

ABSTRACT
Activation of the atrial natriuretic signaling pathway is intrinsic to the pathological responses associated with a range of cardiovascular diseases that stress the heart, especially those involved in sustained cardiac pressure overload which induces hypertrophy and the pathological remodeling that frequently leads to heart failure. We identify transient receptor potential cation channel, subfamily V, member 1, as a regulated molecular component, and therapeutic target of this signaling system. Data show that TRPV1 is a physical component of the natriuretic peptide A, cGMP, PKG signaling complex, interacting with the Natriuretic Peptide Receptor 1 (NPR1), and upon binding its ligand, Natriuretic Peptide A (NPPA, ANP) TRPV1 activation is subsequently suppressed through production of cGMP and PKG mediated phosphorylation of the TRPV1 channel. Further, inhibition of TRPV1, with orally delivered drugs, suppresses chamber and myocyte hypertrophy, and can longitudinally improve in vivo heart function in mice exposed to chronic pressure overload induced by transverse aortic constriction, reversing pre-established hypertrophy induced by pressure load while restoring chamber function. TRPV1 is a physical and regulated component of the natriuretic peptide signaling system, and TRPV1 inhibition may provide a new treatment strategy for treating, and reversing the loss of function associated with cardiac hypertrophy and heart failure.
KEYWORDS: Atrial natriuretic peptide (ANP), heart failure, hypertrophy, ion channel, ionotrophic cannabinoid receptor, member 1 (TRPV1), sildenafil, subfamily V, therapeutic, transient receptor potential cation channel
Introduction
Heart failure prevalence is in excess of 22 million cases worldwide, with an incidence of two million new cases a year. In the United states alone, 670,000 new cases of heart failure are diagnosed each year. Heart failure is the fastest-growing clinical cardiac disease burden in the United States, affecting 2% of the population, accounting for 34% of cardiovascular-related deaths, and representing 1–2% (~$40 billion) of all health care expenditures. Fourteen percent of the Medicare population has HF but they take up a disproportionate amount of Medicare dollars using ~43% of Medicare expenditures [1–6].
Heart failure is a condition that results from the heart’s inability to maintain sufficient cardiac output to meet the metabolic demands of the body. It can be acute or chronic in nature, it can affect either side of the heart and it can be systolic or diastolic. Persistent cardiac pressure overload induces hypertrophy and pathological tissue remodeling, which leads to loss of heart function and often heart failure and death. The progression of cardiac hypertrophy represents the principal risk factor for the development of heart failure and subsequent cardiac death [2]. Cardiac hypertrophy is classically considered to be an adaptive and compensatory response that increases the work output of cardiomyocytes and thus maintains cardiac function despite increased load. Increased resistance, created by pathology such as hypertension or by the aortic constriction technique we use in in vivo experiments, initially compromises left ventricular (LV) function. Subsequently the development of LV hypertrophy begins to restore systolic function, and concentric LV hypertrophy develops, which increases the LV mass. A decline in LV function accompanies LV chamber dilation, apoptosis, myocardial fibrosis and tissue remodeling, which results in eventual heart failure and death [7,8]. Heart failure can result in forced dependency, depression, and the inability to perform activities of daily living. The result of this is most often a drastic reduction in quality of life. There is a need for new drugs that address HF: We need to improve clinical outcomes, specifically maintaining heart function in patients suffering from heart failure, and reducing mortality. As part of a disease management program, such an agent would reduce readmission rates. New targets are needed because extant therapies are not adequately addressing these needs.
The transient receptor potential cation channel subfamily V, member 1 (TRPV1) is an ionotropic nonselective cation channel, initially identified in peripheral sensory neurons and found widespread in the cardiovascular system [9–14]. Studies have implicated the role of endogenous activator anandamide (ANA) in multiple cardiovascular diseases, such as myocardial ischemia reperfusion injury and hypertension [15,16]. Elevated TRPV1 expression is associated with cardiac hypertrophy in mice, and functional knockout of TRPV1 protected heart function in a model of cardiac hypertrophy [17]. Furthermore, we have shown that administration of a TRPV1 antagonist can overcome loss of heart function [9,18,19]. TRPV1 appears to be important in heart failure by virtue of the fact that its genetic knockout or pharmacological inhibition rescues cardiac hypertrophy in the mouse and that an endogenous activator (anandamide) has been implicated in in multiple cardiovascular diseases, including myocardial ischemia reperfusion injury and hypertension. TRPV1 is expressed in cardiac myocytes [20], but we understand relatively little of the potential regulatory coupling of TRPV1 to pathways that control heart physiology, and the longitudinal impact of TRPV1 inhibition in heart health under conditions of applied pathology has some attendant controversies.
Therapeutic or genetic hyperstimulation of guanosine 3′,5′-cyclic monophosphate (cGMP) synthesis counteracts these pathologies [21–23]. Here, we show that the TRPV1 ion channel (transient receptor potential cation channel, subfamily V, member 1), is a component of the natriuretic peptide A, cGMP, PKG signaling complex. It interacts with the natriuretic peptide receptor 1 (NPR1, guanylyl cyclase-A), and upon binding its ligand, natriuretic peptide A (NPPA, ANP) is subsequently suppressed through production of cGMP and PKG mediated phosphorylation. We also show that oral administration of selective TRPV1 antagonists, suppresses chamber and myocyte hypertrophy, and longitudinally reverses pre-established loss of heart function in vivo, in mice exposed to chronic pressure overload induced by transverse aortic constriction. This effect is similar to treatment with the cGMP-specific phosphodiesterase (PDE) inhibitor Sildenafil, and offers a mechanism of action for the efficacy of sildenafil in heart failure; where TRPV1 is a target for the down-stream PKG phosphorylation resulting from the elevated cGMP levels that result from PDE inhibition.
Results
The Natriuretic Peptide Receptor 1 interacts with TRPV1
Expression data and prior in vivo studies suggest that that TRPV1 is ideally positioned to receive stimuli that regulates hypertensive signaling, and thus protect the heart from cardiac hypertrophy [18,24–27]. Interaction trap data using the intracellular TRPV1 amino and carboxy-termini as bait (not shown) was examined for potential regulators of TRPV1 in a cardiovascular context, and suggested that TRPV1 interacts with the natriuretic peptide receptor 1 (NPR1, GC-A), a receptor guanylate cyclase [28,29]. This receptor binds the Atrial Natriuretic Peptide (ANP), the major physiological antagonist of the renin angiotensin system (RAS). This observation led us to propose a testable model (Figure 1). Here we hypothesize that there is a functional physiological interaction between the ion channel TRPV1, and the ANP receptor (NPR1); which upon stimulation causes an inhibitory phosphorylation of TRPV1 via cGMP-dependent protein kinase (PKG) stimulation.
Figure 1.
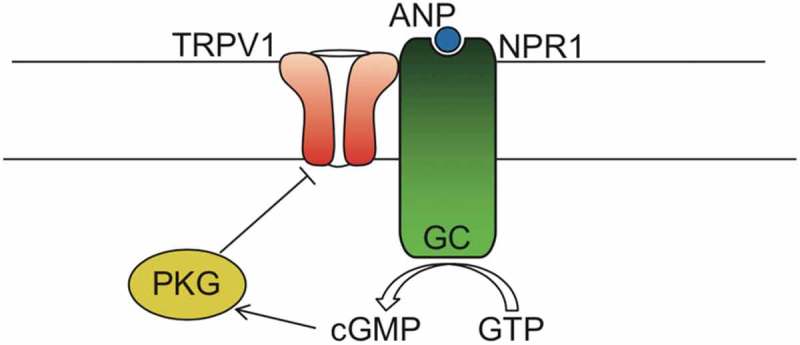
Schematic model of TRPV1 interacting with NPR1.
Our proposed model shows TRPV1 directly interacting with NPR1, which upon stimulation with ANP produces cGMP from GTP, which in turn stimulates PKG phosphorylation of TRPV1, and gating inhibition. (TRPV1, Transient Receptor Potential cation channel subfamily V member 1; ANP, atrial natriuretic peptide; NPR1/GC, Natriuretic peptide receptor A/guanylate cyclase A; PKG, cGMP-dependent protein kinase or Protein Kinase G; GMP, guanosine triphosphate; cGMP, Cyclic guanosine monophosphate).
As a first step in testing this model, we sought to confirm our interaction trap data, through co-immunoprecipitation of TRPV1 with NPR1. The specificity of the ANP receptor (NPR1, GC-A) antibody was first confirmed as follows: A FLAG-tagged NPR1 cDNA was transfected into HEK293 cells. Anti-FLAG immunoprecipitates a 120 kDa protein corresponding to FLAG-NPR1 that is detected by the NPR1 and FLAG antibodies (Figure 2(a)). The same FLAG-tagged protein was also detected in lysates by both the FLAG and NPR1 antibodies (Figure 2(a)). In Figure 2(a) the overexpression of TRPV1 resulted in high over-expression which may lead to non-physiological binding and dysregulated post-translational modification which reads as smears under our Western conditions. TRPV1 expression proved hard to control and so after these initial experiments we moved towards establishing functional relevance of the putative interaction.
Figure 2.
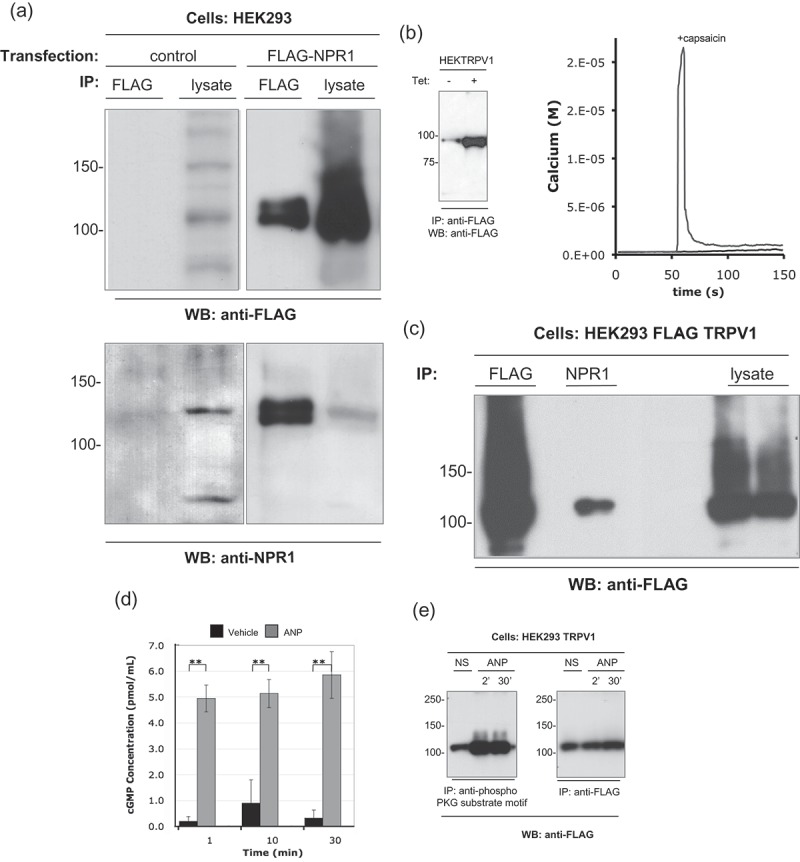
TRPV1 co-immunoprecipitates with NPR1, and is PKG phosphorylated.
(a) The anti-NPR1 antibody specifically immunoprecipitates the NPR1 protein. HEK293 cells were transiently transfected with the FLAG-NPR1 construct or a pcDNA control vector. After 24 h recovery for protein expression, cells (1x107 cells per lane) were harvested and lysed as described in Methods. Lysates were immunoprecipitated with 0.5 µg of anti-FLAG or acetone precipitated to recover total protein. Duplicate membranes were probed with either anti-FLAG (upper panels) or anti-NPR1 (lower panels). Anti-NPR1 specifically recognizes the FLAG-NPR1 proteins. (b) HEK293-FLAG-TRPV1 cells were treated with tetracycline (1μg/ml for 16h) to induce expression of functional FLAG-TRPV1 protein. Left panel shows immunoprecipitated FLAG-TRPV1 protein from non-induced and induced (1μg/ml for 16h) HEK293-FLAG-TRPV1 cells. Right panels show that induction of the cell line produces a capsaicin-sensitive TRPV1 calcium response in single cell Fura-2 assays. (c) TRPV1 interacts with the ANP receptor NPR1. HEK293-FLAG-TRPV1 cells were treated with tetracycline (1μg/ml for 16h) to induce expression of the FLAG-TRPV1 protein. Lysates were prepared (1x107 cells per lane) and immunoprecipitated with the indicated antibodies or acetone precipitated to recover total protein. Immunocomplexes were resolved by SDS-PAGE with separation lanes to prevent sample carry-over. FLAG-TRPV1 is present in anti-NPR1 immunoprecipitations. (d) NPR1 is present and functional in our HEK293 cell line. HEK293 were incubated with either vehicle or ANP (1 µg/ml) for the indicated times. Cytosolic cGMP levels were assayed by competitive immunoassay as described in methods. (e) TRPV1 is a PKG substrate. HEK293-FLAG-TRPV1 cells were treated with tetracycline (1 µg/ml for 16h) to induce expression of the FLAG-TRPV1 protein. Cells were harvested and treated with vehicle or ANP peptide (1 µg/ml) for the indicated times. Lysates were prepared (1x107 cells per lane) and immunoprecipitated with the indicated antibodies. Duplicate membranes were probed with either anti-phospho PKG substrate motif antibody (left panel) or anti-FLAG (right panel). ANP causes a marked increase in the levels of TRPV1 captured by anti-phospho PKG substrate antibody, indicating enhanced PKG-mediated phosphorylation of TRPV1.
To test co-immunoprecipitation, we used an HEK293 cell line that functionally expresses a FLAG epitope-tagged version of TRPV1 under the control of a tetracycline-sensitive transcriptional repressor. FLAG-TRPV1 is inducibly over-expressed by addition of tetracycline to the growth media, (Figure 2(b)-left panel) and functions to allow calcium influx into the cells upon stimulation with the TRPV1 ligand capsaicin (Figure 2(b) Right panel). Anti-FLAG and Anti-NPR1 antibodies both immunoprecipitate a protein corresponding to TRPV1 that is detected by anti-FLAG antibody (Figure 2c). Immunoprecipitation by anti-NPR1 of the FLAG-reactive protein indicates that TRPV1 protein interacts with the ANP receptor, NPR1, supporting one aspect of the proposed model (Figure 1).
Binding of ANP to NPR1 causes phosphorylation of TRPV1 via cGMP/PKG
NPR1 is a guanylate cyclase, catalyzing the conversion of GMP to cyclic GMP (cGMP) [28,29]. We sought to understand whether the production of cGMP that follows ANP binding to NPR1 has any functional consequences for the activity of TRPV1, as per our model (Figure 1). We stimulated the FLAG-TRPV1 HEK293 cell line with vehicle or ANP, confirming that upon stimulation for one minute or more, cGMP concentration was significantly increased many fold over control, indicating the presence of functional NPR1 in the FLAG-TRPV1 HEK293 cell line (Figure 2(d)). PKG is the major cellular target for the actions of cGMP, and previously published data has shown that another cyclic-nucleotide regulated kinase, PKA, affects TRPV1 [30,31], we asked whether TRPV1 is a substrate for PKG in our model system. In Figure 2(e), PKG phosphorylated substrate proteins were immunoprecipitated from untreated, and ANP-treated, FLAG-TRPV1 HEK293 cell lysates, using an anti-phosho-PKG substrate antibody. TRPV1 was represented in the PKG substrate proteins, and quantitatively more TRPV1 was immunoprecipitated via the anti-phospho-PKG substrate motif antibody from lysates derived from ANP-treated cells. These data suggest that TRPV1 is a PKG substrate, and is phosphorylated upon NPR1 ligation by ANP, as per our model.
cGMP/PKG regulates the function of TRPV1
Since we propose that TRPV1 activation regulates calcium-dependent cellular events, and ANP is the primary physiological antagonist to the renin–angiotensin system [32], we asked whether the ANP/PKG/NPR1 pathway would act to oppose the activation of TRPV1. Ratio-metric calcium imaging was undertaken, utilizing a cell-permeable cGMP analog with a greater resistance to hydrolysis by phosphodiesterases than cGMP, and which activates the PKG cGMP-dependent protein kinase. Cells expressing TRPV1 were exposed to increasing concentrations of 8-bromoguanosine 3′,5′-cyclic monophosphate, after which capsaicin was applied, and TRPV1 mediated calcium entry observed as an increase in the Fura2-AM die ratio. (Experimental design is shown in Supplemental Figure 1(a)). Treatment with between 3µM and 30µm 8-bromoguanosine 3′, 5′-cyclic monophosphate significantly reduced the calcium influx observed with subsequent treatment of 100nM capsaicin, as compared to vehicle (DMSO) stimulation (Figure 3(a,b)). We also explored whether the TRPV1-mediated calcium responses initiated by anandamide, using AM404 (N-arachidonoylaminophenol), a TRPV1 activator and endogenous cannabinoid reuptake inhibitor [33]. TRPV1 calcium influx induced by AM404 was also sensitive to suppression by the cell permeant cGMP analog. Figure 3(c) shows that pretreatment with 8-bromoguanosine 3′, 5′-cyclic monophosphate significantly suppresses subsequent TRPV1-mediated calcium responses to AM404 in a statistically significant fashion (Figure 3(d); Experimental design is shown in Supplemental Figure 2(a)).
Figure 3.
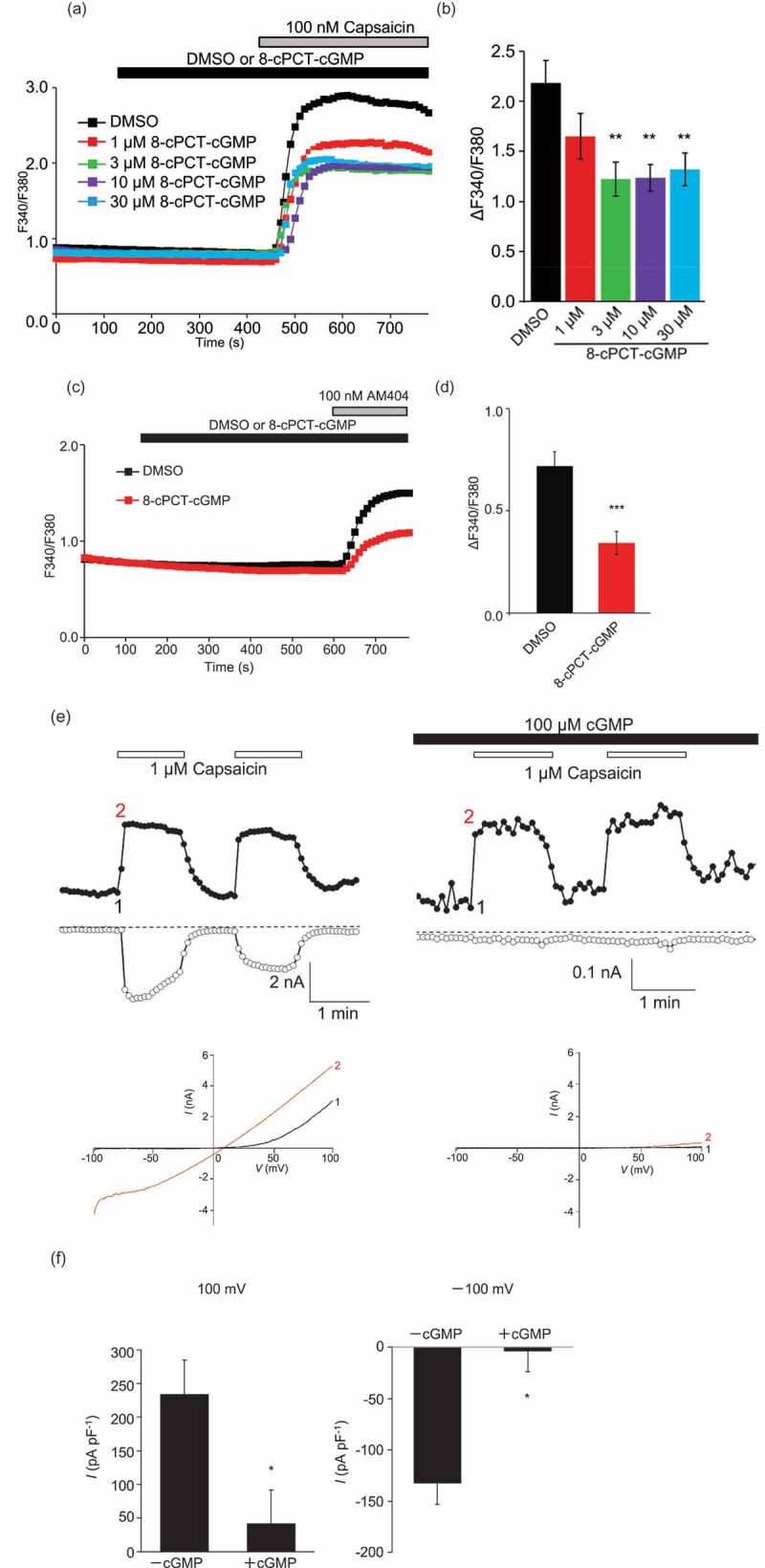
TRPV1 mediated calcium influx and current is inhibited by PKG phosphorylation.
TRPV1 mediated calcium influx was assessed using ratiometric Fura-2 imaging, as described in the methods. (a) Cells expressing TRPV1 were exposed to the indicated concentrations of 8-bromoguanosine 3′,5′-cyclic monophosphate, with subsequent addition of vehicle (DMSO) or 100nM capsaicin. Traces on the graph show averaged TRPV1 mediated calcium entry observed as an increase in the Fura 2-AM dye ratio, treated with vehicle or between 1µM and 100µm 8-bromoguanosine 3′, 5′-cyclic monophosphate. Cells treated with between 3µM and 30µm 8-bromoguanosine 3′, 5′-cyclic monophosphate show reduced calcium influx with 100nM capsaicin treatment. (b) A graph indicating a significant reduction in the calcium influx was observed in cells treated with between 3µM and 30µm 8-bromoguanosine 3′, 5′-cyclic monophosphate. (c) Traces on the graph show averaged AM404 (N-arachidonoylaminophenol) induced, TRPV1 mediated calcium entry observed as an increase in the Fura2-AM dye ratio, treated with vehicle or 100µm 8-bromoguanosine 3′, 5′-cyclic monophosphate. Cells treated with 100µm 8-bromoguanosine 3′, 5′-cyclic monophosphate, show reduced calcium influx. (d) A graph indicating a significant reduction in 100nM AM404-induced calcium influx was observed in cells treated with 100µm 8-bromoguanosine 3′, 5′-cyclic monophosphate, versus vehicle (DMSO). (e) Time-course of Capsaicin-evoked currents at −100 and 100 mV in a TRPV1 transfected HEK293 cells. Without addition of intracellular cGMP (left panels) and with addition of 100 µM cGMP applied intracellularly (right panels), indicating a large reduction in the amount capsaicin induced current. Panels below show the concurrent current-voltage relation of Capsaicin (1μM)-evoked whole-cell currents in TRPV1 transfected HEK cells, again showing a reduction in capsaicin induced current with internal 100 µM cGMP treatment. (f) Mean + S.E.M. data demonstrating the effects of 100µM internal cGMP on current densities at −100 and 100 mV in TRPV1 transfected HEK293 cells.
Electrophysiological demonstration of TRPV1 suppression by cGMP/PKG
We employed a whole cell patch clamp methodology in HEK293 transiently transfected with TRPV1 to assess the effect of cGMP/PKG signaling upon TRPV1 conductances. Experimental design is shown in Supplemental Figure 3. Figure 3(e) shows time-resolved current recordings (upper panels) and corresponding current-voltage relationships (I/V curves, lower panels) for TRPV1 inward and outward currents using sequentially-applied pulses of capsaicin as stimulus. The marked suppression of inward and outward currents in the presence of internally perfused 100 µM cGMP is observed. Note that the patch clamp protocol allows for the use of native cGMP, rather than the cell-permeant analog. The lower panels show the characteristic I/V relationship for TRPV1. Figure 3(f) shows the normalized current sizes in pA per pF at +100 and −100mV, respectively. When normalized to cell size (capacitance, pF), TRPV1-mediated currents are significantly (p < 0.01) suppressed in the presence of perfused cGMP when compared to perfused vehicle.
Orally delivered TRPV1 antagonists restore function in pressure overload cardiac hypertrophy
The data presented above demonstrate a mechanistic linkage between TRPV1 and an ANP/cGMP/PKG signaling circuit that is highly relevant to cardiac physiology and pathophysiology. ANP, and the cGMP signaling it initiates, are suppressive influences on TRPV1 activation. ANP is synthesized, and secreted by cardiac myocytes in the atrial walls of the heart, which is released in response to atrial distension and serves to maintain sodium homeostasis and inhibit activation of the renin-angiotensin-aldosterone system. Circulating atrial natriuretic peptide is greatly increased in congestive heart failure as a result of increased synthesis and release of ANP. This is particularly important in the vasculature, where vascular smooth muscle will bind ANP released as a result of increasing right atrial pressure and will cause the walls of the vasculature to relax. This relaxation decreases peripheral resistance, decreases venous return to the heart, and reduces the preload and resulting in the heart having to do less work [34]. Data in previous publications have shown that mice treated with a TRPV1 antagonist are protected from subsequently induced hypertrophy [19,25]. Moreover, given that the cGMP mobilizing drug Sildenafil (a phosphodiesterase inhibitor) protects and has been shown to reverse hypertrophy, we wished to ask whether TRPV1 inhibition could accomplish similar protection, and especially reversal, in the identically designed in vivo pressure overload model (Figure 4) [22]. Figure 4 shows a schematic timeline of the method used, inducing application of pressure overload surgery, and loss of heart function before delivery of any vehicle, or drug, enabling the identification of any putative beneficial functional reversal following treatment [22,23]. Figure 5(a-e) show percentage ejection fraction (%EF) and percentage fractional shortening (%FS) in sham surgery (Figure 5(a)) or TAC treated (Figure 5(b-e)) mice. The orally-delivered TRPV1 antagonists BCTC, SB366791 and AMG9810 all reverse loss of function in mice modeled by TAC-induced cardiac hypertrophy. Notably these inhibitors target different TRPV1 activation modalities. BCTC inhibits agonist and proton activation modalities [35,36]; SB366791 inhibits agonist, noxious heat but not proton activation modalities [37,38]; AMG9810 inhibits agonist, heat and proton activation modalities [39]; indicating that it is the antagonist modality of TRPV1 activation, and the endocannabinoid anandamide that is responsible for the TRPV1 mediated cardiac hypertrophic pathology observed [26,40]. Figure 5(f) shows that BCTC and SB366971 are also protective when cardiomyocyte cross-sectional area (a measure of cellular hypertrophy) is assessed. These data improves on previously published findings in TRPV1 knockout and agonist treated mice [18,19,25], where the experimental design induced pathology on a background of already negated TRPV1 activation. In the current study, the experimental protocol was to assess the effects of TRPV1 inhibition applied post hoc to the development of the surgically-induced pathology. Thus, the data in Figure 5(a-f) newly document the reversal of pathology and partial regaining of function in the failing heart in response to an orally delivered TRPV1 inhibitor drug.
Figure 4.
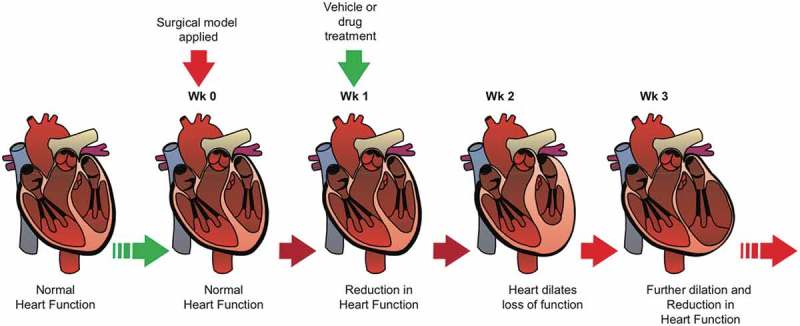
Schematic of in vivo hypertrophic reversal experimental design.
A schematic timeline of the method used, inducing loss of heart function before oral delivery of vehicle, or drug, enabling the identification of any putative beneficial functional reversal following a putative therapeutic intervention.
Figure 5.
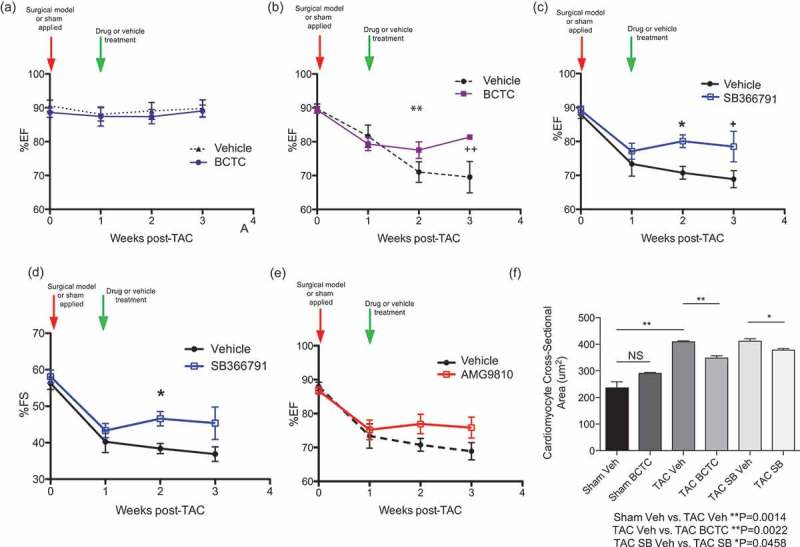
Oral administration of TRPV1 antagonists longitudinal reverses the loss of function, and cardiomyocyte hypertrophy, associated with TAC induced pressure overload.
A TAC induced pressure overload mouse model is allowed to progress for one week, in order to generate pressure overload loss of function, then TRPV1 antagonists are orally administered to ascertain any gain of function over vehicle drug treatment. (a) Vehicle treated sham surgery and BCTC treated sham surgery mice, showing no effect of drug treatment on ejection fraction (%EF), over the treatment period. (NS, n = 7,9). (b) Ejection fraction (%EF) of vehicle treated TAC and BCTC treated TAC mice, at time zero, the time point that TAC is applied; one week after TAC, showing loss of function in both sets of mice; two weeks after TAC, showing continued loss of function in vehicle treated TAC mice, and a moderate gain of function in BCTC treated TAC mice; three weeks after TAC showing continued loss of function in vehicle treated TAC mice, and a significant gain of function in BCTC treated TAC mice (+wk 3 P < 0.05). Overall, oral treatment with BCTC shows a reversal of the loss of function associated with TAC induced pressure overload (**P = 0.0170, n = 5,6). (c) %EF of vehicle treated TAC and SB366791 treated TAC mice (n = x), at time zero, the time point that TAC is applied; one week after TAC, showing loss of function in both sets of mice; two weeks after TAC, showing continued loss of function in vehicle treated TAC mice, and a significant gain of function in SB366791 treated TAC mice (*wk 2 P = 0.0102); three weeks after TAC showing continued loss of function in vehicle treated TAC mice, and a significant gain of function in SB366791 treated TAC mice (+wk 3 P < 0.05). Oral treatment with SB366791 shows a reversal of the loss of function associated with TAC induced pressure overload (n = 10,10) (d) Fraction shortening (%FS) of vehicle treated TAC and SB366791 treated TAC mice, at time zero, the time point that TAC is applied; one week after TAC, showing loss of function in both sets of mice; two weeks after TAC, showing continued loss of function in vehicle treated TAC mice, and a significant gain of function in SB366791 treated TAC mice; three weeks after TAC showing continued loss of function in vehicle treated TAC mice, and a slight loss of function in SB366791 treated TAC mice (*P = 0.0149, n = 10,10). (e) %EF of vehicle treated TAC and AMG9810 treated TAC mice, at time zero, the time point that TAC is applied; one week after TAC, showing loss of function in both sets of mice; between one and three weeks after TAC, showing continued loss of function in vehicle treated TAC mice, and a slight, but not significant, gain of function in AMG9810 treated TAC mice. (wk 2 P = 0.1818, wk 3 P = 0.1017, n = 10,10). (f) Measurement of cardiomyocyte cross sectional area (though staining of plasma membranes with wheat germ agglutinin-Alexa488 in heart tissue sections from week 3 mice), indicated a significant different between vehicle treated TAC mice and BCTC or SB366791(SB) treated TAC mice. Indicating a reversal in the cellular cardiomyocyte hypertrophy associated with TAC induced pressure overload (Sham Vehicle vs. TAC Vehicle **P = 0.0014, TAC Vehicle vs. TAC BCTC **P = 0.0022, TAC SB366791 Vehicle vs. TAC SB366791 *P = 0.0458).
TRPV1 antagonist induced restored function can be longitudinally preserved in pressure overload cardiac hypertrophy
Figure 5(g) documents longitudinal effects of continued drug treatment after TAC. Here, %EF (Figure 5(g) left panel) and %FS (Figure 5(g) right panel) were monitored over a 32 week period. As above, TAC was applied at the start of the experiment, then one week later, and continuously for 31 further weeks, vehicle or the TRPV1 antagonist BCTC were delivered orally, daily. For both %EF and %FS, the protective effect of TRPV1 inhibition was maintained longitudinally and was statistically significantly different to vehicle effects for as long as 32 weeks. Finally, Figure 5(h) shows body weight, an overall health indicator, for vehicle and BCTC treated mice for 224 days (32 weeks) post-TAC. We note that body weight is greater in BCTC-treated mice compared to vehicle controls, indicating an improvement in overall health/QOL.
Figure 5.
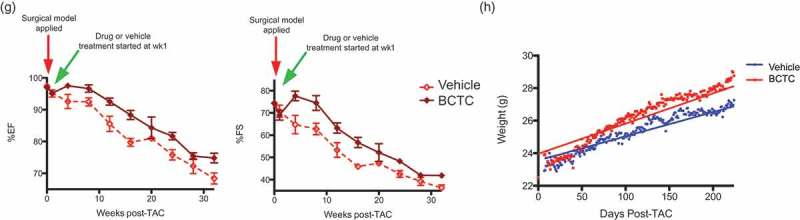
(g) Left panel, %EF of vehicle treated TAC (n = 5) and BCTC treated TAC mice (n = 5), at time zero, the time point that TAC is applied; one week after TAC, showing loss of function in both sets of mice; between one and three weeks after TAC, showing continued loss of function in vehicle treated TAC mice, and a gain of function in BCTC treated TAC mice; between three and thirty two weeks after TAC both vehicle and BCTC treated TAC mice show the loss of function, but BCTC treated mice maintain their initial gain in function. (n = 5,5, wk 4 P = 0.069, wk 8 P = 0.3911, wk 12 P = 0.0295 *, wk 16 P = 0.0031 **, wk 20 P = 0.3667, wk 24 P = 0.0184 *, wk 28 P = 0.2682, wk 32 P-0.0243 *). Right panel, %FS of vehicle treated TAC (n = 5) and BCTC treated TAC mice (n = 5), at time zero, the time point that TAC is applied; one week after TAC, showing loss of function in both sets of mice; between one and three weeks after TAC, showing continued loss of function in vehicle treated TAC mice, and a gain of function in BCTC treated TAC mice; between three and thirty two weeks after TAC both vehicle and BCTC treated TAC mice show the loss of function, but BCTC treated mice maintain their initial gain of function (n = 5,5, wk 4 P = 0.0245 *, wk 8 P = 0.3979, wk 12 P = 0.0405 *, wk 16 P = 0.0056 **, wk 20 P = 0.2664, wk 24 P = 0.0214 *, wk 28 P = 0.2158, wk 32 P = 0.0172 *). (h) Body weight, an overall health indicator, for vehicle and BCTC treated mice for 224 days (32 weeks) starting 1 week post-TAC. We note that body weight is significantly greater in BCTC-treated mice compared to vehicle controls, indicating improved overall health P < 0.0001 (n = 5,5).
TRPV1 is most likely a downstream target of cGMP-specific phosphodiesterase (PDE) inhibitor Sildenafil
The in vivo effect shown in our data, is similar to treatment with the cGMP-specific phosphodiesterase (PDE) inhibitor Sildenafil [22], and offers a mechanism of action for the efficacy of sildenafil in heart failure; where TRPV1 is a target for the down-stream PKG phosphorylation resulting from the elevated cGMP levels that result from PDE inhibition (Figure 6).
Figure 6.
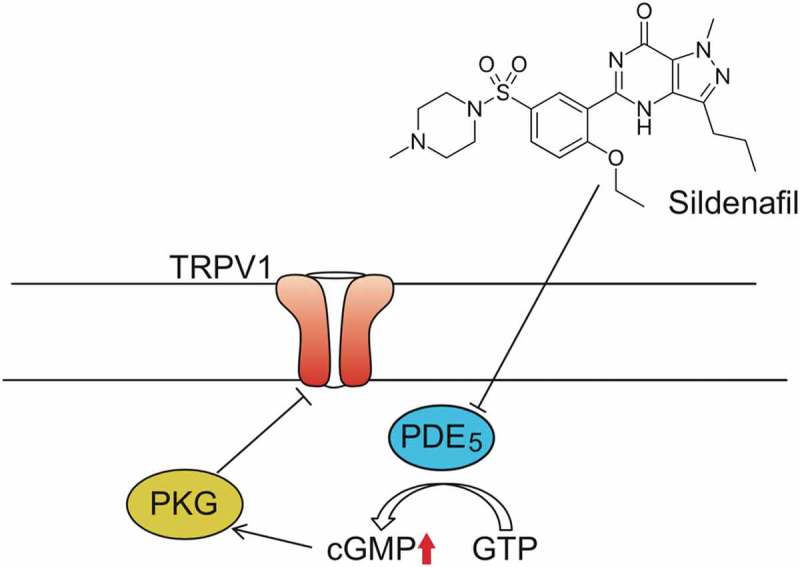
Schematic model of Sildenafil/PDE5 mediated TRPV1 inhibitory phosphorylation.
A proposed model showing TRPV1 inhibition though Sildenafil treatment. (TRPV1, Transient Receptor Potential cation channel subfamily V member 1; PDE5, cGMP-specific phosphodiesterase type 5; PKG, cGMP-dependent protein kinase or Protein Kinase G; GMP, guanosine triphosphate; cGMP, Cyclic guanosine monophosphate).
Discussion
The transient receptor potential cation channel subfamily V, member 1 (TRPV1) is an ionotropic nonselective cation channel. It was initially identified in peripheral sensory neurons and later found to be widespread in the cardiovascular system [9–14]. TRPV1 appears to be important in the heart by virtue of the fact that its genetic knockout or pharmacological inhibition rescues cardiac hypertrophy in the mouse [18,19,25]. TRPV1 is expressed in cardiac myocytes [20], but as we started the current study we understood relatively little of the potential regulatory coupling of TRPV1 to pathways that control heart physiology, and the longitudinal impact of TRPV1 inhibition in heart health under conditions of applied pathology had not been explored. In the current paper we show that TRPV1 interacts with the natriuretic peptide receptor 1, NPR1, and that activation of NPR1 with the atrial natriuretic peptide pathway causes cGMP stimulated PKG phosphorylation of TRPV1, causing an inhibition of TRPV1 activation and reduced surface currents. We also show that orally available TRPV1 antagonists can reverse the loss of function associated with heart failure, in an in vivo pressure overload model of heart failure. Our data also show that this rescued function can be maintained longitudinally with continued drug treatment.
These data place TRPV1 as component of the atrial natriuretic peptide/PKG signaling system, which is central to volume control in the heart. There are established connections between cGMP, PKG and heart health [41–45]. ANP is produced in cardiomyocytes of the atrial wall, which contain volume receptors that are stretch-responsive. Knockout of the ANP receptor, NPR1, has been shown to be protective in cardiac hypertrophy, improving outcomes, with NPR1 knockout experiments effectively evidencing the existence of an ANP/NPR1 anti-hypertrophic pathway in the heart [46]. Here we show that ANP/NPR1/PKG leads to regulatory phosphorylation of TRPV1, and since TRPV1 KO, like that of NPR1 is antagonistic to hypertrophy, we can assume this is a negative regulatory phosphorylation and that TRPV1 activity and function are involved in hypertrophic processes [47]. Thus the ANP/NPR1/cGMP/PKG/TRPV1 linkage, which is novel, provides a target and effector molecule (TRPV1) for the anti-hypertrophic effects of ANP/NPR1. Open questions still remain, however, including clarification of the effect of PKG phosphorylation on TRPV1 (soluble Guanylate Cyclase/PKG. An NO-regulated PKG pathway does negatively regulate TRPV1 via PKG phosphorylation and our data now support a negative regulatory NPR1/PKG/TRPV1 phosphorylation, but overall the effects of PKG phosphorylation on TRPV1 are less well studied than those of PKA and PKC [47–49].
One limitation of these studies is the fact that future experiments need to use ANP as an applied ligand, in whole cell patch clamp, measure that there is an effect in terms of an increase in a potentially short-lived cGMP flux and then assess impact on TRPV1 currents or the resulting calcium fluxes. The experiments showing 8-bromo cGMP inhibition of TRPV1 responses would benefit from addition of a washout step in future studies. A further limitation is the need to demonstrate a native NPR1-TRPV1 complex, possibly by FRET or co-IP. While these two proteins are indeed co-expressed in cardiomyocytes, the regulation of TRPV1 via NPR1-generated cGMP does not necessarily require NPR1 and TRPV1 to be physically co-associated in a complex [20]. Our data suggesting interaction in an overexpression system needs to be followed up to establish if the physical association is a component of the regulatory pathway.
The second major finding of the present report is the longitudinal impact of TRPV1 inhibition on heart health under conditions of applied pathology. Here we show that TRPV1 antagonists can reverse the loss of function associated with heart failure, in the in vivo pressure overload model of heart failure. Moreover, this restoration of function can be maintained longitudinally with continued drug treatment. These findings are of potential clinical significance and are open to rapid translation given the extensive TRPV1 pharmacopeia that has been developed for pain therapy. Of course, these findings need to be extended beyond murine models, but we note that the effect of TRPV1 antagonism on functional recovery could be much greater in the human clinical situation, in which there is not a continued surgically produced aortic constriction and pressure overload applied. In humans therefore, there may be the opportunity to recover to an actually improved ejection fraction, higher than the initially identified symptomatic disease state.
One of the intriguing questions arising from our studies is the identity of the cell types that might contain functional TRPV1 and effect protection in the context of heart failure [16,17,20,24,50–57]. As our data is generated in an in vivo model that effects systemic inhibition of TRPV1 during heart failure progression, the exact combination of cell and tissue type cannot be pinned down. We hypothesize however that a major cell type effected is most likely cardiomyocytes. TRPV1, along with TRPA1, expression has been shown localized to z-discs, costameres and intercalated discs in cardiac muscle [20], as well as being functionally regulated in cultured cardiomyocytes, where its activation increased cell size and elevated expression of atrial natriuretic peptide mRNA and intracellular calcium [57].
The data presented here provides new possibilities in the treatment and reversal of heart failure, and further hope for patient survival and life extension. TRPV1 is a focus of multiple drug discovery efforts in both pre-clinical and clinical stages [27,58,59]. The TRPV1 agonist capsaicin is used as a treatment for pain through its effect in desensitization of TRPV1 channels in neurons [60]. Since the therapeutic goal in both pain, and hypertrophy, is to decrease TRPV1 activity, then either direct antagonism or indirect suppression through agonism and desensitization could be viable approaches. Early TRPV1 agonists showed adverse effects during clinical development including mild hyperthermia and impaired noxious heat sensation, impairing their progression to the clinic as analgesics. However, in the context of imminent heart failure, mild hyperthermia might be viewed as an acceptable side effect. Moreover, at least seven later generation TRPV1 compounds that are orally active have progressed into clinical development with reduced side effect profiles [59]. For example, GRC 6211 (Eli Lilly), an orally available TRPV1 antagonist, is currently in phase IIb clinical trials [61].
Materials & methods
Cell culture and transfections
HEK293 stably transfected with the pcDNA6TR (Invitrogen, Temecula, CA) plasmid (encoding the tetracycline-sensitive TRex repressor protein), were maintained in DMEM + 10% fetal bovine serum (inactivated at 55°C for 1h), + 2 mM glutamine in humidified 5% CO2 atmosphere at 37°C. Selection pressure on these HEK TRex 293 cells was maintained by continuous culture in 10µg/ml Blasticidin (Sigma, St Louis, MO). For production of TRex HEK293 cells with inducible expression of FLAG-tagged TRPV1, parental pcDNA6/TR transfected cells were electroporated with the TRPV1 cDNA in the pcDNA4TO vector and clonal cell lines were selected by limiting dilution in the presence of 400 μg/ml Zeocin (Invitrogen). Expression of the TRPV1 channel was induced using 1 μg/ml tetracycline for 16 h at 37°C. Stable lines were screened for inducible protein expression using anti-FLAG Western blot. Transient expression of pcDNA5/TO NPR1-FLAG was achieved by lipid-mediated transfection (LT1 reagent, Mirus, Madison, WI).
Antibody and reagent suppliers
Mouse monoclonal anti-FLAG was from Sigma (St Louis, MO). HRP-conjugated secondary antibodies were from Amersham (Piscataway, NJ). Alexa fluorescently coupled secondary antibodies were from ThermoFisher (Waltham, MA). Capsaicin from MP Biomedicals (Solon, OH). BCTC, SB366791, and AMG9810 from TOCRIS (Minneapolis, MN). Atrial natriuretic peptide was from American Peptide Company (Sunnyvale, CA). Anti-phospho PKG substrate motif antibody, 8-pCPTcGMP was from Calbiochem (San Diego, CA).
Cell lysis, acetone precipitation and immunoprecipitation
Cells were pelleted (2000g, 2 min) and washed once in ice cold PBS. Approximately 107 cells were lysed (ice/30 minutes) in 350μl of lysis buffer (50mM Hepes pH 7.4, 75mM NaCl, 20mM NaF, 10mM iodoacetamide, 0.5% (w/v) Triton X100, 1mM PMSF (phenylmethylsulfonyl fluoride), 500 μg/ml Aprotinin, 1.0 mg/ml Leupeptin and 2.0 mg/ml chymostatin). Lysates were clarified (10,000g, 5 min). For preparation of total protein, lysates were acetone precipitated (1.4 volumes acetone for 1h at – 20°C, followed by centrifugation at 10,000g for 5 min). For immunoprecipitation, supernatants were tumbled (4°C/2h) with the indicated antibody, covalently coupled (dimethylpimelidate/ethanolamine) to Protein G sepharose.
Western blot
Samples were incubated (95°C/8 min) in SDS-PAGE loading buffer and resolved by SDS-PAGE in 25 mM Tris, 192 mM glycine, 0.05% (w/v) SDS, pH 8.8. Resolved proteins were electro-transferred to PVDF. For Western blotting, membranes were blocked using 5% non-fat milk or BSA in PBS (1h/RT). Primary antibodies were dissolved in PBS/0.05% Tween-20/0.05% NaN3 and incubated with membrane for 16 h at 4°C. Developing antibodies (anti-rabbit or anti-mouse IgGs conjugated to horseradish peroxidase) were diluted to 0.1 μg/ml in PBS/0.05 % Tween-20 and incubated with membranes for 45 min at RT. Standard washes (4 x 5 min in 50 ml PBS/0.1% Tween-20 at RT) occurred between primary and secondary incubations.
Cyclic AMP and cyclic GMP determination assays
Cytosolic cAMP and cGMP levels were determined by the Direct EIA kits from Assay Designs, Inc. (Ann Arbor, MI) according to the manufacturer’s instructions. Cells were stimulated as described. Reactions were halted by cell lysis in 0.1 M HCl/0.5 % Triton X-100 (37°C, 10 min). Cell lysates were centrifuged (10,000 g, 1 min, RT) and cAMP or cGMP levels were measured from these cell lysates using Correlate-EIATM Direct cAMP or cGMP assays (Assay Designs, Ann Arbor, MI). Absorbance A405 for each sample was converted to pmol cyclic nucleotide, relative to a cAMP or cGMP standard curve that was obtained for each run of the experiment. Two hour acetylation reactions were performed to optimize sensitivity.
Calcium measurements
Transfected HEK293 cells were subjected to [Ca2+]i measurement 3–16 h after plating onto poly-L-lysine-coated glass coverslips. The Fura-2 (Dojindo, Japan) fluorescence was measured in HEPES-buffered saline containing the following: 107 mM NaCl, 6 mM KCl, 1.2 mM MgSO4, 2 mM CaCl2, 11.5 mM glucose, and 20 mM HEPES (pH adjusted to 7.4 with NaOH). Fluorescence images of the cells were recorded and analyzed with the video image analysis system AQUACOSMOS (Hamamatsu Photonics, Shizuoka, Japan) according to the manufacturer instructions. Fura-2 measurements were carried out at room temperature in HEPES-buffered saline. The 340:380-nm ratio images were obtained on a pixel-by-pixel basis.
Single cell fura-2 calcium assay
Cells were seeded onto glass coverslips and loaded with 4 μM Fura-2 AM (Molecular Probes) in standard Ringer buffer with 1 mM CaCl2, ~330 mOsm, for 45 min at 37°C. After washing, single cell calcium assay was performed as described [62]. Stimuli were perfused extracellularly using a wide-tipped application pipette (Figure 2(b)).
Electrophysiology
For electrophysiological measurements, coverslips with cells were placed in dishes containing bath solutions. Currents from cells were recorded at room temperature using patch-clamp techniques of whole-cell mode with EPC-10 (Heka Elektronik, Lambrecht/Pfalz, Germany) patch clamp amplifier. The patch electrode prepared from borosilicate glass capillaries had a resistance of 2–4 megaohms. Current signals were filtered at 2.9 kHz with a 4-pole Bessel filter and digitized at 10 kHz. Patchmaster (Heka Elektronik) software was used for command pulse control, data acquisition, and data analysis. The series resistance was compensated (50–70%) to minimize voltage errors. Ramp pulses were applied every 5 seconds from −100 to + 100 mV at a speed of 0.4 mV ms−1 after a 50 ms step to −100 mV from a holding potential of 0 mV. The external solution contained 100 mM NaCl, 5 mM KCl, 2 mM BaCl2, 5 mM MgCl2, 25 mM HEPES, and 30 mM glucose (pH 7.3 adjusted with NaOH, and osmolarity adjusted to 320 mosM with D-mannitol). The pipette solution contained 140 mM CsCl, 4 mM MgCl2, 10 mM EGTA, 10 mM HEPES (pH 7.3 adjusted with CsOH and osmolarity adjusted to 300 mosM with D-mannitol).
Animal care
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Hawaii.
Animals
C57BL/6J mice (Jackson Labs, ME) were used. Mice were housed under a 12-hour light/dark cycle and fed with standard diet and water ad libitum. Ten-week-old male mice were used for all experiments. Mice were euthanized by CO2 asphyxiation for tissue analysis.
Transverse Aortic Constriction (TAC)
Transverse aortic constriction was performed as described by Rockman, producing left ventricular hypertrophy by constriction of the aorta [63]. The left side of the chest was depilated with Nair and a baseline 2-D echocardiogram was obtained as described below. Mice were then deeply anesthetized with a mixture of ketamine and xylazine. The transverse aorta between the brachiocephalic and left carotid artery was banded using 6–0 silk ligature around the vessel and a 26G blunt needle, after which the needle was withdrawn. Sham surgeries were identical apart from the constriction of the aorta [22,64–68].
In vivo drug treatment
All drugs were administered by oral gavage. AMG9810 30mg/kg (Tocris) delivered at 10 mg/ml in 20% hydroxypropyl-β-cyclodextrin/PBS/corn oil, SB366791 3mg/Kg (Abcam) delivered at 3mg/ml in 20% hydroxypropyl-β-cyclodextrin/PBS/corn oil; BCTC at 20mg/Kg delivered at 3mg/ml in 20% hydroxypropyl-β-cyclodextrin/PBS/corn oil.
Doppler echocardiography
Doppler echocardiography was performed one week post TAC to measure the level of constriction. Mice were anesthetized lightly with isofluorene gas and shaved. Doppler was performed using the Visualsonics Vevo 770 system. In the parasternal short-axis view, the pulsed wave Doppler sample volume was placed in the transverse aorta just proximal and distal to the site of banding. Peak velocity was traced using Vevo 770 software, and the pressure gradient was calculated using the simplified Bernoulli equation [22].
Transthoracic echocardiography
Baseline and post TAC transthoracic echocardiography were used to assess changes in mouse heart dimensions and function. Briefly, after two days of acclimatization and depilation, unanesthetized transthoracic echocardiography was performed using a 30-Mhz transducer (Vevo 770, VisualSonics). High quality two-dimensional images and M-mode images of the left ventricle were recorded. Measurements of left ventricular end-diastolic (LVIDd) and end-systolic (LVIDs) internal dimensions were performed by the leading edge to leading edge convention adopted by the American Society of Echocardiography. The left ventricular ejection fraction (%EF) was calculated as (LV Vol; d-LV Vol;s/LV Vol; d x 100) (Visualsonics Inc.) [66–68].
Tissue preparation for histology
Mice were euthanized by CO2 asphyxiation, and hearts were collected for tissue analysis. For histology, hearts were perfused with phosphate-buffered saline and 10% formalin in situ, collected immediately, and fixed overnight in 10% formalin at 4°C. Tissues were then cut in a sagittal orientation, embedded in paraffin, mounted on glass slides, and stored until use.
Cardiomyocyte cross sectional area
Heart sections were deparaffinized and permeabilized, then stained with wheat germ-agglutinin conjugated to Alexa488 (WGA-Alexa488, Invitrogen, W11261) at a concentration of 50μg/mL to identify sarcolemmal membranes and measure cardiomyocyte cross sectional area.
Cardiomyocyte cross sectional area, image collection and analysis
Fluorescent and bright field images were collected on an epifluorescence-microscope (Axioscope, Zeiss). Cross-sectional cardiomyocyte area were quantified using ImageJ software (NIH). Perivascular tissue was excluded from this calculation. Three heart sections from each animal were imaged at five images per heart. Images were averaged for each animal and graphed in Prism GraphPad. Cardiomyocytes from WGA stained sections were randomly selected in a blinded fashion then traced to determine the cross sectional area of individual myocytes (n = 100). Perivascular tissue was excluded from this calculation. Three heart sections from each animal were imaged at five images per heart. Images were averaged for each animal and graphed in Prism GraphPad. All images were captured and analyzed in a double-blind manner.
Statistics
Statistical significance of echocardiography data was evaluated using 2-way ANOVA, with a Bonferroni post hoc test. and linear regression. Histology and molecular data were evaluated using the 2-tailed Student’s t-test. Evaluations were performed using PRISM software (La Jolla, CA) with P < 0.05 regarded as significant. All data are shown as mean ± SD.
Funding Statement
Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM113134; and the Hawaii Community Foundation, 16CON-78925.
Acknowledgments
The authors would like to thank Dr. Kimberly A. del Carmen, Dr. Michaela Kuhn, (Physiological Institute, University of Würzburg, Germany); Dr. Hanjun Wang, and Dr. Irving H. Zucker, (Dept. of Cellular & Integrative Physiology, University of Nebraska Medical Center, Omaha, NE).
Dedication
This work is dedicated to Dr. Ruth Gates, an amazing scientist and friend. We will miss you.
Disclosure statement
AJS is founder of Makai Biotechnology, LLC, a cardiovascular focused biotechnology company. HT is a member of the scientific advisory board of GBSciences, Inc. ALSH is employed by GBSciences, Inc.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics–2012 update: a report from the American heart association. Circulation. 2012. January 3;125(1):e2–e220. PubMed PMID: 22179539; eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics–2011 update: a report from the American heart association. Circulation. 2011. February 1;123(4):e18–e209. PubMed PMID: 21160056; eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lloyd-Jones D, Adams RJ, Brown TM, et al. Executive summary: heart disease and stroke statistics–2010 update: a report from the American heart association. Circulation. 2010. February 23;121(7):948–954. PubMed PMID: 20177011; eng. [DOI] [PubMed] [Google Scholar]
- [4].McElroy PA, Janicki JS, Weber KT.. Cardiopulmonary exercise testing in congestive heart failure. Am J Cardiol. 1988. July 11;62(2):35A–40A. PubMed PMID: 3133938; eng. [DOI] [PubMed] [Google Scholar]
- [5].Packham N, Brasseaux H, Rotter H, et al. Removal of iodine for spacecraft applications. Warrendale, PA: SAE International; 1999. (SAE technical paper series,; 1999-01-2118). [Google Scholar]
- [6].Aschermann M. 2001. Nestabilní angina pectoris. 1. vyd. ed Praha: Galen; (Trendy soudobé kardiologie; s 3) [Google Scholar]
- [7].Patten RD, Hall-Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail. 2009. March;2(2):138–144. . PubMed PMID: 19808329; eng. [DOI] [PubMed] [Google Scholar]
- [8].Lygate C. Surgical models of hypertrophy and heart failure: myocardial infarction and transverse aortic constriction. Drug Discov. Today Dis. Models. 2006;3(3):283–290. [Google Scholar]
- [9].Alfulaij N, Meiners F, Michalek J, et al. Cannabinoids, the heart of the matter. J Am Heart Assoc. 2018. July 13;7(14). PubMed PMID: 30006489; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cao E, Liao M, Cheng Y, et al. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013. December 5;504(7478):113–118. PubMed PMID: 24305161; PubMed Central PMCID: PMCPMC4023639. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ferreira LG, Faria RX. TRPing on the pore phenomenon: what do we know about transient receptor potential ion channel-related pore dilation up to now? J Bioenerg Biomembr. 2016. February;48(1):1–12. PubMed PMID: 26728159; eng. [DOI] [PubMed] [Google Scholar]
- [12].Samways DS, Tomkiewicz E, Langevin OM, et al. Measurement of relative Ca(2)(+) permeability during sustained activation of TRPV1 receptors. Pflugers Arch. 2016. February;468(2):201–211. PubMed PMID: 26490461; eng. [DOI] [PubMed] [Google Scholar]
- [13].Caterina MJ, Schumacher MA, Tominaga M, et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997. October 23;389(6653):816–824. PubMed PMID: 9349813. [DOI] [PubMed] [Google Scholar]
- [14].Tominaga M, Caterina MJ, Malmberg AB, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998. September;21(3):531–543. PubMed PMID: 9768840. [DOI] [PubMed] [Google Scholar]
- [15].Peng J, Li YJ. The vanilloid receptor TRPV1: role in cardiovascular and gastrointestinal protection. Eur J Pharmacol. 2010. February 10;627(1–3):1–7. PubMed PMID: 19879868; eng. [DOI] [PubMed] [Google Scholar]
- [16].Smith SA, Leal AK, Williams MA, et al. The TRPv1 receptor is a mediator of the exercise pressor reflex in rats. J Physiol. 2010. April 1;588(Pt 7):1179–1189. PubMed PMID: 20142275; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhong B, Wang DH. Protease-activated receptor 2-mediated protection of myocardial ischemia-reperfusion injury: role of transient receptor potential vanilloid receptors. Am J Physiol Regul Integr Comp Physiol. 2009. December;297(6):R1681–90. PubMed PMID: 19812353; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Buckley CL, Stokes AJ. Mice lacking functional TRPV1 are protected from pressure overload cardiac hypertrophy. Channels (Austin). 2011. Jul-Aug;5(4):367–374. PubMed PMID: 21814047; PubMed Central PMCID: PMCPMC3225734. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Horton JS, Buckley CL, Stokes AJ. Successful TRPV1 antagonist treatment for cardiac hypertrophy and heart failure in mice. Channels (Austin). 2013. January 01;7(1):17–22. . PubMed PMID: 23221478; PubMed Central PMCID: PMCPMC3589277. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Andrei SR, Sinharoy P, Bratz IN et al. TRPA1 is functionally co-expressed with TRPV1 in cardiac muscle: co-localization at z-discs, costameres and intercalated discs. Channels (Austin). 2016. May 4 PubMed PMID: 27144598; Eng DOI: 10.1080/19336950.2016.1185579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang M, Koitabashi N, Nagayama T, et al. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008. December;20(12):2231–2236. PubMed PMID: 18790048; PubMed Central PMCID: PMCPMC2601628. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005. February;11(2):214–222. PubMed PMID: 15665834; eng. [DOI] [PubMed] [Google Scholar]
- [23].Lewis GD, Shah R, Shahzad K, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007. October 02;116(14):1555–1562. PubMed PMID: 17785618; eng. [DOI] [PubMed] [Google Scholar]
- [24].Thilo F, Liu Y, Schulz N, et al. Increased transient receptor potential vanilloid type 1 (TRPV1) channel expression in hypertrophic heart. Biochem Biophys Res Commun. 2010. October 8;401(1):98–103. PubMed PMID: 20833132; eng. [DOI] [PubMed] [Google Scholar]
- [25].Lu S, Xu D. Cold stress accentuates pressure overload-induced cardiac hypertrophy and contractile dysfunction: role of TRPV1/AMPK-mediated autophagy. Biochem Biophys Res Commun. 2013. December 6;442(1–2):8–15. PubMed PMID: 24211590; eng. [DOI] [PubMed] [Google Scholar]
- [26].Hiley CR. Endocannabinoids and the heart. J Cardiovasc Pharmacol. 2009. April;53(4):267–276. PubMed PMID: 19276990; PubMed Central PMCID: PMCPMC2728560. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kaneko Y, Szallasi A. Transient receptor potential (TRP) channels: a clinical perspective. Br J Pharmacol. 2014. May;171(10):2474–2507. PubMed PMID: 24102319; PubMed Central PMCID: PMCPMC4008995. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuhn M. Molecular physiology of natriuretic peptide signalling. Basic Res Cardiol. 2004. March;99(2):76–82. PubMed PMID: 14963665. [DOI] [PubMed] [Google Scholar]
- [29].Tremblay J, Desjardins R, Hum D, et al. Biochemistry and physiology of the natriuretic peptide receptor guanylyl cyclases. Mol Cell Biochem. 2002. January;230(1–2):31–47. PubMed PMID: 11952095. [PubMed] [Google Scholar]
- [30].Mohapatra DP, Nau C. Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem. 2003. December 12;278(50):50080–50090. PubMed PMID: 14506258. [DOI] [PubMed] [Google Scholar]
- [31].Varga A, Bolcskei K, Szoke E, et al. Relative roles of protein kinase A and protein kinase C in modulation of transient receptor potential vanilloid type 1 receptor responsiveness in rat sensory neurons in vitro and peripheral nociceptors in vivo. Neuroscience. 2006. June 30;140(2):645–657. PubMed PMID: 16564637. [DOI] [PubMed] [Google Scholar]
- [32].Johnston CI, Hodsman PG, Kohzuki M, et al. Interaction between atrial natriuretic peptide and the renin angiotensin aldosterone system. Endogenous antagonists. Am J Med. 1989. December 26;87(6b):24s–28s. PubMed PMID: 2532457; eng. [DOI] [PubMed] [Google Scholar]
- [33].Zygmunt PM, Chuang H, Movahed P, et al. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000. May 12;396(1):39–42. PubMed PMID: 10822052; eng. [DOI] [PubMed] [Google Scholar]
- [34].Brandt RR, Wright RS, Redfield MM, et al. Atrial natriuretic peptide in heart failure. J Am Coll Cardiol. 1993. October;22(4 Suppl A):86a–92a. PubMed PMID: 8376700; eng. [DOI] [PubMed] [Google Scholar]
- [35].Pomonis JD, Harrison JE, Mark L, et al. N-(4-Tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl)tetrahydropyrazine −1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: II. in vivo characterization in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;306(1):387–393. Epub 2003 Apr 29. [DOI] [PubMed] [Google Scholar]
- [36].Valenzano KJ, Grant ER, Wu G, et al. N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine −1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: I. in vitro characterization and pharmacokinetic properties. J Pharmacol Exp Ther. 2003;306(1):377–386. Epub 2003 Apr 29. [DOI] [PubMed] [Google Scholar]
- [37].Varga A, Nemeth J, Szabo A, et al. Effects of the novel TRPV1 receptor antagonist SB366791 in vitro and in vivo in the rat. Neurosci Lett. 2005. September 9;385(2):137–142. PubMed PMID: 15950380; eng. [DOI] [PubMed] [Google Scholar]
- [38].Gunthorpe MJ, Rami HK, Jerman JC, et al. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology. 2004. January;46(1):133–149. PubMed PMID: 14654105; eng. [DOI] [PubMed] [Google Scholar]
- [39].Gavva NR, Tamir R, Qu Y, et al. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005. April;313(1):474–484. PubMed PMID: 15615864; eng. [DOI] [PubMed] [Google Scholar]
- [40].Duerr GD, Heinemann JC, Dunkel S, et al. Myocardial hypertrophy is associated with inflammation and activation of endocannabinoid system in patients with aortic valve stenosis. Life Sci. 2013. May 30;92(20–21):976–983. PubMed PMID: 23567807; eng. [DOI] [PubMed] [Google Scholar]
- [41].Lee DI, Zhu G, Sasaki T, et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015. March 26;519(7544):472–476. PubMed PMID: 25799991; PubMed Central PMCID: PMCPMC4376609. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu Z, Lee S, Han J. Dual role of cyclic GMP in cardiac cell survival. Int J Biochem Cell Biol. 2013. August;45(8):1577–1584. PubMed PMID: 23660294; eng. [DOI] [PubMed] [Google Scholar]
- [43].Janssen W, Schermuly RT, Kojonazarov B. The role of cGMP in the physiological and molecular responses of the right ventricle to pressure overload. Exp Physiol. 2013. August;98(8):1274–1278. . PubMed PMID: 23873899; eng. [DOI] [PubMed] [Google Scholar]
- [44].Takimoto E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ J. 2012;76(8): 1819–1825. PubMed PMID: 22785374; eng. [DOI] [PubMed] [Google Scholar]
- [45].Vemula P, Gautam B, Abela GS, et al. Myocardial ischemia/reperfusion injury: potential of TRPV1 agonists as cardioprotective agents. Cardiovasc Hematol Disord Drug Targets. 2014;14(1):71–78. PubMed PMID: 24304232; eng. [DOI] [PubMed] [Google Scholar]
- [46].Molkentin JD. A friend within the heart: natriuretic peptide receptor signaling. J Clin Invest. 2003. May;111(9):1275–1277. . PubMed PMID: 12727915; PubMed Central PMCID: PMCPMC154452. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jin Y, Kim J, Kwak J. Activation of the cGMP/protein kinase G pathway by nitric oxide can decrease TRPV1 activity in cultured rat dorsal root ganglion neurons. Korean J. Physiol. Pharmacol.. 2012. June;16(3):211–217. . PubMed PMID: 22802704; PubMed Central PMCID: PMCPMC3394925. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bhave G, Hu HJ, Glauner KS, et al. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc Natl Acad Sci U S A. 2003. October 14;100(21):12480–12485. PubMed PMID: 14523239; PubMed Central PMCID: PMCPMC218783. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem. 2005. April 8;280(14):13424–13432. PubMed PMID: 15691846; eng. [DOI] [PubMed] [Google Scholar]
- [50].Basu S, Srivastava P. Immunological role of neuronal receptor vanilloid receptor 1 expressed on dendritic cells. Proc Natl Acad Sci U S A. 2005. April 5;102(14):5120–5125. PubMed PMID: 15793000; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gunthorpe MJ, Szallasi A. Peripheral TRPV1 receptors as targets for drug development: new molecules and mechanisms. Curr Pharm Des. 2008;14(1): 32–41. PubMed PMID: 18220816; eng. [DOI] [PubMed] [Google Scholar]
- [52].Earley S. Vanilloid and melastatin transient receptor potential channels in vascular smooth muscle. Microcirculation. 2010. May;17(4):237–249. PubMed PMID: 20536737; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Peng G, Lu W, Li X, et al. Expression of Store-operated Ca2+ entry and transient receptor potential canonical and vanilloid-related proteins in rat distal pulmonary venous smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2010. August 6 PubMed PMID: 20693314; Eng DOI: 10.1152/ajplung.00176.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang YX, Wang J, Wang C, et al. Functional expression of transient receptor potential vanilloid-related channels in chronically hypoxic human pulmonary arterial smooth muscle cells. J Membr Biol. 2008. June;223(3):151–159. PubMed PMID: 18787888; eng. [DOI] [PubMed] [Google Scholar]
- [55].Bratz IN, Dick GM, Tune JD, et al. Impaired capsaicin-induced relaxation of coronary arteries in a porcine model of the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2008. June;294(6):H2489–96. PubMed PMID: 18390821; eng. [DOI] [PubMed] [Google Scholar]
- [56].Mohapatra SS. Role of natriuretic peptide signaling in modulating asthma and inflammation. Can J Physiol Pharmacol. 2007. July;85(7):754–759. PubMed PMID: 17823639; eng. [DOI] [PubMed] [Google Scholar]
- [57].Chen M, Xin J, Liu B, et al. Mitogen-activated protein kinase and intracellular polyamine signaling is involved in TRPV1 activation-induced cardiac hypertrophy. J Am Heart Assoc. 2016. July 29;5(8). PubMed PMID: 27473037; PubMed Central PMCID: PMCPMC5015292. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huang S, Szallasi A. Transient Receptor Potential (TRP) channels in drug discovery: old concepts & new thoughts. Pharmaceuticals (Basel). 2017. July 6;10(3). PubMed PMID: 28684697; PubMed Central PMCID: PMCPMC5620608. eng DOI: 10.3390/ph10030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Magdalene MM. TRP channels as potential drug targets. Annu Rev Pharmacol Toxicol. 2018;58(1):309–330. [DOI] [PubMed] [Google Scholar]
- [60].Smith H, Brooks JR. Capsaicin-based therapies for pain control. Prog. Drug Res. 2014;68: 129–146. PubMed PMID: 24941667; eng. [DOI] [PubMed] [Google Scholar]
- [61].Charrua A, Cruz CD, Narayanan S, et al. GRC-6211, a new oral specific TRPV1 antagonist, decreases bladder overactivity and noxious bladder input in cystitis animal models. J Urol. 2009. January;181(1):379–386. PubMed PMID: 19010489; eng. [DOI] [PubMed] [Google Scholar]
- [62].Stokes AJ, Shimoda LM, Lee JW, et al. Fcepsilon RI control of Ras via inositol (1,4,5) trisphosphate 3-kinase and inositol tetrakisphosphate. Cell Signal. 2006. May;18(5):640–651. PubMed PMID: 16005187; eng. [DOI] [PubMed] [Google Scholar]
- [63].Rockman HA, Ono S, Ross RS, et al. Molecular and physiological alterations in murine ventricular dysfunction. Proc Natl Acad Sci U S A. 1994. March 29;91(7):2694–2698. PubMed PMID: 8146176; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Breckenridge R. Heart failure and mouse models. Dis Model Mech. 2010. Mar-Apr;3(3–4):138–143. PubMed PMID: 20212081; eng. [DOI] [PubMed] [Google Scholar]
- [65].Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, et al. Animal models of cardiovascular diseases. J Biomed Biotechnol. 2011;2011:497841 PubMed PMID: 21403831; PubMed Central PMCID: PMCPmc3042667. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kreusser MM, Lehmann LH, Keranov S, et al. Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation. 2014. October 7;130(15):1262–1273. PubMed PMID: 25124496; PubMed Central PMCID: PMCPMC4316667. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ling H, Zhang T, Pereira L, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009. May;119(5):1230–1240. PubMed PMID: 19381018; PubMed Central PMCID: PMCPMC2673879. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009. February 17;106(7):2342–2347. PubMed PMID: 19179290; PubMed Central PMCID: PMCPMC2650158. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.