

Abstract
We have previously shown that protein kinase Cε (PKCε) is involved in mitochondrial dysfunction in renal proximal tubular cells (RPTC). This study examined mitochondrial targets of active PKCε in RPTC injured by the model oxidant tert-butyl hydroperoxide (TBHP). TBHP exposure augmented the levels of phosphorylated (active) PKCε in mitochondria, which suggested translocation of PKCε to mitochondria after oxidant exposure. Oxidant injury decreased state 3 respiration, ATP production, ATP content, and complex I activity. Further, TBHP exposure increased ΔΨm and production of reactive oxygen species (ROS), and induced mitochondrial fragmentation and RPTC death. PKCε activation by overexpressing constitutively active PKCε exacerbated decreases in state 3 respiration, complex I activity, ATP content, and augmented RPTC death. In contrast, inhibition of PKCε by overexpressing inactive PKCε mutant restored state 3 respiration, respiratory control ratio, complex I activity, ΔΨm, and ATP production and content, but did not prevent decreases in F0F1-ATPase activity. Inhibition of PKCε prevented oxidant-induced production of ROS and mitochondrial fragmentation, and reduced RPTC death. We conclude that activation of PKCε mediates: 1) oxidant-induced changes in ΔΨm, decreases in mitochondrial respiration, complex I activity and ATP content, 2) mitochondrial fragmentation, and 3) RPTC death. In contrast, oxidant-induced inhibition of F0F1-ATPase activity is not mediated by PKCε. These results show that, in contrast to the protective effects of PKCε in the heart, PKCε activation is detrimental to mitochondrial function and viability in RPTC and mediates oxidant-induced injury.
Keywords: Mitochondria, Protein kinase C-ε, Renal proximal tubular cells, Respiratory chain, ATP synthase, Oxidant injury, Reactive oxygen species, Mitochondrial membrane potential
Acute renal injury is a common result of ischemic or nephrotoxic kidney damage. Renal proximal tubular cells (RPTC) are a major target of ischemia and nephrotoxicants due to their dependence on oxidative metabolism and mitochondria for ATP production. Oxidative stress is one of major mechanisms mediating renal cell injury caused by different drugs, toxicants, hypoxia, and ischemia/reperfusion (Koyner et al., 2008; Cachofeiro et al., 2008). Disruption of mitochondrial electron transfer through the respiratory chain often leads to increased production of reactive oxygen species (ROS) and initiates oxidative stress (Orrenius et al., 2007). Activation of protein kinase C-ε (PKCε) has been implicated in a variety of pathological processes including ischemia/reperfusion, hypoxia, and oxidative stress in cardiomyocytes and pulmonary artery smooth muscle cells (Gopalakrishna et al., 2000; Rathore et al., 2006; Rathore et al., 2008). Numerous reports have demonstrated that activation of PKCε mediates protection from ischemia/reperfusion injury in the heart (Liu et al., 1999; Mochly-Rosen et al., 2000; Ping et al., 1999a, 1999b, 2001, 2002; Baines et al., 2002; Gray et al., 1997, 2004; Budas et al., 2010; Jaburek et al., 2006). In ventricular myocytes, active PKCε is present in mitochondria and colocalizes with mitochondrial ATP-dependent K+ channels (mitoKATP), which leads to their opening and increases in K+ flux into mitochondria (Jaburek et al., 2006). PKCε interacts with the mitochondrial permeability transition pore (MPTP), inhibits its opening, and stabilizes cardiac mitochondria by preventing the loss of potential across the inner mitochondrial membrane (Baines et al., 2003; Ogbi et al., 2004, 2006; Guo et al., 2007). Finally, PKCε associates with the cytochrome oxidase and this interaction increases cytochrome oxidase activity (Baines et al., 2003; Ogbi et al., 2004, 2006; Guo et al., 2007).
In contrast, the role of PKCε and its subcellular targets in renal injury are less known. It has been shown that PKCε activation inhibits Na+ transport in the cortical collecting duct (DeCoy et al., 1995) We have demonstrated that, in contrast to the protective role of PKCα, PKCε activation is involved in mitochondrial dysfunction and decreases in active Na+ transport induced by oxidants in RPTC (Nowak, 2002, 2003; Nowak et al., 2004, 2011). Further, sustained activation of PKCε in non-injured RPTC mediates mitochondrial dysfunction, production of reactive oxygen species (ROS), and results in cell death (Nowak et al., 2011). The model oxidant tert-butyl hydroperoxide (TBHP) activates PKCε in RPTC and the translocation of active PKCε to mitochondria is followed by mitochondrial dysfunction and decreases in active Na+ transport and Na+/K+-ATPase activity (Nowak et al., 2004). Inhibiting PKCε and preventing PKCε translocation to mitochondria without blocking PKCε activation reduces mitochondrial dysfunction and accelerates recovery of mitochondrial function and active Na+ transport in injured RPTC (Nowak et al., 2004). However, specific targets of PKCε in mitochondria of injured RPTC are not known. The goals of this study were to: 1) examine whether selective activation and inhibition of PKCε alters mitochondrial function, ATP production and levels, mitochondrial morphology, and RPTC viability, and 2) determine mitochondrial functions and complexes targeted by PKCε in oxidant-injured RPTC.
MATERIALS AND METHODS
Animals and materials
Female New Zealand White rabbits (2.0–2.5 kg) were purchased from Myrtle’s Rabbitry (Thompson Station, TN). All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. The cell culture media were purchased from MediaTech Cellgro (Herndon, VA). Phospho-PKCε and PKCε antibodies were purchased from Upstate Biotechnology (Lake Placid, NY) and BD Transduction Laboratory (San Diego, CA), respectively. The sources of the other reagents have been described previously (Nowak and Schnellmann, 1995, 1996; Nowak et al., 2004).
Isolation and culture of renal proximal tubular cells
Renal proximal tubules were isolated from rabbit kidneys by the iron-oxide perfusion method and cultured in 35-mm culture dishes in improved conditions as previously described (Nowak and Schnellmann, 1995, 1996). The culture medium was a 50:50 mixture of DMEM and Ham’s F-12 nutrient mix without phenol red, pyruvate, and glucose, supplemented with 15 mM NaHCO3, 15 mM N-2-hydroethylpiperazine-N’−2-ethanesulfonic acid (Hepes), 6 mM lactate (pH 7.4, 295 mosmol/kg), human transferrin (5 μg/ml), selenium (5 ng/ml), hydrocortisone (50 nM), bovine insulin (10 nM), and L-ascorbic acid-2-phosphate (50 μM).
Adenoviral constructs and amplification
Adenoviral vector encoding dominant negative (inactive) PKCε (dnPKCε) was constructed by mutations at the ATP-binding site (replacement of lysine with arginine at position 436) and the pseudosubstrate domain (replacement of alanine with glutamate at position 159) as described previously (Ping et al., 1999). These mutations destroyed the construct’s kinase activity without changing the active conformation of PKC-ε (Ping et al., 1999). An aliquot of the adenovirus encoding dnPKCε was provided by Dr. Peipei Ping (UCLA, Los Angeles, CA). PKCε was made constitutively active by deletion of residues 154–163 of its inhibitory pseudosubstrate domain (Wotton et al., 1993). The adenoviral vector encoding caPKCε was constructed as described previously (Strait and Samarel., 2000) and provided by Dr. Allen Samarel (Loyola University Medical Center, Maywood, IL). Adenoviruses were amplified in AD293 and HEK293 cells and purified as described previously (Nowak et al., 2011; Shaik et al., 2007). The multiplicity of infection (MOI) was determined by a viral dilution assay in HEK-293 cells.
TBHP treatment of RPTC
Adenoviral infections of confluent, quiescent RPTC were carried out 48 h prior to TBHP exposure (350 μM, 45 min). PKCε was inhibited in RPTC by overexpressing dnPKCε using adenoviral vector encoding dnPKCε (MOI=50). PKCε activation was initiated by overexpressing caPKCε using adenoviral vector encoding caPKCε mutant (MOI=25). Infection with adenovirus encoding an empty vector (MOI = 25) was used as a negative control. At 45 min after TBHP treatment of RPTC cultures, the oxidant exposure was terminated by washing the monolayer with fresh, warm (37°C) culture medium. Control RPTC cultures were treated with the diluent (dimethyl sulfoxide, 0.1%). RPTC samples were taken at 4 and 24 hours after TBHP exposure to assess mitochondrial functions, cell viability, and changes in protein levels using immunoblotting.
LDH release
Lactate dehydrogenase (LDH) release from RPTC into the culture medium was used as a marker of plasma membrane permeabilization (cell lysis). LDH activity was determined spectrophotometrically at 340 nm by measuring NADH (0.3 mM) oxidation in the presence of 1.8 mM pyruvate as a substrate as described previously (Nowak et al., 2008, 2011).
Apoptosis
Apoptosis was evaluated by measuring phosphatidylserine externalization on the plasma membrane using the annexin V/propidium iodide-binding assay as previously described (Nowak et al., 2003).
Production of Reactive Oxygen Species
The carboxy-derivative of fluorescein, 5-(and-6)-carboxy-2´,7´-dichlorodihydro-fluorescein (carboxy-H2DCFDA), was used to assess oxidant generation in RPTC as described previously.[26] RPTC were loaded with 5 μM carboxy-H2DCFDA followed by treatment with TBHP for 45 minutes. Following incubation, the fluorescence of RPTC monolayers was analyzed by fluorometry. Final results were calculated and presented as % of fluorescence in control RPTC.
State 3 respiration
Oxygen consumption was measured polarographically using a Clark-type electrode as described previously.[25, 44, 50] State 3 respiration in RPTC energized by electron donors to the respiratory complexes I (5 mM glutamate + 5 mM malate), II (10 mM succinate + 0.1 μM rotenone), and IV (1 mM ascorbate + 1 mM N,N,N’,N’-tetramethyl-p-phenylenediamine) was measured at 37°C in digitonin-permeabilized RPTC (0.01% final concentration) in an assay buffer resembling an intracellular electrolyte milieu as described previously.[26, 47, Shaik et al., 2008 50, 51] At the concentration of 0.01%, digitonin permeabilizes RPTC plasma membrane without affecting mitochondrial membranes.[Shaik et al., 2008] State 3 respiration was initiated by addition of 0.4 mM ADP. State 4 respiration was measured following addition of oligomycin (0.6 μg/ml) to RPTC respiring at state 3. Uncoupled respiration was measured by adding FCCP (0.5 μM final concentration) to RPTC respiring at state 3.
ATP production rate and intracellular content
ATP production rate was measured in digitonin-permeabilized RPTC (0.01% final concentration) respiring at state 3 using the method of Borkan et al. (1993), modified as described previously (Nowak et al., 2006). The assay was carried out using 5 mM glutamate + 5 mM malate or 10 mM succinate + 0.1 μM rotenone as the energizing substrates. The reaction was terminated by adding an aliquot of ice-cold perchloric acid (3% final concentration) and the suspension was spun down (15,000×g for 1 min). The supernatant was neutralized to pH 7.5 and analyzed for ATP content using the luciferase method and ATP Bioluminescence Assay Kit HS II (Roche, Mannheim, Germany).
Intracellular ATP content was measured in freshly prepared RPTC lysates using the luciferase method and ATP Bioluminescence Assay Kit HS II as described previously (Nowak et al., 2004).
Mitochondrial membrane potential (ΔΨm)
ΔΨm was assessed by flow cytometry (FACSCalibur; BD Biosciences) as described previously using JC-1, a cationic dye that exhibits potential-dependent accumulation and formation of red fluorescent J-aggregates in mitochondria, which is indicated by the fluorescence shift from green (525 nm) to red (590 nm) (Nowak, 2002; Nowak et al., 2006). Fluorescence was measured using excitation by a 488-nm argon-ion laser. JC-1 monomer (green) and the mitochondrial J-aggregates (red) were detected separately in FL1 (emission, 525 nm) and FL2 (emission, 590 nm) channels, respectively. ΔΨm is presented as the ration of red to green fluorescence intensity.
Isolation of RPTC mitochondria
RPTC were homogenized in the ice-cold isolation buffer (10 mM Hepes, 225 mM mannitol, 75 mM sucrose, 2 mM EGTA, and 0.1% BSA, fatty acid free, pH 7.4). The homogenate was spun down at 1,000 g for 5 min at 4°C and the resulting supernatant was centrifuged at 15,000 g for 15 min at 4°C. The mitochondrial pellet was washed twice using the isolation buffer and centrifuged at 15,000 g for 10 min at 4°C. The final mitochondrial pellet was resuspended in the isolation buffer and used to measure state 3 respiration or in the assay buffer (10 mM KH2PO4, 5 mM MgCl2, pH 7.2) used to determine the activities of respiratory complexes and F0F1-ATPase.
Activity of respiratory complexes
Activities of complexes of the electron transport chain were measured in isolated mitochondria as described previously (Nowak et al., 2011). Complex I (NADH:Ubiquinone Oxidoreductase) activity was assayed spectrophotometrically at 30°C by following the oxidation of NADH (0.25 mM) at 340 nm in the assay buffer (10 mM KH2PO4, 5 mM MgCl2, 0.25% BSA; pH 7.2) containing 62.5 μM ubiquinone, antimycin A (2 μg/ml), and mitochondria. The decrease in absorbance due to NADH oxidation was recorded for 3 min, rotenone (10 μg/ml) added, and the absorbance was recorded for another 2 min. Complex I activity was calculated as the rotenone-sensitive NADH:Ubiquinone Oxidoreductase activity. Complex II (Succinate:Ubiquinone Oxidoreductase) activity was measured at 30°C by following the reduction of dichlorophenolindophenol (0.25 mM) at 590 nm in the presence of 20 mM succinate, antimycin A (2 μg/ml), rotenone (10 μg/ml), 0.25% BSA, and ubiquinone (62.5 μM). Complex III (Ubiquinol:Cytochrome c Oxidoreductase) activity was assessed at 30°C, by following the reduction of cytochrome c (60 μM) at 550 nm in the assay buffer containing rotenone (10 μg/ml), decylubiquinol (50 μM), 0.25% BSA, and KCN (0.24 mM). The increase in absorbance due to the reduction of cytochrome c was recorded in the absence and presence of antimycin A (2 μg/ml). Complex III activity was calculated as the antimycin A-sensitive activity. Complex IV (cytochrome oxidase) activity was assessed by following the oxidation of reduced cytochrome c (90 μM) at 550 nm (30°C) in the assay buffer containing 10% BSA, antimycin A (2 μg/ml) in the presence and absence of KCN (0.24 mM). Complex IV activity was calculated as KCN-sensitive cytochrome oxidase activity.
F0F1-ATPase activity
ATPase activity of the ATP synthase was determined in freshly isolated mitochondria by measuring the release of Pi from ATP by the method of Law et al.(1995) as described previously (Nowak et al., 2011). Each sample was run in the absence and presence of oligomycin (10 μg/ml) and the oligomycin-sensitive ATPase activity of F0F1-ATPase was calculated.
Mitochondrial morphology
RPTC monolayers were loaded with 100 nM MitoTracker Red 580 for 30 minutes at 37°C. The live monolayers were examined under a Zeiss fluorescent microscope (Axioscop) using a water immersion objective (63x) as described previously (Nowak et al., 2011). The images were captured using Zeiss AxioCam digital camera.
Immunoblotting
Phosphorylation and protein levels of PKCε in mitochondria isolated from RPTC were assessed by immunoblot analysis as described previously (Nowak et al., 2004).
Protein concentration was determined using bicinchoninic acid assay with bovine serum albumin as the standard.
Statistical analysis
Data are presented as means ± S.E. and were analyzed for significance by the two-sided Student t test for independent samples or by ANOVA. Multiple means were compared using Fisher’s protected least significance difference (LSD) test with a level of significance of p<0.05. Renal proximal tubular cells isolated from an individual rabbit represented one experiment (n=1) consisting of data obtained from two to ten culture plates.
RESULTS
PKCε translocates to mitochondria in oxidant-injured RPTC
Figure 1 shows that oxidant exposure in RPTC is associated with phosphorylation (indicative of activation) of PKCε shortly after the exposure (Fig. 1A) and translocation of the phosphorylated PKCε (p-PKCε) to mitochondria (Fig. 1B). The levels of phosphorylated (active) PKCε in RPTC decreased at 24 hours after oxidant exposure, but it remained at an elevated level in mitochondria of injured RPTC (Fig. 1A,B). Adenoviral delivery of the constitutively active PKCε mutant (caPKCε) to RPTC resulted in increased levels of total and phosphorylated PKCε in mitochondria (Fig. 1C). TBHP exposure further increased the mitochondrial levels of active PKCε suggesting increased translocation of caPKCε to mitochondria in response to oxidant exposure (Fig. 1C). Overexpressing the inactive PKCε mutant (dnPKCε) in RPTC did not change the mitochondrial levels of PKCε and blocked PKCε translocation to mitochondria after TBHP treatment (Fig. 1C), which suggests that the non-phosphorylatable (permanently inactive) form of PKCε does not localize in mitochondria of RPTC. These data show that oxidant exposure induces activation of PKCε in RPTC and that the active PKCε localizes to mitochondria in oxidant-injured RPTC.
Fig. 1.
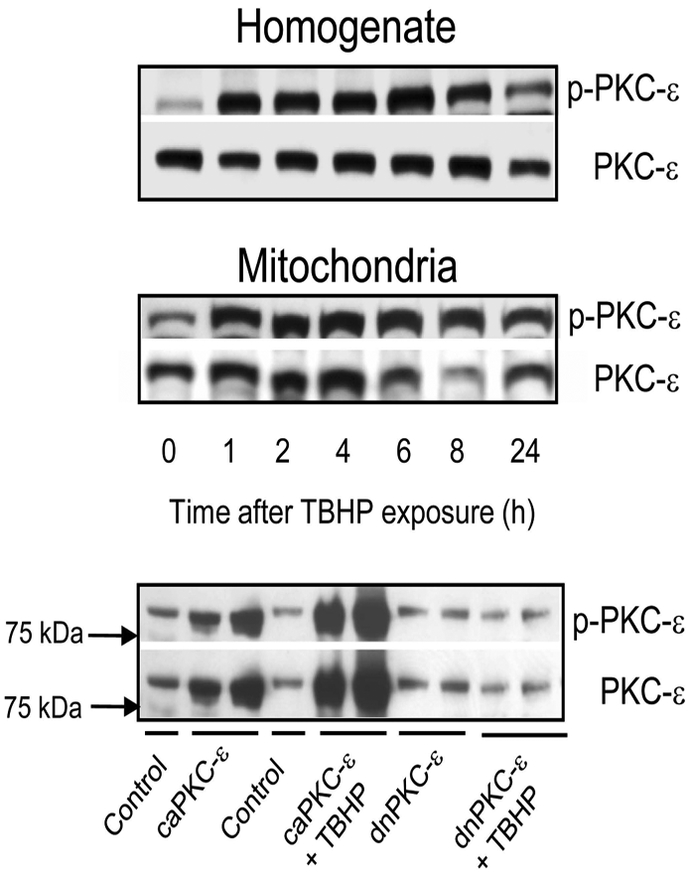
Protein levels of phosphorylated PKCε (p-PKCε) and total PKCε (PKCε) in homogenates (upper panel) and mitochondria (middle panel) isolated from RPTC at different time points after tert-butyl hydroperoxide (TBHP; 0.35 mM, 45 min) exposure. Lower panel: Protein levels of phosphorylated PKCε (p-PKCε) and total PKCε (PKCε) in mitochondria of non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε at 4 hours after TBHP exposure. The blots are representative of 3 independent experiments.
PKCε activation mediates oxidant-induced cell death
TBHP exposure increased LDH release from RPTC (19 ± 3% and 46 ± 6% of total LDH activity at 4 and 24 hours after TBHP exposure vs. 2.5 ± 1% in controls) (Fig. 2A). These data demonstrate the plasma membrane permeabilization in oxidant-injured RPTC indicative of cell death by oncosis. Expressing the constitutively active mutant of PKCε (caPKCε) increased LDH release to 16 ± 1% in non-injured RPTC and exacerbated LDH release to 78 ± 2% in TBHP-injured RPTC (Fig. 2A). Overexpressing the inactive mutant of PKCε (dnPKCε) and adenovirus carrying the empty vector had no effect on LDH release in non-injured RPTC (5.7 ± 1.1%) (Fig. 2A; data not shown). Overexpressing dnPKCε blocked LDH release at 4h and reduced it at 24h following TBHP exposure (Fig. 2A). No apoptosis was observed in TBHP-injured RPTC regardless of PKCε overexpression (Fig. 2B). These data show that PKCε activation mediates oxidant-induced RPTC membrane permeabilization and lysis, and that inhibition of PKCε activation offers protection against oxidant-induced cell death in RPTC.
Fig. 2.
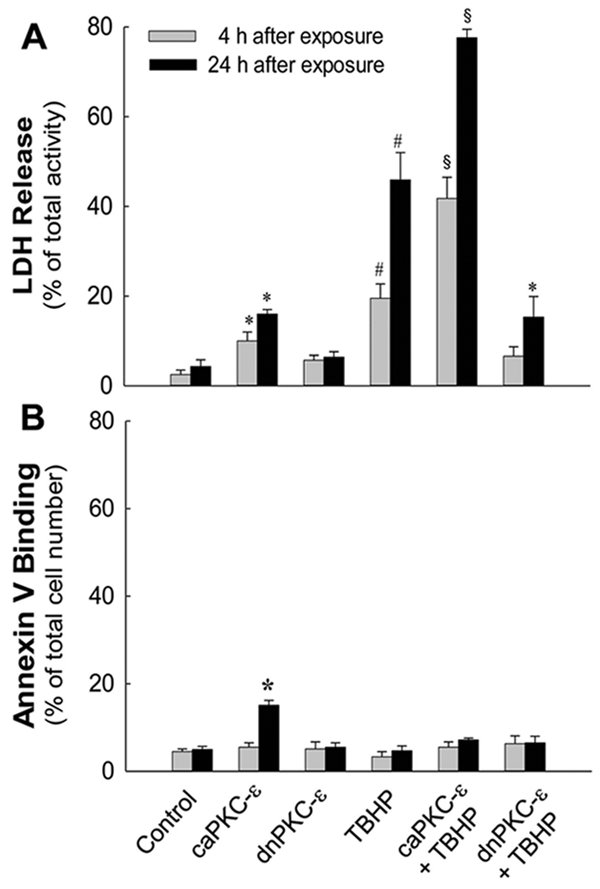
Lactate dehydrogenase (LDH) release (A) and annexin V binding (B) non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε at 4 and 24 hours following TBHP (0.35 mM, 45 min) exposure. RPTC were subjected to adenoviral infection for 24 hours, the adenovirus was removed by aspirating media, and RPTC were incubated for subsequent 24 hours in fresh media prior to the exposure to TBHP. Cells positive for annexin V and negative for propidium iodide were considered apoptotic. Cells positive for propidium iodide and negative for annexin V were considered necrotic. Results are the average ± S. E. of 6–8 independent experiments (RPTC isolations). Values with superscripts are significantly different (P<0.05) from their respective controls at 4 and 24 hours. Values with dissimilar superscripts at a given time point are significantly different from each other.
Inhibition of PKCε prevents oxidant generation
We demonstrated that TBHP exposure induces oxidative stress in RPTC (Nowak et al. 2008) and that sustained activation of PKCε stimulates ROS production in non-injured RPTC (Nowak et al., 2011). Here, we tested whether PKCε activation mediates TBHP-induced generation of ROS in RPTC. Overexpression of caPKCε in non-injured RPTC increased ROS production 1.6-fold whereas TBHP exposure induced a 1.8-fold increase in ROS production in RPTC (Fig. 3). Overexpressing caPKCε had no additional effect on TBHP-induced oxidant production. In contrast, inhibition of PKCε activation by expressing dnPKCε blocked ROS production in RPTC exposed to TBHP (Fig. 3). These data show that PKCε activation mediates ROS generation and that blocking PKCε activation prevents ROS production in oxidant-injured RPTC.
Fig. 3.
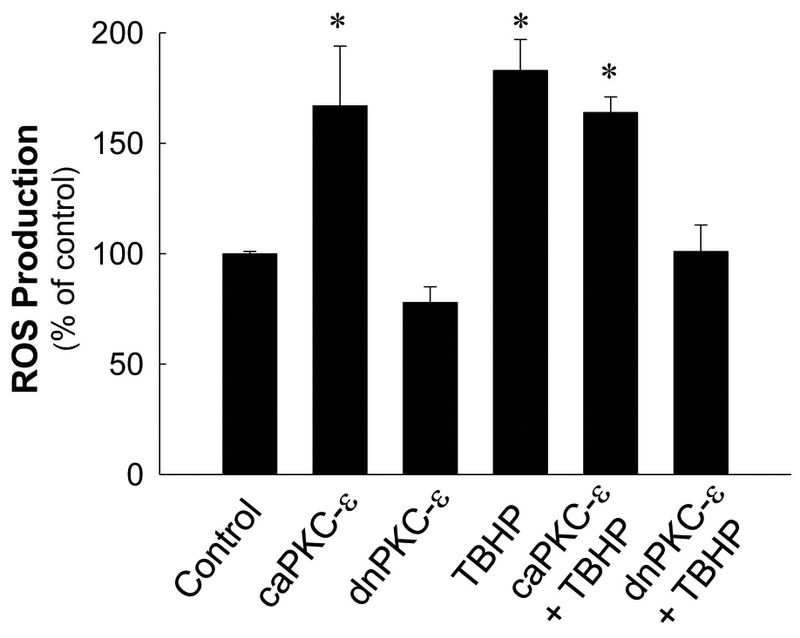
Reactive oxygen species (ROS) production in non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε. RPTC were subjected to adenoviral infection for 24 hours, the adenovirus was removed by aspirating media, and RPTC were incubated in fresh media for subsequent 24 hours prior to the exposure to TBHP. Results are the average ± S.E. of 5 independent experiments (RPTC isolations). * significantly different (p<0.05) from controls.
PKCε activation disrupts integrity of the electron transport chain
Disruptions of mitochondrial electron transport chain are major contributors to the production of superoxide and reactive oxygen species derived from superoxide. We have previously shown that sustained activation of PKCε in non-injured RPTC disrupts the integrity of the respiratory chain.[26] Here, we tested whether PKCε activation is involved in oxidant-induced inhibition of the respiratory chain in injured RPTC. We also assessed which respiratory complexes are targeted by active PKCε. Uncoupled respiration (used as a marker of integrity of the electron transport chain) in TBHP-injured RPTC decreased to 35% of controls following TBHP exposure whereas overexpressing caPKCε in non-injured RPTC decreased uncoupled respiration to 57% of controls (Fig. 4A). Overexpressing caPKCε exacerbated TBHP-induced decreases in uncoupled respiration (to 14% of controls) whereas expressing dnPKCε blocked them (Fig. 4A). Expressing the empty vector had no effects on TBHP-induced decreases in uncoupled respiration (data not shown). These data show that PKCε activation in RPTC decreases electron transport rate through the respiratory chain and that PKCε activation mediates TBHP-induced disruption of respiratory chain. In addition, these data suggest that PKCε activation also mediates oxidant-independent decreases in the electron transfer through the respiratory chain.
Fig. 4.
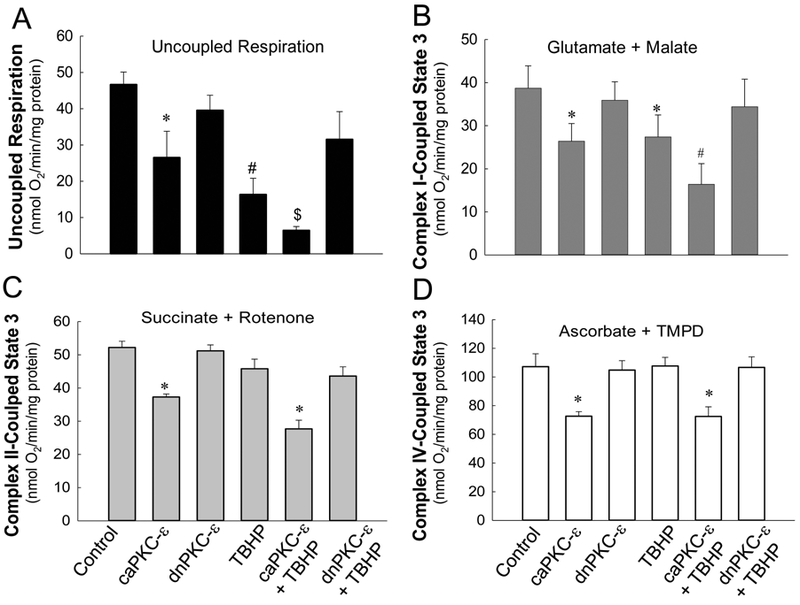
A. Uncoupled respiration in non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε. B-D: State 3 respiration coupled to the oxidation of electron donors to complex I (B), complex II (C), and complex IV (D) of the electron transport chain in non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε. State 3 was measured in digitonin-permeabilized RPTC energized with 5 mM glutamate + 5 mM malate (B), 10 mM succinate + 0.1 μM rotenone (C), and 1 mM ascorbate + 1 mM N,N,N’,N’-tetramethyl-p-phenylenediamine (D) at 4 hours after TBHP (0.35 mM) exposure (45 min). Results are the average ± S.E. of 3–7 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (p<0.05) from controls and from each other.
To identify specific complexes of the respiratory chain that are targeted by PKCε, we assessed state 3 respiration in the presence of different oxidative substrates that enter the respiratory chain through different complexes. TBHP exposure reduced (30% decrease in comparison with controls) state 3 respiration coupled to oxidation of substrates through complex I (Fig. 4B). Expressing caPKCε in non-injured RPTC decreased complex I-coupled state 3 respiration by 32% in comparison with controls (Fig. 4B). Furthermore, expressing caPKCε exacerbated (58% decrease in comparison with controls) whereas expressing dnPKCε prevented TBHP-induced decreases in complex I-coupled state 3 respiration in injured RPTC (Fig. 4B). TBHP had no effect on state 3 respiration coupled to oxidation of substrates through complexes II and IV (Fig. 4C-D). However, activation of PKCε by overexpressing caPKCε decreased state 3 respiration coupled to complexes II and IV in both non-injured and TBHP-injured RPTC (Fig. 4C-D). Expressing the empty vector had no effect on state 3 respiration in non-injured and injured RPTC (data not shown).
State 4 respiration was not affected by TBHP exposure and/or PKCε activation status (data not shown). TBHP exposure decreased the respiratory control ratio (the ratio of state 3/state 4 respiration; RCR) by 46% (2.4 ± 0.1 vs. 4.5 ± 0.3 in TBHP-treated and control RPTC, respectively). Inhibition of PKCε activation restored RCR in TBHP injured RPTC (Fig. 6A).
Fig. 6.
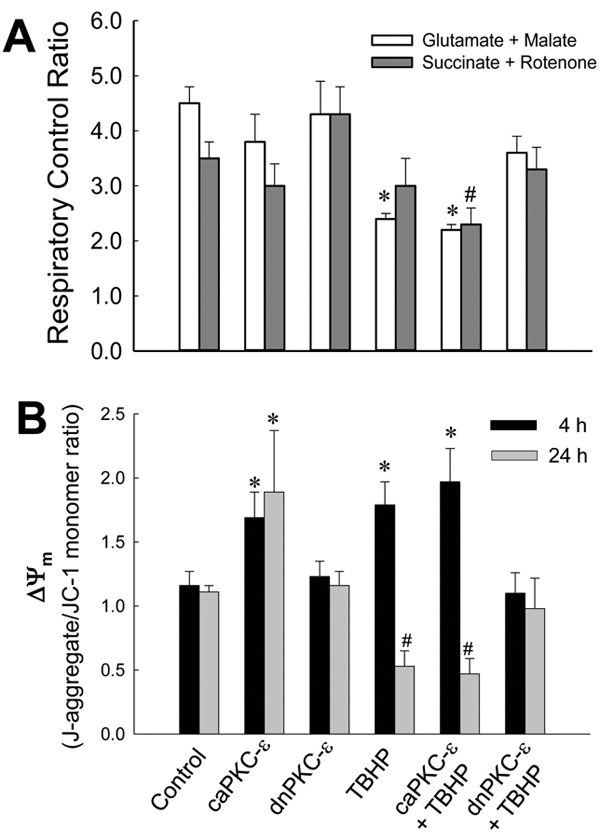
Respiratory control ratio (A) and mitochondrial membrane potential (ΔΨm, B) in non-injured and TBHP-injured RPTC expressing caPKCε or dnPKCε mutants of PKCε at 4 hours (A) and 4 and 24 hours following TBHP-induced injury. ΔΨm is expressed as the ratio of J-aggregate (red fluorescence) to monomeric form of JC-1 (green fluorescence). RPTC were subjected to adenoviral infection for 24 hours, the adenovirus was removed by aspirating media, and RPTC were incubated in fresh media for subsequent 24 hours prior to the exposure to TBHP. Results are the average ± S.E. of 6–9 independent experiments (RPTC isolations). Values with dissimilar superscripts are significantly different (p<0.05) from their respective controls and from each other.
These results demonstrate that 1) PKCε activation decreases oxidation of substrates coupled to respiratory complexes I, II, and IV, 2) PKCε activation mediates oxidant-induced decreases in oxidation of substrates through the respiratory complex I, and 3) activation of PKCε exacerbates inhibitory effects of oxidant-injury on mitochondrial respiration coupled to complex I.
PKCε activation mediates inhibition of complexes I and IV and oxidant-induced decreases in activity of complex I.
TBHP exposure decreased the activity of complex I by 40%, but had no effect on activities of other complexes (Fig. 5). Overexpressing caPKCε exacerbated (81% decrease) and expressing dnPKCε blocked TBHP-induced decreases in the activity of complex I (Fig. 5A). Furthermore, sustained activation of PKCε by overexpressing caPKCε decreased the activity of complex IV in non-injured and TBHP-injured RPTC (49% and 44% decreases, respectively) (Fig. 5D). Expressing caPKCε had no effect on activities of complexes II and III (Fig. 5B-C). Overexpressing dnPKCε had no effect on activities of complexes of the respiratory chain (Fig. 5A-D). These results demonstrate that transient activation of PKCε mediates oxidant-induced decreases in activity of complex I and that sustained activation of PKCε exacerbates TBHP effects on activities of complexes I and IV.
Fig.5.
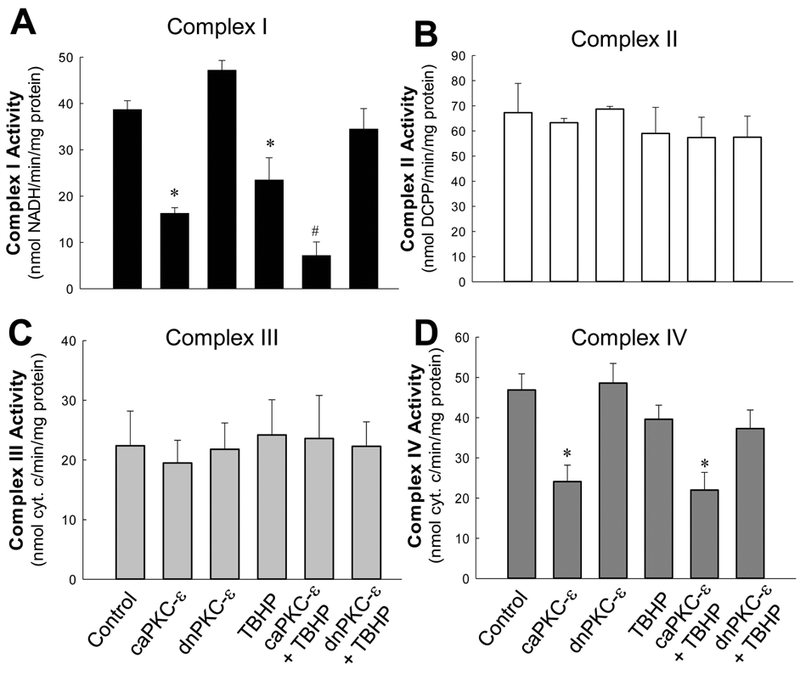
Activities of NADH:ubiquinone oxidoreductase (complex I, A), succinate:ubiquinone oxidoreductase (complex II, B), ubiquinol:cytochrome c oxidoreductase (complex III, C) and cytochrome oxidase (complex IV, D) in mitochondria isolated from control and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKC-ε at 4 hours after TBHP (0.35 mM) exposure (45 min). RPTC were subjected to adenoviral infection for 24 hours, the adenovirus was removed by aspirating media, and RPTC were incubated in fresh media for subsequent 24 hours prior to the exposure to TBHP. Results are the average ± S.E. of 3–9 independent experiments (mitochondrial isolations). Values with dissimilar superscripts are significantly different (p<0.05) from controls and from each other.
PKCε activation mediates oxidant-induced changes in mitochondrial membrane potential (ΔΨm)
J-aggregate/JC-1 monomer ratio in RPTC was used as a marker of ΔΨm. Active PKCε levels in mitochondria of TBHP-injured RPTC were the highest at 4h after injury (Fig. 1). This event was accompanied by 1.8-fold increase in J-aggregate/JC-1 monomer ratio, indicative of mitochondrial hyperpolarization, and decreased RCR (Fig. 6A, B). Mitochondrial hyperpolarization was followed by decreases in ΔΨm and RCR at 24h after TBHP exposure (Fig. 6B, data not shown). Blocking PKCε activation prevented TBHP-induced increases in ΔΨm at 4h and maintained ΔΨm at control levels at 24h after injury (Fig. 6B). This protective effect was associated with improved state 3 respiration, complex I activity, and viability in TBHP-injured RPTC (Figs. 2, 4 and 5). In contrast, persistent activation of PKCε induced hyperpolarization of the mitochondrial membrane in non-injured RPTC (Fig. 6B). Thus, PKCε activation and translocation to mitochondria mediates the hyperpolarization of the mitochondrial membrane and ΔΨm increases whereas PKCε inhibition blocks these changes after oxidant exposure in RPTC.
PKCε activation mediates oxidant-induced decreases in ATP production and content
Because the data show that PKCε activation mediates mitochondrial dysfunction and cell death in RPTC, we tested whether PKCε is also involved in the decreases in ATP production and content in oxidant-injured RPTC. TBHP exposure decreased ATP production to 60% of controls whereas sustained activation of PKCε decreased ATP production to 55% of controls (Fig. 7A). Blocking PKCε activation by expressing dnPKCε had no effect on ATP production in non-injured RPTC, but it ameliorated the decreases in ATP production in TBHP-injured RPTC (Fig. 7A). In contrast, ATP production remained decreased (64% of controls) at 24h after TBHP exposure in RPTC overexpressing caPKCε, whereas it returned to control levels in RPTC expressing the empty vector (data not shown). Thus, these data show that the active PKC-ε is involved in oxidant-induced decreases in ATP production and blocks recovery of ATP production in oxidant-injured RPTC.
Fig. 7.
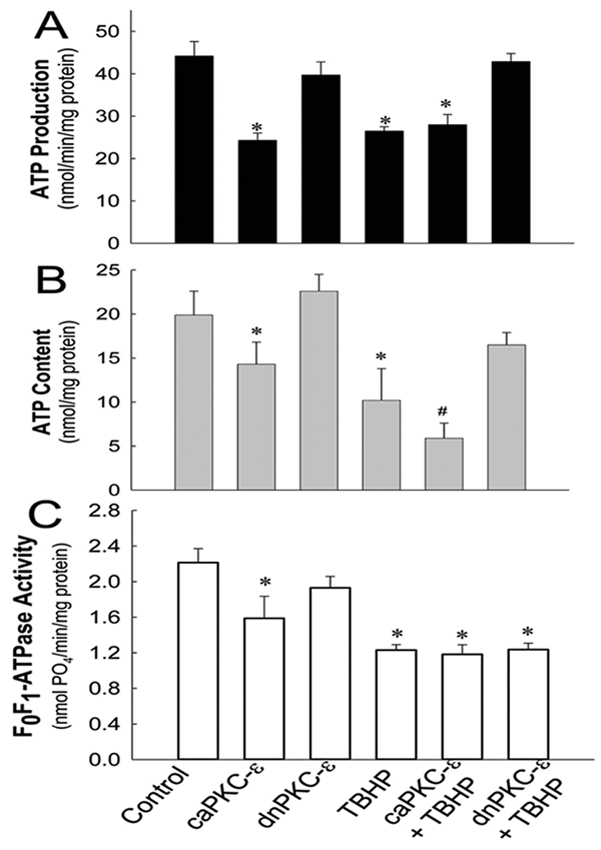
ATP production rate (A), intracellular ATP content (B) and F0F1-ATPase activity (C) in non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε at 4 hours after TBHP (0.35 mM) exposure (45 min). RPTC were subjected to adenoviral infection for 24 hours, the adenovirus was removed by aspirating media, and RPTC were incubated in fresh media for subsequent 24 hours prior to the exposure to TBHP. ATP production was measured in the presence of 2 mM ADP in digitonin-permeabilized RPTC energized with 5 mM glutamate and 5 mM malate. Results are the average ± S.E. of 5–7 independent experiments (RPTC isolations). Values with dissimilar superscripts on a given day are significantly different (P<0.05) from each other.
TBHP exposure decreased intracellular ATP content to 50% and 41% of controls at 4h and 24h, respectively, after TBHP exposure (Fig. 7B, data not shown). Overexpressing caPKCε decreased ATP content in non-injured RPTC and exacerbated the decreases in ATP content in TBHP-injured cells (Fig. 7B). In contrast, blocking PKCε activation improved ATP content at 4h (78% of controls) after oxidant exposure and restored it at 24h after injury (Fig. 7B, data not shown). These results demonstrate that PKCε activation mediates decreases in ATP content in oxidant-injured RPTC.
Subsequently, we tested whether F0F1-ATPase is a target of PKCε and if the decreases in ATP production and content are mediated by PKCε-dependent reduction in F0F1-ATPase activity in oxidant-injured RPTC. F0F1-ATPase activity in oxidant-injured RPTC declined to 55% of controls whereas activation of PKCε reduced F0F1-ATPase activity to 72% of controls (Fig. 7C). Neither inhibition nor activation of PKCε had any effect on TBHP-induced decreases in F0F1-ATPase activity (Fig. 7C). These data demonstrate that PKCε does not mediate decreases in F0F1-ATPase activity in oxidant-injured RPTC. Together, our data show that PKCε mediates mitochondrial dysfunction and decreases in ATP production in oxidant-injured RPTC through a mechanism independent of changes in F0F1-ATPase activity.
PKCε activation mediates oxidant-induced mitochondrial fragmentation
Morphological examination of control RPTC revealed a network of elongated mitochondria distributed throughout the cytoplasm with a higher density of mitochondria around the nucleus (Fig. 8A). Exposure to TBHP resulted in fragmentation of mitochondria (Fig. 8B). Expressing caPKCε in non-injured RPTC produced short, punctuated, and brightly stained mitochondria that often concentrated around the nucleus (Fig. 8C). These mitochondria had similar morphology as those in TBHP-injured RPTC (Fig. 8D). Expressing caPKCε exacerbated TBHP-induced fragmentation of mitochondria, disrupted their network and subcellular distribution, and produced irregular clusters of fragmented mitochondria (Fig. 8D). In contrast, expressing dnPKCε did not affect mitochondrial morphology in non-injured RPTC (Fig. 8E) and prevented TBHP-induced mitochondrial fragmentation and maintained the mitochondrial network similar to that observed in controls (Fig. 8F). These data demonstrate that PKCε activation mediates mitochondrial fragmentation induced by oxidant exposure in RPTC.
Fig. 8.
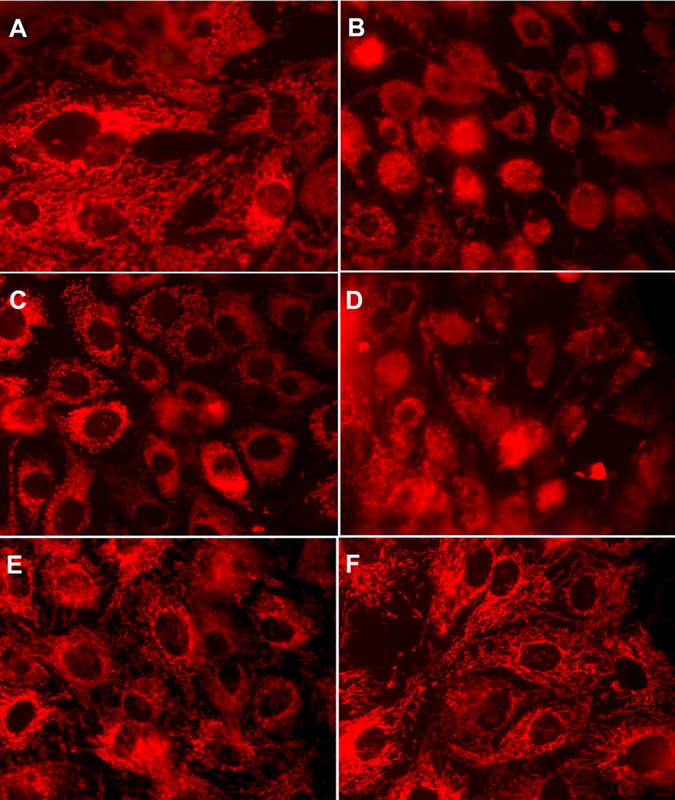
Mitochondrial morphology in non-injured and TBHP-injured RPTC expressing the constitutively active (caPKCε) or inactive (dnPKCε) mutants of PKCε at 4 hours after TBHP (0.35 mM) exposure (45 min). A: Controls. B: TBHP-injured RPTC. C: non-injured RPTC expressing caPKCε. D: TBHP-injured RPTC expressing caPKCε. E: non-injured RPTC expressing dnPKCε. F: TBHP-injured RPTC expressing dnPKCε. RPTC were subjected to adenoviral infection for 24 hours, the adenovirus was removed by aspirating media, and RPTC were incubated in fresh media for subsequent 24 hours prior to the exposure to TBHP. Representative images for each experimental group are shown. Original magnification, x630.
DISCUSSION
It has been well established that PKCε activation is involved in the mechanisms of cardioprotection and neuroprotection against injury caused by ischemia-reperfusion and hypoxia (Liu et al., 1999; Ping et al., 1999a; Gray et al., 1997; Mackay and Mochly-Rosen, 2001; Budas and Mochly-Rosen, 2007; Di-Capua et al., 2003). However, PKCε activation is not protective in all cell types. In contrast to the protective actions of PKCε in cardiac, neuronal, and cancer cells, activation of PKCε mediates injury in epithelial cells that carry out trans-epithelial transport functions. This includes injury induced by nitric oxide to intestinal epithelial mucosal cells, vasopressin-stimulated decreases in active Na+ transport in renal cortical collecting ducts, and injury to RPTC (DeCoy et al., 1995; Nowak et al., 2004; Tepperman et al., 1999). Thus, the effect of PKCε activation on specific cellular functions and cell survival appears to be cell type-dependent. Previously, we have shown that sustained activation of PKCε in RPTC induces accumulation of PKCε in mitochondria, mitochondrial dysfunction and fragmentation, energy deficits, and cell death (Nowak et al., 2011). We have also demonstrated that PKC regulates RPTC injury and recovery after injury and that PKCε activation is involved in oxidant-induced changes in cellular respiration, active Na+-transport, and Na+/K+-ATPase activity in RPTC (Nowak et al., 2004). Inhibition of PKCε activation accelerates recovery of active Na+-transport and Na+/K+-ATPase activity following oxidant injury in RPTC (Nowak et al., 2004). Those studies utilized pharmacological inhibitors and activators of PKCε that are also known to affect other protein kinases and signaling pathways. The present study utilized constitutively active and inactive mutants of PKCε to test whether oxidant-induced mitochondrial dysfunction and fragmentation, energy deficits, and RPTC death are mediated by PKCε.
Active PKCε associates with RPTC mitochondria. Activation and association of PKCε with mitochondria in TBHP-injured RPTC is transient, subsides within 24 h after oxidant exposure, and is followed by the recovery of cellular respiration and ATP production (Nowak et al., 1998, 2004). Therefore, we tested whether the electron transport chain, ΔΨm, and/or F0F1-ATPase (three major components required for oxidative phosphorylation of ADP) are the targets of PKCε. Our previous report shows that the respiratory chain is a target of active PKCε in non-injured RPTC (Nowak et al., 2011. This study shows that TBHP-induced decreases in uncoupled respiration (a marker of the respiratory chain’s integrity) are mediated by PKCε and that specific inhibition of PKCε improves respiratory chain’s integrity and mitochondrial maximum respiration in injured RPTC. Transient activation of PKCε mediates TBHP-induced decreases in oxidation of substrates through complex I, but not oxidation through other complexes. The decreases in respiration are due to the inhibitory effect of active PKCε on the activity of complex I. They are abrogated by inhibition of PKCε and exacerbated by activation of PKCε (overexpression of caPKCε). PKCε-mediated decrease in complex I activity is not due to reduced protein levels of complex I in the mitochondria because sustained activation of PKCε does not change the levels of crucial subunits of responsible for the assembly and function of complex I (Nowak et al., 2011). It is still unknown whether PKCε phosphorylates complex I or has an indirect effect on complex I, for example, by decreasing substrate availability for complex I. Proteomic analysis did not reveal any evidence of phosphorylation of complex I by PKCε (data not shown). However, the large number of peptides making up complex I (46 peptides) and instability of phosphorylation residues of certain sites during sample processing and proteomic analysis makes this analysis difficult and the negative results of the analysis do not preclude an existence of such phosphorylation(s) in the living cell. Nevertheless, this is the first report demonstrating functional data documenting that PKCε mediates oxidant-induced decreases in complex I activity.
In contrast to transient activation of PKCε that reduces activity of complex I, sustained activation of the kinase by overexpressing its active mutant (caPKCε) reduces substrate oxidation through complexes I, II and IV, and decreases activities of complexes I and IV (cytochrome oxidase). Furthermore, active PKCε prevents recovery of mitochondrial function after oxidant injury. Thus, we conclude that transient activation of PKCε mediates disruption of the respiratory chain’s integrity in TBHP-injured RPTC through decreasing the activity of complex I and reducing oxidation of substrates through complex I. In contrast, sustained activation of PKCε leads to the inhibition of complex IV in addition to inhibition of complex IV and prevents recovery of their activity after oxidant injury.
Translocation of protons from the mitochondrial matrix into the intermembrane space by respiratory complexes generates ΔΨm and the proton-motive force, both of which serve as the driving force for F0F1-ATPase rotation and ATP synthesis from ADP. Oxidant-induced PKCε activation decreased the activity of F0F1-ATPase, ATP production, and ATP content in RPTC. Inhibition of F0F1-ATPase reduces utilization of the proton-motive force and leads to proton accumulation in the intermembrane space and increases in ΔΨm. Electrochemical gradient becomes too great for the complexes to pump protons against their concentration gradient and activities of complexes, electron flow, and respiration decrease. This is often followed by superoxide and ROS generation and subsequent collapse of ΔΨm. Dysfunction of complex I has particularly profound effects on ΔΨm as complex I provides approximately 40% of the proton-motive force for ATP synthesis (Brandt, 2006). Our results show decreases in F0F1-ATPase activity, mitochondrial hyperpolarization, and ROS generation at 4h followed by decreases in ΔΨm at 24h after TBHP exposure. TBHP-induced hyperpolarization of mitochondria was accompanied by decreases in complex I activity, state 3 respiration, ATP production, and ATP content. Thus, decreases in F0F1-ATPase activity could have been the initiating factor leading to dysfunction of the respiratory chain. However, increases in ROS production and ΔΨm and decreases in respiration, complex I activity, and ATP production were alleviated by PKCε inhibition whereas the decreases in F0F1-ATPase activity were independent of PKCε. These facts suggest that F0F1-ATPase inhibition is not the initiating mechanism of TBHP- and PKCε-mediated mitochondrial dysfunction and that the mitochondrial permeability pore, voltage-dependent anion channel (VDAC) mediating the transport of oxidative substrates, and/or the respiratory chain are the targets of active PKCε in TBHP-injured RPTC. It has been shown that PKCε associates with the cardiac mitochondrial permeability transition pore (MPTP) and inhibits pore opening (Baines et al., 2003).
Our data show that TBHP injury and PKCε activation are associated with mitochondrial fragmentation. In contrast, PKCε inhibition prevents this event suggesting that PKCε activation mediates mitochondrial fragmentation in TBHP-injured RPTC. Recently, we have shown that PKCε activation induces accumulation of the dynamin-related protein DRP1 in RPTC mitochondria and that this event precedes mitochondrial fragmentation (Nowak et al., 2011). DRP1 localization to mitochondria is required for mitochondrial fission and occurs after stress associated with apoptosis (Smirnowa et al., 2001; Van der Bliek, 2009). Interestingly, TBHP exposure in RPTC caused oncosis and not apoptosis, which demonstrates that PKCε-dependent mitochondrial fission occurs during oncosis as well. Thus, PKCε regulates mitochondrial morphology and dynamics in oxidant-injured RPTC.
TBHP exposure produces RPTC oncosis, which is mediated by PKCε activation. Blocking PKCε activation by expressing dnPKCε prevents PKCε association with mitochondria, reduces mitochondrial dysfunction and fragmentation, and decreases RPTC death. In contrast, overexpressing caPKCε exacerbates mitochondrial dysfunction and RPTC death. Interestingly, overexpressing caPKCε using the same adenoviral vector protects endothelial cells against apoptosis (Steinberg et al., 2007). The role of PKCε activation in mitochondrial dysfunction and RPTC injury remains in contrast with the protective function of PKCε in cardiac tissue (Rathore et al., 2006; Liu et al., 1999; Ping et al., 1999a, 2001, 2002; Jaburek et al., 2006; Korzik et al., 2007; Liu et al., 2002; McCarthy et al. 2005). Several targets of PKCε have been identified in cardiomyocytes to explain PKCε-mediated cardioprotection, including cardiac Ca2+ and Na2+ channels (Hu et al., 2000; Xiao et al., 2001), mitoKATP channels (Jaburek et al., 2006; Liu et al., 2002; Zhang et al., 2005; Ohnuma et al., 2002), adenine nucleotide translocase, VDAC, cytochrome oxidase and MPTP (Baines et al., 2003; Ogbi et al., 2004, 2006; Guo et al., 2007; McCarthy et al., 2005). Cardiomyocytes originate from mesodermal cells and are excitable cells. RPTC are epithelial and non-excitable cells whose main function is trans-epithelial transport. We speculate that PKCε targets different mitochondrial proteins in RPTC or the phosphorylation of the same targets by PKCε has different outcomes in RPTC mitochondria. We hypothesize that VADC is one of the likely targets for PKCε in RPTC mitochondria. Phosphorylation of this channel could be carried out during a transient translocation and association of PKCε with mitochondria, such as observed after the exposure of RPTC to TBHP, and would not require importing PKCε into the mitochondrial matrix. Phosphorylation induces VDAC closure, which leads to reduced import of substrates oxidized by the citric acid cycle and the complexes of the electron transport chain. Our unpublished data show that the deletion of VDAC1 reduces the electron transport rate, complex I activity, and ATP content in renal tubular cells (data not shown). We speculate that overexpression of caPKCε results in persistent association of active PKCε with mitochondria, possibly and import of caPKCε into this organelle, which leads to decreases in activities of complex I and complex IV. This further limits the ability of mitochondria to maintain ATP production and exacerbates mitochondrial dysfunction and RPTC injury.
In conclusion, our study demonstrates that PKCε activation in RPTC mediates mitochondrial dysfunction, ROS production, energy deficits, mitochondrial fragmentation, and oxidant-induced cell death. The respiratory chain, specifically, complex I and ΔΨm are major mitochondrial targets of PKCε and their disruption leads to decreases in ATP production and content in oxidant-injured RPTC. Persistent activation of PKCe and its association with mitochondria exacerbates dysfunction of the electron transport chain by inhibiting complexes I and IV of the chain. In contrast to cardiomyocytes, PKCε activation in RPTC exacerbates injury and PKCε inhibition offers protection against RPTC injury by improving mitochondrial function, decreasing energy deficits, and increasing cell survival.
ACKNOWLEDGEMENTS
This work was supported by a grant from National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, DK59558. UAMS Translational Research Institute supported by the National Institutes of Health National Center for Research Resources grant UL1 RR029884 provided partial funding for Flow Cytometry Core at UAMS.
We thank Dr. Peipei Ping (University of California at Los Angeles; Los Angeles, CA) for providing an aliquot of adenovirus carrying the dominant negative PKCε mutant cDNA and Dr. Allen M. Samarel (Loyola University Medical Center, Maywood, IL) for providing an aliquot of adenovirus carrying the constitutively active PKCε mutant cDNA. Adenoviral vector carrying the constitutively active PKCε (caPKCε) was constructed using caPKCε cDNA kindly provided by Drs. Peter Parker and Peter Sugden, Imperial College London (London, UK).
Footnotes
The authors declare no conflict of interest.
REFERENCES
- Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. 2002. Mitochondrial PKCε and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCε-MAPK interactions and differential MAPK activation in PKCε-induced cardioprotection. Circ Res 90:390–397. [DOI] [PubMed] [Google Scholar]
- Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. 2003. Protein kinase Cε interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92:873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkan SC, Emami A, Schwartz JH. 1993. Heat stress protein-associated cytoprotection of inner medullary collecting duct cells from rat kidney. Am J Physiol 265:F333–41. [DOI] [PubMed] [Google Scholar]
- Brandt U 2006. Energy Converting NADH: Quinone Oxidoreductase (Complex I). Annu Rev Biochem 75:69–92. [DOI] [PubMed] [Google Scholar]
- Budas GR, Mochly-Rosen D. 2007. Mitochondrial protein kinase Cε (PKCε): emerging role in cardiac protection from ischaemic damage. Biochem Soc Trans 35:1052–1054. [DOI] [PubMed] [Google Scholar]
- Budas GR, Disatnik M, Chen C, Mochly-Rosen D. 2010. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase Cε (PKCɛ) knockout mice. J Mol Cell Cardiol 48:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoy DL, Snapper JR, Breyer MD. 1995. Anti-sense DNA down-regulates proteins kinase C-ε and enhances vasopressin-stimulated Na+ absorption in rabbit cortical collecting duct. J Clin Invest 95:2749–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di-Capua N, Sperling O, Zoref-Shani E. 2003. Protein kinase C-ε is involved in the adenosine-activated signal transduction pathway conferring protection against ischemia-reperfusion injury in primary rat neuronal cultures. J Neurochem 84:409–412. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R, Jaken S. 2000. Protein kinase C signaling and oxidative stress. Free Radic Biol Med 28:1349–1361. [DOI] [PubMed] [Google Scholar]
- Gray MO, Karliner JS, Mochly-Rosen D. 1997. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem 272:30945–30951. [DOI] [PubMed] [Google Scholar]
- Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. 2004. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C ε. J Biol Chem 279:3596–3604. [DOI] [PubMed] [Google Scholar]
- Guo D, Nguyen T, Ogbi M, Tawfik H, Ma G, Yu Q, Caldwell RW, Johnson JA. 2007. Protein kinase C-ε coimmunoprecipitates with cytochrome oxidase subunit IV and is associated with improved cytochrome-c oxidase activity and cardioprotection. Am J Physiol Heart Circ Physiol 293:H2219–30. [DOI] [PubMed] [Google Scholar]
- Hu K, Mochly-Rosen D, Boutjdir M. 2000. Evidence for functional role of epsilonPKC isozyme in the regulation of cardiac Ca(2+) channels. Am J Physiol Heart Circ Physiol 279:H2658–64. [DOI] [PubMed] [Google Scholar]
- Jaburek M, Costa ADT, Burton JR, Costa CL, Garlid KD. 2006. Mitochondrial PKCε and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ Res 99:878–883. [DOI] [PubMed] [Google Scholar]
- Korzick DH, Kostyak JC, Hunter JC, Saupe KW. 2007. Local delivery of PKCε-activating peptide mimics ischemic preconditioning in aged hearts through GSK-3β but not F1-ATPase inactivation. Am J Physiol Heart Circ Physiol 293:H2056–2063. [DOI] [PubMed] [Google Scholar]
- Koyner JL, Sher Ali R, Murray PT. 2008. Antioxidants. Do they have a place in the prevention or therapy of acute kidney injury? Nephron Exp Nephrol 109:e109–17. [DOI] [PubMed] [Google Scholar]
- Law RH, Manon S, Devenish RJ, Nagley P. 1995. ATP synthase from Saccharomyces cerevisiae. Methods Enzymol 260:133–163. [DOI] [PubMed] [Google Scholar]
- Liu GS, Cohen MV, Mochly-Rosen D, Downey JM. 1999. Protein kinase C-ε is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol 31:1937–1948. [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang HY, Zhu X, Shao Z, Yao Z. 2002. Preconditioning blocks cardiocyte apoptosis: role of K(ATP) channels and PKC-ε. Am J Physiol Heart Circ Physiol 282:H1380–6. [DOI] [PubMed] [Google Scholar]
- Mackay K, Mochly-Rosen D. 2001. Localization, anchoring, and functions of protein kinase C isozymes in the heart. J Mol Cell Cardiol 33:1301–1307. [DOI] [PubMed] [Google Scholar]
- McCarthy J, McLeod CJ, Minners J, Essop MF, Ping P, Sack MN. 2005. PKCε activation augments cardiac mitochondrial respiratory post-anoxic reserve - a putative mechanism in PKCε cardioprotection. J Mol Cell Cardiol 38:697–700. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D, Wu G, Hahn H, Osinska H, Liron T, Lorenz JN, Yatani A, Robbins J, Dorn GW 2nd. 2000. Cardiotrophic effects of protein kinase Cε: analysis by in vivo modulation of PKCε translocation. Circ Res 86:1173–1179. [DOI] [PubMed] [Google Scholar]
- Nowak G, Aleo MD, Morgan JA, Schnellmann RG. 1998. Recovery of cellular functions following oxidant injury. Am J Physiol 274:F509–15. [DOI] [PubMed] [Google Scholar]
- Nowak G, Price PM, Schnellmann RG. 2003. Lack of a functional p21WAF1/CIP1 gene accelerates caspase-independent apoptosis induced by cisplatin in renal cells. Am J Physiol Renal Physiol 285:F440–50. [DOI] [PubMed] [Google Scholar]
- Nowak G, Schnellmann RG. 1996. L-ascorbic acid regulates growth and metabolism of renal cells: improvements in cell culture. Am J Physiol 271:C2072–80. [DOI] [PubMed] [Google Scholar]
- Nowak G, Schnellmann RG. 1995. Improved culture conditions stimulate gluconeogenesis in primary cultures of renal proximal tubule cells. Am J Physiol 268:C1053–61. [DOI] [PubMed] [Google Scholar]
- Nowak G 2003. Protein kinase C mediates repair of mitochondrial and transport functions after toxicant-induced injury in renal cells. J Pharmacol Exp Ther 306:157–165. [DOI] [PubMed] [Google Scholar]
- Nowak G 2002. Protein kinase C-α and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem 277:43377–43388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Bakajsova D, Clifton GL. 2004. Protein kinase C-ε modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am J Physiol Renal Physiol 286:F307–16. [DOI] [PubMed] [Google Scholar]
- Nowak G, Clifton GL, Bakajsova D. 2008. Succinate ameliorates energy deficits and prevents dysfunction of complex I in injured renal proximal tubular cells. J Pharmacol Exp Ther 324:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Clifton GL, Godwin ML, Bakajsova D. 2006. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol 291:F840–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Bakajsova D, Samarel AM. 2011. Protein kinase C-ε activation induces mitochondrial dysfunction and fragmentation in renal proximal tubules. Am J Physiol Renal Physiol 301:F197–F208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogbi M, Chew CS, Pohl J, Stuchlik O, Ogbi S, Johnson JA. 2004. Cytochrome c oxidase subunit IV as a marker of protein kinase Cε function in neonatal cardiac myocytes: implications for cytochrome c oxidase activity. Biochem J 382:923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogbi M, Johnson JA. 2006. Protein kinase Cε interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem J 393:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Gogvadze V, Zhivotovsky B. 2007. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183. [DOI] [PubMed] [Google Scholar]
- Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, Wu W, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R. 2002. Formation of protein kinase C(ε)-Lck signaling modules confers cardioprotection. J Clin Invest 109:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafonova Z, Bolli R. 1999. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res 84:587–604. [DOI] [PubMed] [Google Scholar]
- Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y, Clerk A, Sugden P, Han J, Bolli R. 1999. PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Physiol 277:H1771–85. [DOI] [PubMed] [Google Scholar]
- Ping P, Zhang J, Pierce WM Jr, Bolli R. 2001. Functional proteomic analysis of protein kinase C ε signaling complexes in the normal heart and during cardioprotection. Circ Res 88:59–62. [DOI] [PubMed] [Google Scholar]
- Rathore R, Zheng Y, Li X, Wang Q, Liu Q, Ginnan R, Singer HA, Ho Y, Wang Y. 2006. Mitochondrial ROS-PKCε signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem Biophys Res Commun 351:784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore R, Zheng Y, Niu C, Liu Q, Korde A, Ho Y, Wang Y. 2008. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCɛ signaling axis in pulmonary artery smooth muscle cells. Free Rad Biol Med 45:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik ZP, Fifer EK, Nowak G. 2007. Protein kinase B/Akt modulates nephrotoxicant-induced necrosis in renal cells. Am J Physiol Renal Physiol 292:F292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland D, van der Bliek AM. 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg R, Harari OA, Lidington EA, Boyle JJ, Nohadani M, Samarel AM, Ohba M, Haskard DO, Mason JC. 2007. A protein kinase Cε-anti-apoptotic kinase signaling complex protects human vascular endothelial cells against apoptosis through induction of Bcl-2. J Biol Chem 282:32288–32297. [DOI] [PubMed] [Google Scholar]
- Strait JB, Samarel AM. 2000. Isoenzyme-specific protein kinase C and c-Jun N-terminal kinase activation by electrically stimulated contraction of neonatal rat ventricular myocytes. J Mol Cell Cardiol, 32:1553–1566. [DOI] [PubMed] [Google Scholar]
- Tepperman BL, Chang Q, Soper BD. 1999. The involvement of protein kinase C in nitric oxide-induced damage to rat isolated colonic mucosal cells. Br J Pharmacol 128:1268–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Bliek AM. 2009. Fussy mitochondria fuse in response to stress. The EMBO J 28:1533–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Ways DK, Parker PJ, Owen MJ. 1993. Activity of both Raf and Ras is necessary for activation of transcription of the human T cell receptor beta gene by protein kinase C, Ras plays multiple roles. J Biol Chem 268:17975–17982. [PubMed] [Google Scholar]
- Xiao GQ, Qu Y, Sun ZQ, Mochly-Rosen D, Boutjdir M. 2001. Evidence for functional role of εPKC isozyme in the regulation of cardiac Na(+) channels. Am J Physiol Cell Physiol 2001; 281:C1477–86. [DOI] [PubMed] [Google Scholar]