50%; therefore, the procedure was stopped [Figure 3].
Figure 3.
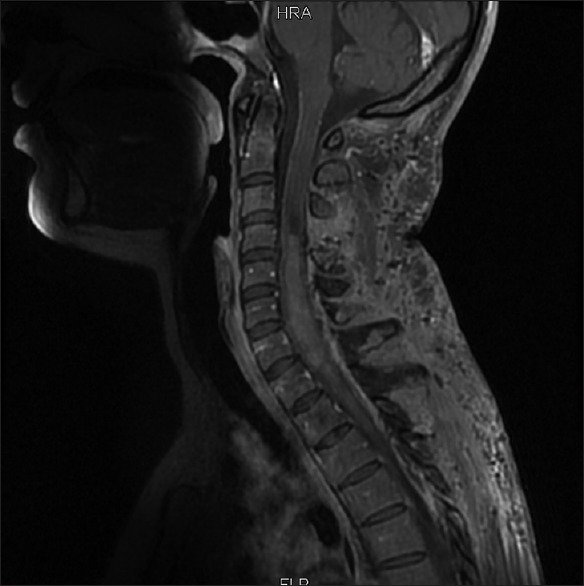
MRI of spinal cord at 3-month follow-up
The gross description of the lesion intraoperatively was a grayish mass with both solid and cystic components. Histopathological analysis confirmed the diagnosis of medullary cellular ependymoma (WHO Grade II) [Figure 4]. Ten days postoperatively, she regained both light touch and pain sensations. On the 30th postoperative day, her leg power was 4/5. However, she remained in hospital for quite some time as she developed right leg deep venous thrombosis. Her long-term plan included referral to physiotherapy and follow-up with the neurosurgeon to arrange for another surgery for complete the removal of the tumor.
Figure 4.
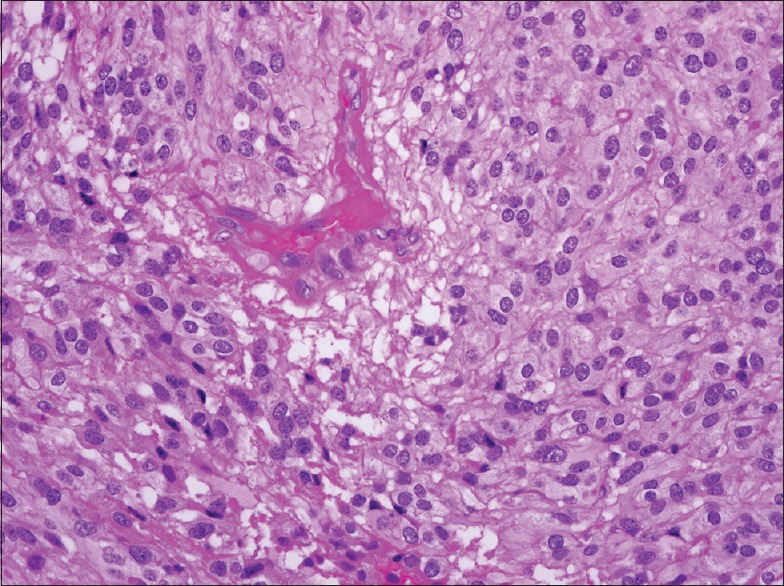
Histopathology of ependymoma with moderately cellular glial neoplasm with low-grade nuclear features
DISCUSSION
SMA disorders are characterized by degeneration of the anterior horn cells in the spinal cord and motor nuclei in the lower brainstem. These diseases are classified based on the age of onset and clinical course into four types. Proximal SMA4 is the adult-onset form with a prevalence estimated at 1/300,000. SMA4 usually manifests in the second or third decade of life.[1,2,3] The muscle weakness predominantly affects the legs and hip muscles and then progresses to the shoulders and arms. The clinical picture is similar to that seen in SMA3, but the motor weakness is less severe in SMA4.[2,6,7,8] Humans have two nearly identical copies of SMN gene: SMN1 and SMN2. SMA has been associated with deletions in the SMN1 gene (5q12.2-q13.3) encoding the SMN protein. Although there is some variation, disease severity in SMA is inversely correlated with the number of copies of the second SMN gene (SMN2; 5q13.2) and some studies have shown that patients with the mild SMA4 form have multiple SMN2 copies. Another gene is NAIP; its functional role in the pathogenesis of SMA has not been fully elucidated.[9,10] SMA4 is the mildest form of SMA and, in general, the disease course is benign with patients having a normal life expectancy.[9]
Ependymomas are the most common intramedullary tumors in adults, with a peak age at presentation between 30 and 40 years.[4,5] The incidence of spinal cord astrocytoma is 0.8–2.5 in 100,000 a year. Localized pain is usually the earliest presenting complaint and may precede the neurological deficit by months. Neurological deficits are similar to that of other spinal cord lesions, with weakness, sphincter abnormalities and sensory changes.[11] Ependymomas tend to occur centrally in the spinal cord, and the cervical region is the most common site. These lesions enhance intensely on MRI. Optimal management consists of gross total resection. Although these are infiltrative tumors, a total or near-total resection can frequently be achieved without causing further neurologic deficits.[12,13]
Ependymomas are considered as benign tumors, but their slow evolution course causes a delay in the diagnosis and there is an important functional risk of extensive disabilities.[4,5] It is well-known that SMA is a motor neuron disease; therefore, the recognition of an unusual complaint, such as the presence of sensory and/or sphincter abnormalities, should always raise the suspicion of another differential diagnosis. A detailed medical history and thorough neurological examination are mandatory for clinical diagnosis. There are only a few case reports of SMA with malignancy.[14,15] SMA3 was reported with alveolar rhabdomyosarcoma and SMA1 with neuroblastoma.[14,15,16] To the best of our knowledge, this is the first time that the coexistence of SMA4 and ependymoma has been reported.
CONCLUSION
Both SMA and astrocytoma are relatively rare diseases, and the probability of their co-existence is very low. In such circumstances, the diagnosis of these two conditions in the same individual may be challenging. Presentation and a timeline of events highlight the importance of detailed clinical history, physical examination and necessary investigation. In chronic patients, new and unusual pathological symptoms should initiate a differential diagnosis of another, coexisting disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Prior TW, Russman BS. Spinal muscular atrophy. [Last accessed on 2014 Nov 13];GeneReviews. www.ncbi.nlm.nih.gov/books/NBK1352/ [Google Scholar]
- 2.Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. 2014;137(Pt 11):2879–96. doi: 10.1093/brain/awu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–33. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 4.Van Goethem JW, van den Hauwe L, Ozsarlak O, De Schepper AM, Parizel PM. Spinal tumors. Eur J Radiol. 2004;50:159–76. doi: 10.1016/j.ejrad.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 5.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. Classification of Tumours of the Nervous System. Lyon, France: IARC Press; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroksmark AK, Beckung E, Tulinius M. Muscle strength and motor function in children and adolescents with spinal muscular atrophy II and III. Eur J Paediatr Neurol. 2001;5:191–8. doi: 10.1053/ejpn.2001.0510. [DOI] [PubMed] [Google Scholar]
- 7.Finkel RS, Crawford TO, Swoboda KJ, Kaufmann P, Juhasz P, Li X, et al. Candidate proteins, metabolites and transcripts in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7:e35462. doi: 10.1371/journal.pone.0035462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haaker G, Fujak A. Proximal spinal muscular atrophy: Current orthopedic perspective. Appl Clin Genet. 2013;6:113–20. doi: 10.2147/TACG.S53615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauke J, Riessland M, Lunke S, Eyüpoglu IY, Blümcke I, El-Osta A, et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. 2009;18:304–17. doi: 10.1093/hmg/ddn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sedghi M, Behnam M, Fazel E, Salehi M, Ganji H, Meamar R, et al. Genotype-phenotype correlation of survival motor neuron and neuronal apoptosis inhibitory protein genes in spinal muscular atrophy patients from Iran. Adv Biomed Res. 2014;3:74. doi: 10.4103/2277-9175.125872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SH, Bak KH, Kim DW, Kang TH. Primary intramedullary spinal sarcoma: A case report and review of the current literatures. J Korean Neurosurg Soc. 2010;48:448–51. doi: 10.3340/jkns.2010.48.5.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang ZY, Sun JJ, Xie JC, Li ZD, Ma CC, Liu B, et al. Comparative analysis on the diagnosis and treatments of multisegment intramedullary spinal cord tumors between the different age groups. Neurosurg Rev. 2012;35:85–92. doi: 10.1007/s10143-011-0345-2. [DOI] [PubMed] [Google Scholar]
- 13.Kyoshima K, Akaishi K, Tokushige K, Muraoka H, Oikawa S, Watanabe A, et al. Surgical experience with resection en bloc of intramedullary astrocytomas and ependymomas in the cervical and cervicothoracic region. J Clin Neurosci. 2004;11:623–8. doi: 10.1016/j.jocn.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Yaris N, Kutluk T, Topaloglu H, Akçören Z, Büyükpamukçu M. Disseminated alveolar rhabdomyosarcoma in a child with spinal muscular atrophy. J Pediatr Hematol Oncol. 2002;24:508–9. doi: 10.1097/00043426-200208000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Rudnik-Schöneborn S, Anhuf D, Koscielniak E, Zerres K. Alveolar rhabdomyosarcoma in infantile spinal muscular atrophy: Coincidence or predisposition? Neuromuscul Disord. 2005;15:45–7. doi: 10.1016/j.nmd.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Sag E, Sen HS, Haliloglu G, Yalcin B, Kutluk T. Neuroblastoma in a patient with spinal muscular atrophy type I: Is it just a coincidence? J Child Neurol. 2015;30:1075–8. doi: 10.1177/0883073814542950. [DOI] [PubMed] [Google Scholar]