

Abstract
Inspired by nature, poly(acrylic acid) (PAA) and other polyelectrolytes have been used as noncollagenous proteins (NCPs) surrogates for biomimetic intrafibrillar mineralization of collagen fibrils and thus, to model the ultrastructure of bone, to study the mechanism of bone mineralization and, more scarcely to fabricate scaffolds for hard tissue engineering. The objective of this study was to systematically investigate the effect of the molecular weight (MW) and the concentration of PAA on the rate and pattern of biomineralization of collagen matrices. Densified type I collagen films were mineralized in supersaturated PAA-stabilized amorphous calcium-phosphate (PAA-ACP) solutions containing increasing MW (2 kDa, 50 kDA, 450 kDa) and concentrations (10, 25, 50 mg/L) of PAA up to 7 days. The stability and physical properties of collagen-free PAA-ACP solutions were also investigated. In our system, lowering PAA MW and increasing PAA concentration resulted in solutions with increasing stability. Over stable PAA-ACP solutions that fully inhibited mineralization of the collagen matrices were achieved using PAA 2k-50. Conversely, unstable solutions were obtained using high PAA MW at low concentrations. Nucleation and growth of significant amount of extrafibrillar minerals on the collagen fibrils was obtained using these solutions. In a wide range of combined MW and concentration of PAA we obtained intrafibrillar mineralization of collagen with hydroxyapatite crystals aligned parallel to the collagen fibril as in natural tissues. Intrafibrillar mineralization was correlated with PAA-ACP stability and growth of the PAA-ACP particles in solution. Our results support using PAA to surrogate NCPs function as selective inhibitors or promoters of biological mineralization and provide parameters to manufacture new biomimetic scaffolds and constructs for bone and dentin tissue engineering.
Keywords: Poly(Acrylic Acid), Molecular Weight, Intrafibrillar Biomineralization, Amorphous Calcium Phosphate, Stabilization
Graphical Abstract
1. Introduction
Bone is a natural organic-inorganic hybrid composite with unique hierarchical structure and mechanical properties1–3. The basic building block of bone is a mineralized nanostructure which is comprised of collagen fibrils with platelets of carbonated hydroxyapatite (HAp) nanocrystals. HAp nanocrystals in bone are around 2–6 nm thick, 30–50 nm wide, and 60–100 nm long4 with their c-axis preferentially aligned with the long axis of the collagen fibrils, leading to an interpenetrating organic-inorganic nanocomposite2, 5–6. The highly ordered structure of intrafibrillar mineralization at the nanoscale is considered the foundation of the biomechanical properties of bone6–7.
Even though the nanostructure and hierarchy of bone are well understood, the process by which minerals precipitate within the collagen fibril is still under debate. Ex vivo observations of bone formation have demonstrated that the mineral associates with collagen at an early stage in a transient metastable amorphous calcium phosphate phase (ACP)8–10. It has been proposed that ACPs are delivered to collagen at the bone growing front and infiltrate into the fibril interstices where they transform into apatite crystals9. However, collagen fibrils cannot initiate and mediate this organized calcium phosphate mineralization when only mineralizing ions or simulated body fluids are used as the reaction media11–14. The intrafibrillar mineralization of collagen is believed to be regulated through interactions between the ACPs with the collagen matrix and noncollagenous proteins (NCPs). NCPs are intrinsically-disordered proteins that can sequester ions in solution to form stabilized ACPs that mediate bone mineralization. Many of the NCPs are highly negatively charged with abundant carboxylate groups from aspartic and glutamic acid residues and/or phosphorylated amino acids, such as phosphoserine15–16. Although the specific role of each NCP in bone mineralization is still under debate, mineralization studies using NCPs have indicated that they have multiple potential roles as nucleators, inhibitors, growth modifiers, anchoring molecules, or scaffolds for mineral deposition8, 17–23. These studies have also suggested that the degree of phosphorylation of NCPs plays a crucial role on the intrafibrillar mineralization of collagen in bone24–25.
Due to the unavailability of NCPs or their recombinant versions in practice, negatively-charged polyelectrolytes such as poly-L-aspartic acid (pAsp), poly-L-glutamic acid (pGlu), polyvinylphosphonic acid (PVPA), and poly(acrylic acid) (PAA) have been used as analogs of NCPs in in vitro biomimetic models for collagen mineralization13, 26–31. The polymer-induced liquid precursor process (PILP) that uses pAsp to form pAsp-ACP precursors has been the most widely used method to produce biomimetic bone mineralization in vitro28, 32–34. This process and its variants using alternative poly-anions have been adopted to model the ultrastructure of bone, to study the mechanism of bone mineralization13, 35 and to fabricate scaffolds for bone and dentin tissue engineering36–38.
Discordances in the literature about the role of pAsp in the biomineralization of collagen fibers have been reported and discussed in relation to the mechanism of bone mineralization (promoter/inhibitor effect) and rate and pattern of mineralization31–32. Those discrepancies might be attributed to differences in the relevant variables of the poly-anions, namely the molecular weight (MW) of the polymer, and the concentration used; which in turn determine the [-COO−]/[Ca2+] ratio in solution. An initial attempt to investigate the effect of MW of pAsp (MW: 10.3 kDa or 32.2 kDa) on intrafibrillar collagen mineralization was reported by Jee et al.28. Using 32.2 kDa pAsp yielded faster and more mature mineralization than using 10.3 kDa pAsp, which was attributed to a higher stability of pAsp-ACP precursors produced with higher-MW pAsp. The above study was limited to a narrow range of pAsp MW and did not investigate effects of the polyelectrolyte concentration on biomineralization or the combined effects of MW and concentration on crystal morphologies and orientation.
Here, we studied the effects of MW and concentration of PAA, a polyelectrolyte NCP analog, on the rate and pattern of biomineralization of collagen matrices. Specifically, PAA was selected for this work due to its availability in a broad range of MWs, namely 2 to 4,000 kDa. The molecular structure of PAA contains carboxylate groups that provide abundant negative charges to the molecule at physiological pH and temperature. PAA has been successfully applied in in vitro mineralizing processes for controlling the dimensions of the amorphous phases, modulating the mineral morphology and modifying crystal growth of calcium-based minerals14, 33, 39–40. However, the relation between the stability of mineralizing PAA-ACP, as a function of the MW and concentration of PAA, and collagen biomimetic mineralization with calcium phosphates has yet to be determined, which is also an objective of this work.
2. Materials and Methods
2.1. Fabrication of collagen matrix
PureCol collagen solution (97% bovine dermal type I collagen) was purchased from Inamed Biomaterials (Fremont, CA, USA). Collagen matrices were prepared as described previously31. To reconstitute the fibrils, 48 mL of type-I collagen solution (2.9 mg/ml) was mixed with 12 ml of a 10× PBS buffer and 8 ml of 0.1 N NaOH solution. The mixture was incubated at 30 °C for three days and compressed to form a thin sheet. Cross-linked collagen matrices were obtained by immersing the collagen sheets in a solution of 50 mM 2-(N-morpholino)ethanesulfonic acid hydrate (MES, Sigma-Aldrich, St. Louis, MO, USA) (pH 7) with 50 mM 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC, Sigma-Aldrich, St. Louis, MO) and 25 mM N-hydroxysuccinimide (NHS, Sigma-Aldrich, St. Louis, MO, USA) overnight. The reaction was quenched in a solution of 0.1 M Na2HPO4 and 2 M NaCl for 2 hours. The collagen matrix was then rinsed with deionized (DI)-water and lyophilized for mineralization.
2.2. Mineralization of collagen matrix
9 mM CaCl2 (Sigma-Aldrich, St. Louis, MO, USA) and 4.2 mM K2HPO4 (Sigma-Aldrich, St. Louis, MO, USA) solutions were prepared in tris-buffered saline (TBS) at pH 7.4 and 37 °C. PAA of three different MWs (2 kDa (Sigma-Aldrich, St. Louis, MO, USA), 50 kDa (Polysciences, Inc, Warrington, PA, USA) or 450 kDa (Sigma-Aldrich, St. Louis, MO, USA)) were used as process-directing agents32. Each PAA was dissolved in the phosphate solution at three different concentrations (20, 50 or 100 mg/L) before being mixed with an equal volume of the calcium counterion solution. Thus, nine different experimental conditions were tested and named “MW-PAA final concentration in mineralizing solution”. For example, a solution with PAA-2kDa at 50 mg/L concentration was named “2k-50”. Mineralization was conducted by incubating cross-linked collagen matrices in the aforementioned mineralizing solutions28, which were refreshed every 3 days. After 1, 3, and 7 day(s), the collagen matrices were rinsed with DI-water and lyophilized for characterization.
2.3. Scanning Electron Microscopy (SEM)
The morphologies of the specimens were imaged with a Field Emission Gun SEM (JEOL 6500, Tokyo, Japan) operated at an accelerating voltage of 5 kV. All specimens were sputter-coated with a 5-nm thick platinum layer. For elemental analysis of mineralized samples, energy dispersive X-ray spectroscopy (EDS) analysis was performed during SEM examination. EDS line scans across the cross-sections and mapping of Ca for samples incubated in 450k-50, 450k-10 and 50k-10 (MW in Da-concentration in mg/L) solutions were collected with back-scattered electron (BSE) imaging at 15 kV (Hitachi SU8230, Tokyo, Japan).
2.4. Transmission Electron Microscopy (TEM)
Mineralized collagen matrices were crushed to fine-grained powders in liquid nitrogen, dispersed in ethanol and dropped on a lacey carbon coated copper TEM grid with 200 mesh size. Samples were analyzed using a TEM (FEI Tecnai 12, Thermo Fisher Scientific, Waltham, MA, USA) operated at 120 kV in bright-field (BF), dark-field (DF), and selected-area electron diffraction (SAED) modes.
2.5. Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR)
FTIR analysis of the collagen films before and after mineralization was performed by a FT-IR spectrometer (Nicollet iS50, Thermo Fisher Scientific, Waltham, MA, USA), equipped with a built-in diamond attenuated total reflection (ATR) for single-spot ATR measurement. Each spectrum was the result of signal-averaging of 32 scans at a resolution of 2 cm−1 with wavenumber ranged from 400 to 4000 cm−1.
2.6. X-ray Diffractometry (XRD) Analysis
The crystal structure of mineralized samples was characterized using a micro-diffractometric system with a two-dimensional area detector (AXS, Bruker, Billerica, MA, USA) operated at 40 kV and 35 mA. The incident angle was 15° and the detector position was fixed at 30°, which covered a 2θ = 15°−45° range. The data collection time was 1500 s and the results were analyzed using JADE8 software (JADE, Materials Data Inc., Livermore, CA, USA).
2.7. Thermogravimetric and Derivative Thermogravimetric Analysis (TG/DTG).
The mineral content of collagen films was determined by TG/DTG (Stare V9.30, Mettler Toledo, Columbus, OH, USA). Lyophilized collagen films were cut into small pieces of about 10 mg and placed in a 900 μL Alumina pan. The temperature was raised from 30 to 800 °C at a heating rate of 5 °C min−1 in air. Air flow was controlled to be 100 mm3/min. The mineral content was determined by the remaining weight at 700 °C after the organic portion of the samples was fully combusted.
2.8. Stability assessment of PAA-stabilized amorphous calcium-phosphate (PAA-ACP) solutions
The effect of concentration and MW of PAA on the stability and growth rate of the amorphous calcium-phosphate precursor was examined. The solutions were placed in an incubator at 37 °C for 7 days. Optical density (OD) measurements at a fixed wavelength of 650 nm were taken at 0 h, 24 h, and 2, 3, 4, 5, 6 and 7 days with a Beckman Coulter AD 340 Plate Reader (Indianapolis, IN, USA). Three independent measurements were taken at each time point.
Subsequent OD measurements were conducted every 2 hours on selected solutions to get a detailed precipitation curve and to calculate the precipitation half-time, t1/2, of each reaction. Curves of OD as a function of time (t) were fitted to a logistic function:
where A is the final OD at t → ∞; B is the rate of OD increase; and t1/2 is the midpoint of the logistic curve (or 1/2 peak height). A sum of squares method and the Excel Solver Function was used to minimize the sum of squared residuals.
Particle sizes of the mineralizing solutions were then analyzed using a Particle Size Analyzer (Nanotrac Flex, Microtrac, Montgomeryville, PA, USA) with the dynamic light scattering technique (DLS). The measurements were taken with the refractive index mode and the polymer shape was simplified to be spherical. Zeta potential of nanoparticles suspended in solutions was measured with a Zeta potential analyzer (Stabino, Particle Metrix, Meerbusch, Germany) using the Current Status mode. Particle size and charge was determined after 1, 24 and 48 h in solution. Three independent measurements were taken at each time point.
3. Results
3.1. Effect of concentration and MW of PAA on mineralization of collagen films
Mineralization of the densified collagen films was conducted in PAA-ACP solutions for 7 days with PAA of different MWs (2, 50 and 450 kDa) and concentrations (10, 25 and 50 mg/L) (Fig. 1).
Figure 1:

SEM images of type I collagen fibrils mineralized for 7 days with 2k-10 (A), 2k-25 (B), 2k-50 (C), 50k-10 (D), 50k-25 (E), 50k-50 (F), 450k-10 (G), 450k-25 (H), and 450k-50 (I) PAA solutions. IM: fibrils with intrafibrillar mineralization. EM: extrafibrillar mineralization of fibrils. Arrows in (B) and (C): collagen fibrils not mineralized showing the characteristic banding pattern of native collagen. Scale bar: 200 nm.
Low-MW (2 kDa) PAA at high concentration (50 mg/L) (2k-50) inhibited both extrafibrillar and intrafibrillar mineralization (Fig. 1C) of the collagen matrix. This was the only experimental condition tested here that impeded both forms of mineralization of collagen. After the mineralization period, collagen fibrils in the above solution displayed the same morphologies as those before mineralization, showing the characteristic banding pattern of native collagen (Fig. 1C arrow, S1). The lack of mineralization was further verified by quantification of mineral content by TG (Fig. 4A).
Intrafibrillar mineralization of the collagen matrices was achieved with solutions containing low-MW (2 kDa) PAA at low (10 mg/L) and medium (25 mg/L) concentrations (2k-10, 2k 25) as well as solutions containing medium- (50 kDa) and high-MW (450-kDa) PAA at medium (25 mg/L) and high (50 mg/L) concentrations (50k-25, 50k-50, 450k-25, 450k-50). When intrafibrillar mineralization was achieved in these systems, collagen fibrils with uniform diameters and surfaces with no apparent banding pattern or extrafibrillar minerals were obtained (Fig. 1A, 1B, 1E, 1F, 1H, 1I). Intrafibrillar mineralization of the collagen matrices started after just 1 day in solution (Fig. S2, S5A–C). The width of the fibrils increased due to the infiltration of minerals, which replaced the water, during mineralization (Fig. S3B, S3). All collagen matrices that were intrafibrillarly mineralized contained more than 50 wt% mineral, with the highest mineral contents (63–65 wt%) obtained in solutions with 50 kDa and 450 kDa PAAs (Fig. 1, 4B, 4C). In the least mineralized samples (2k-25), some of the non-mineralized thinner fibrils were still seen with the apparent banding pattern (Fig. 1B, arrow).
Intra- and extrafibrillar mineralization of fibrils in collagen matrices were obtained with 50 kDa and 450 kDa PAA solutions at low (10 mg/L) concentration (Fig. 1D, 1G). Platelet-like nanocrystals were found randomly distributed on the fibrils’ surface. Fibrils on the outer surface of the collagen matrix were also infiltrated with nanocrystals. However, deep inside the collagen matrices exposed to these two PAA solutions, intrafibrillar mineralization of the fibrils was not achieved, even after 7 days in solution (Fig. S4B, S4D, arrows). This most likely was due to the thick layer of extrafibrillar apatite crystals deposited on the surface of the collagen matrix (Fig. S4A, S4C, stars) which hindered passage of PAA-ACP particles and/or ions to deeper areas of the matrix.
BSE cross-sectional images and EDS Ca line scans and mappings showed different mineralization depths of the collagen matrices treated with 450k-50 and 450k-10 solutions (Fig. 2). Uniform and full mineralization occurred across the sample treated with the 450k-50 solution as the Ca signal was evenly distributed throughout the cross-section (Fig. 2A, 2B). However, Ca was detected only in areas close to the surfaces of matrices mineralized with the 450k-10 solution. The inner region had a significant drop in Ca content due to the sparse mineral penetration (Fig. 2C, 2D). This mineral-depleted area was more prominent in matrices mineralized with the 450k-10 solution than those with the 50k-10 solution (Fig. S6). This was also in agreement with the relatively lower mineral content in the matrices mineralized with the 450k-10 solution (53 wt%) as compared to those mineralized in the 50k-10 solution (62 wt%) and the ones with only intrafibrillar mineralization obtained with 50 kDa and 450 kDa PAA at medium and high concentrations (63–65 wt%).
Figure 2:

BSE images with EDS Ca line-scans (A, C) and Ca mapping (B, D) of the cross-section of densely packed collagen matrices mineralized for 7 days with 450k-50 (A, B) and 450k-10 (C, D) PAA solutions. Scale bar: 50 μm.
3.2. Characterization of minerals
SEM and TEM micrographs confirmed that after 7 days in mineralizing PAA solutions, intrafibrillar crystalline HAp was formed (Fig. 3A). A cross-sectional view of a mineralized collagen fibril (Fig. 3Ai) shows nanocrystals through the thickness of the collagen fibril. SAED analysis of the obtained crystals showed characteristic HAp patterns with arcing of the (002) planes, indicating that the minerals were aligned with the [001] direction parallel to the longitudinal axis of the fibrils (Fig. 3Aii). Dark-field TEM micrographs for the (002) planes also showed high infiltration of these aligned crystals in the collagen fibrils (Fig. 3Aiii). ATR-FTIR spectra for mineralized collagen samples after 7 days in selected mineralizing solutions showed characteristic absorbance bands for carbonated apatite (Fig. 3B). Bands at 960 and 1020 cm−1 were assigned to the stretching modes of υ3PO4 and υ1PO4. The peaks at 560 and 602 cm−1 were identified as υ4 and υ2 O-P-O bending modes. The peaks at 873 cm−1 were identified as the υ2 C-O stretching mode of carbonate substitution in the apatite lattice. No specific bands assigned to the minerals were detected in collagen mineralized with the 2k-50 PAA solution and the control pure collagen matrix. XRD spectra of collagen matrices mineralized with the 2k-10 solution and 450 kDa PAA solutions at high (50 mg/L) and low (10 mg/L) concentrations for 7 days showed characteristic peaks of HAp from the (002), (210), (211), (300), (310) and (311) planes (Fig. 3C). The broad peak around 2θ=32° was composed of overlapping peaks from the (211), (112), and (300) planes, as was similarly found in XRD spectra for native trabecular bone28, 31, 41.
Figure 3:

(A) SEM (i), TEM bright-field (ii) and TEM dark-field (iii) micrographs of collagen fibrils mineralized with 450k-50 PAA solution for 7 days. Insert in Aii is the SAED pattern of the mineralized fibril shown in Aii and Aiii. Scale bar: 100nm. (B) ATR-FTIR spectra of collagen matrices mineralized for 7 days with 450k-10, 450k-50, 2k-10, 2k-50 PAA solutions and control pure type I collagen. Characteristic absorption bands of carbonated HAp are labeled. (C) XRD spectra of collagen matrices mineralized for 7 days with 2k-10, 450k-10 and 450k-50 PAA solutions. Characteristic peaks of HAp for the (002), (210), (211), (300), (310), (311) planes are labeled.
3.3. Quantification of mineralization
The almost identical mineral content in control type-I collagen (5 wt%) and matrices in PAA 2k-50 solutions (4 wt%) further confirmed that neither extrafibrillar nor intrafibrillar mineralization was achieved for this experimental condition (Fig. 4A). For the mineralized collagen matrices, mineral contents range between 53–67 wt%. Lower mineral contents were obtained in collagen matrices mineralized in solutions with the lowest MW, 2 kDa i.e., those with delayed intrafibrillar mineralization. Collagen matrices with significant extrafibrillar mineralization, as the ones obtained using PAA 450k-10 solutions (Fig.4C), also showed mineral contents in the lower range. This was probably due to the fact that early extrafibrillar crystal deposition hindered further mineral infiltration into the collagen fibrils. Five wt% residue was detected in control unmineralized collagen matrices, which is likely due to the presence of unwashed salts during sample preparation.
Figure 4:

TG (A-C) and DTG (D-F) curves of collagen matrices mineralized for 7 days with solutions PAA 2-kDa (A, D), 50-kDa (B, E), and 450-kDa (C, F) and different concentrations. The mineral after decomposition up to 700 °C and decomposition temperature are noted on each curve.
In the case of pure collagen, three characteristic weight loss peaks were observed in the DTG curves. These weight losses correspond to the loss of physisorbed water (under 100 °C), followed by decomposition (260–360 °C) and then combustion (450–600 °C) of the organic matrix42. The intrafibrillarly mineralized collagen matrices had a slightly higher thermal decomposition temperature for collagen than the unmineralized collagen samples (from 300 °C to 334 °C). As we have reported previously, this change in decomposition temperature reflects the intimate structural relationship between collagen and HAp31, which suggests the majority of the crystals are embedded within the collagen fibrils. This has been previously reported for collagen in bone, dentin and turkey tendon42–43. It is worth noting that the matrices mineralized in PAA 2k-50 solutions also showed a peak near 334oC, which is not present in the pure collagen matrices, and has lower relative intensity than in the matrices that were fully intrafibrillarly mineralized. This might be an indication of an initial stabilization of the collagen due to ionic interactions in the system and thus, a longer period of exposure to the 2k-50 solution might result in a in a mineralized matrix. The high-temperature peak (536 °C) present in the pure collagen matrices almost disappeared in the mineralized specimens, demonstrating that the mineralization of collagen strongly altered its thermal behavior.
3.4. Characterization of the PAA-ACP system
The stability of the PAA-ACP solutions with different PAA MWs and concentrations was studied by measuring the solution OD (Fig. 5A) and the size and charge of the PAA-stabilized mineral precursors (Fig. 5B, 5C), and the calculated precipitation half-time (Table 1). Overall, the lower the PAA MW or the higher the PAA concentration, the higher the stability of the mineralizing solution. For each PAA MW, t1/2 decreased as PAA concentration decreased; and for each PAA concentration, t1/2 decreased with increasing PAA MW (Table 1). Only solutions containing 50 mg/L of 2 kDa or 50 kDa PAA were stable, with no mineral precipitation over the period tested. The least stable solution was 450k-10 with t1/2 = 3.4 h.
Figure 5:

(A) OD profiles of PAA-ACP solutions with 2k-Da (i), 50-kDa (ii) and 450-kDa (iii) PAA and different PAA concentrations. (B) Mean particle size and (C) mean charge of PAA-ACP particles in mineralizing solutions with 2k-Da (i), 50k-Da (ii) and 450k-Da (iii) PAA and different PAA concentrations after 1, 24 and 48 hour(s). *: Particles, if in solution, had sizes below the limit of detection (1 nm). #: Solutions with precipitated minerals.
Table 1.
Precipitation half-time (t1/2, h) for PAA-ACP solutions with PAA of different MWs and concentrations.
| PAA MW (kDa) | PAA concentration | ||
|---|---|---|---|
| 10 mg/L | 25 mg/L | 50 mg/L | |
| 2 | 29.0 | 75.2 | >168.0 |
| 50 | 8.0 | 15.6 | >168.0 |
| 450 | 3.4 | 7.1 | 43.9 |
Particle-size analysis with DLS showed that PAA-ACP particles grew faster when formed in solutions with higher-MW PAA and/or lower PAA concentration. Larger particle sizes were found in solutions with higher-MW PAA. The mean particle sizes of all 2 kDa PAA solutions at 1 h were below the limit of detection (1 nm). The particles in the 2k-50 solution remained under the limit of detection for the period tested. However, particles formed in 2k-25 and 2k-10 solutions grew to a size detectable by DLS, this being about 50 nm and 300 nm (most likely agglomerates of smaller particles), after 48 h and 24 h, respectively (Fig. 5Bi). Particles formed in the 50 kDa or 450 kDa PAA solutions were larger than those formed in the 2 kDa PAA solutions when compared at the same time points and PAA concentrations (i.e. 50 mg/L at 1 h), and, except for 50k-50, precipitated after 48 h. The precipitated particles were not detectable by DLS (Fig. 5Bii, 5Biii). These results agreed with those from the turbidity tests.
The charge of the PAA-ACP particles was not influenced by the PAA MW or PAA concentration and did not change over time with a value of near −25 mV for the detectable particles (Fig. 5C).
4. Discussion
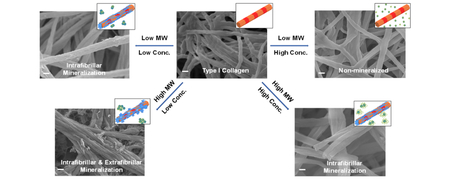
The main goal of this study was to investigate the effect of MW and concentration of PAA on the process and outcome of intrafibrillar mineralization of collagen. Previous studies have demonstrated that PAA, as is the case for pAsp, stabilizes fluidic ACP nanoprecursors17, 44–48 and that concentration of PAA in solution can modulate the rate of mineralization of demineralized dentin matrices49. Also, the stabilizing strength of PAA on calcium carbonate crystals is weaker with a longer molecular chain length or a lower ratio of [-COO−]/[Ca2+] in solution50. Fig. 6 schematically summarizes our results and shows the main relationships found in this study for MW and concentration of PAA, PAA-ACP particles formed, and the pattern of mineralization of collagen fibrils. In our system, lowering PAA MW and increasing PAA concentration resulted in PAA-ACP solutions with increasing stability. The most stable PAA-ACP solution that fully inhibited mineralization of the collagen matrices was achieved using the combination 2k-50. Conversely, the least stable solutions were obtained using high PAA MW at low concentrations. Significant amount of extrafibrillar minerals on the collagen fibrils was obtained using these unstable solutions. Thus, the stability of the PAA-ACP solutions and the intrafibrillar mineralization of collagen in these solutions were related and controlled by the combined effects of the PAA MW and concentration. Solutions with intermediate stability seemed to be the most effective in inducing purely intrafibrillar mineralization of the collagen.
Figure 6:

A schematic representation of the influence that MW and concentration of PAA has on formation of PAA-ACP in solution and collagen fibril mineralization. PAA-ACP, HAp nanocrystals, and collagen fibrils are not in the same scale.
Our results support using PAA as a surrogate for the proposed dual functions of some NCPs as being selectively inhibitors or promoters of biological mineralization. We showed that biomimetic intrafibrillar mineralization of collagen was obtained in mineralization solutions containing PAA with a wide range of MW, provided that each polymer was in the appropriate concentration so that neither full inhibition nor extrafibrillar mineralization occurred. NCPs, as it is also the case of PAA in this study, have a wide range of MW, are intrinsically-disordered and highly negatively charged, which provides their ability to stabilize ACPs51. The level of expression of NCPs is regulated in living systems to avoid pathological events so that each specific protein performs the role of inhibitor or promoter of mineralization that is required at each body location and point of time51.
Increasing PAA concentration increases the [-COO−]/[Ca2+] ratio, retards ACP crystallization to HAp in solution, and hinders initial ACP aggregation49. The latter was confirmed here as the growth of the PAA-ACP particles/agglomerates was slower in solutions with increasing concentration of PAA for each MW. Adsorption of PAA on the surface of ACP to form PAA-ACPs would hinder ACP aggregation, as it has been previously shown for PAA-silicon nitride colloidal systems52. The ability to delay PAA-ACP agglomeration was also more effective with decreasing PAA MW, which highlights the interactive effects between the two PAA variables evaluated here on stability of the solution. Others have shown that lowering PAA MW provided more stable amorphous calcium carbonates formed using the delayed addition method50. The authors speculated that the higher mobility of PAA with a lower MW in water may cause faster adsorption on the precursors to inhibit their growth. The very small size of the PAA-ACPs found in our 2k-50 PAA solution while it was stable supports this proposition. Also, the length of the polyelectrolyte chains may affect the binding efficiency between–COO− and Ca2+ such that PAA with low to medium MW are more effective in inhibiting the growth and agglomeration of PAA-ACPs.
It seems that the ability of the very stable 2k-50 PAA solution to inhibit PAA-ACP growth could also inhibit mineralization of collagen altogether. On the other hand, the equally stable 50k-50 PAA solution, which had faster-growing and thus, larger PAA-ACP particles, was able to induce purely intrafibrillar mineralization of collagen. The 2k-50 combination therefore represented the limit, in terms of the lowest PAA MW and the highest PAA concentration, beyond which no intrafibrillar mineralization could occur.
In vitro studies have provided evidence that minerals within collagen fibers start as stable amorphous mineral particles that eventually transform into HAp crystallites49, 53. In a solution supersaturated with respect to more than one phase of a mineral, crystallization is thought to start from the most soluble (least stable) phase which then transforms to the most stable phase through a series of intermediate phases of different crystal structures17, 54. In the non-classical mineralization pathway, the multistep kinetically driven mineralization reaction depends on the relative heights of the Gibbs free energy barriers (ΔGi) between each of the metastable phases. In our system, we speculate that the key feature that PAA provides in transiently-stabilizing ACPs is its polyanionic character to produce a PAA-ACP precursor phase with the appropriate physical properties to enter the collagen matrix. The affinity of PAA to ACP increases the ΔGT of phase transformation from ACP to HAp. This was shown in all PAA-ACP solutions with longer t1/2 than the supersaturated calcium phosphate solution without PAA, which precipitated immediately after preparation. In the presence of collagen, although the exact mechanism is still elusive, ΔGT can be reduced by the collagen microfibril as a substrate promoting HAp solidification/nucleation53, 55 or by the nanoscale confinement effect56–57. Others have also proposed alternative mechanisms, such as the ‘osmotic pressure theory’30. This is in accordance with our results as the initial intrafibrillar mineralization of the collagen fibrils occurred under experimental conditions where PAA-ACP solutions showed stability and controlled growth of PAA-ACP agglomerates in the absence of collagen.
In solutions containing a very low concentration of PAA with high MW, minerals quickly aggregated, crystallized and precipitated. These large solid crystals were not able to penetrate the collagen fibrils and, as a result, extrafibrillar mineralization was formed. Both 50k-10 and 450k-10 PAA solutions induced intra- and extrafibrillar collagen mineralization, which suggests that the size and rate of growth of the mineral agglomerates in these solutions was near the limit of stability that prevents crystallization and precipitation of HAp and, thus, allows penetration into the collagen fibrils.
A full infiltration of HAp minerals with their crystal structure oriented in the collagen fibril direction as in the natural tissues was obtained; that is, the composition, distribution, structure, and orientation of the minerals within the collagen matrices closely resembled the ones in bone at the nano level. The maximum mineral content obtained for the intrafibrillarly mineralized collagen matrices indicates that these fibrils were fully mineralized, as 65% mineral and 35% collagen content is well within the range of values for natural bones58. Moreover, when extrafibrillar mineralization was prevented in this biomineralization system, the collagen matrices were fully mineralized through thickness. This was not the case when extrafibrillar mineralization occurred, probably because the mineral layer formed on the surface of the collagen matrix constituted a diffusion barrier for ions or mineral precursors to penetrate through its thickness. The full penetration of minerals to the inner parts of the matrices and to the inside of the collagen fibrils differentiate these HAp-collagen composites from others obtained by either the use of ionic supersaturated mineralization solutions or manufacturing processes that simply mix the two phases. The biomimetic character of the method used here for obtaining mineralized collagen matrices emphasizes its potential to manufacture scaffolds and constructs for tissue engineering with exceptional mechanical properties that match those of bone.
5. Conclusion
In our study PAA was used as a surrogate of NCPs as stabilizers of ACPs for collagen biomimetic mineralization. Densified type-I collagen matrices were mineralized in PAA-ACP solutions containing different PAA concentrations and MW. Together, our results showed that PAA MW and concentration modulated intrafibrillar mineralization of collagen fibrils by providing solution stability and control over the growth of the PAA-ACP precursor agglomerates. This biomimetic mineralization system produced collagen matrices with potential for hard tissue engineering applications as they resembled the ultrastructure of bone and were fully mineralized through thickness with high mineral contents. This system could also be used as a model for mineralized tissues such as bone and dentin.
Supplementary Material
Acknowledgments
Funding
This work was supported by the National Nature Science Foundation of China [grant number 81400506]; the Medical Science and Technology Research Fund, Guangdong Province [grant number A2015304]; the Grant for Young Teachers, Sun Yat-sen University [grant number 14ykpy34], and the National Institute for Dental and Craniofacial Research of the National Institutes of Health [grant number RO1DE026117]. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding bodies had not role in study design, the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Supporting information Available
The following files are available free of charge:
Figure S1 contains a representative SEM image of type I collagen fibrils before exposure to mineralizing solutions; Figures S2 and S3 contain SEM images of type I collagen fibrils mineralized for 1 day and 3 day, respectively in all mineralization solutions; Figure S4 contains SEM images of extrafibrillar crystals on collagen fibrils from the inner regions of collagen matrices mineralized for 7 days with 50k-10 and 450k-10 PAA solutions; Figure S5 contains atomic percentages of Ca and P from EDS analysis in collagen matrices mineralized in all solutions; and Figure S6 contains backscattered scanning electron images with corresponding EDS Ca line-scans and Ca mapping of the cross-section of densely packed collagen matrices mineralized for 7 days with 50k-10 PAA solution.
References
- 1.Weiner S; Traub W, BONE-STRUCTURE - FROM ANGSTROMS TO MICRONS. Faseb J. 1992, 6 (3), 879–885. [PubMed] [Google Scholar]
- 2.Traub W; Arad T; Weiner S, Three-dimensional ordered distribution of crystals in turkey tendon collagen fibers. Proc Natl Acad Sci U S A 1989, 86 (24), 9822–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landis WJ; Song MJ; Leith A; McEwen L; McEwen BF, Mineral and organic matrix interaction in normally calcifying tendon visualized in three dimensions by high-voltage electron microscopic tomography and graphic image reconstruction. J Struct Biol 1993, 110 (1), 39–54. DOI: S1047–8477(83)71003–8 [pii] 10.1006/jsbi.1993.1003. [DOI] [PubMed] [Google Scholar]
- 4.Beniash E, Biominerals-hierarchical nanocomposites: the example of bone. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2011, 3 (1), 47–69. DOI: 10.1002/wnan.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landis WJ; Hodgens KJ; Arena J; Song MJ; McEwen BF, Structural relations between collagen and mineral in bone as determined by high voltage electron microscopic tomography. Microsc Res Techniq 1996, 33 (2), 192–202. [DOI] [PubMed] [Google Scholar]
- 6.Weiner S; Wagner HD, The material bone: Structure mechanical function relations. Annu Rev Mater Sci 1998, 28, 271–298. [Google Scholar]
- 7.Gao HJ; Ji BH; Jager IL; Arzt E; Fratzl P, Materials become insensitive to flaws at nanoscale: Lessons from nature. P Natl Acad Sci USA 2003, 100 (10), 5597–5600. DOI: DOI 10.1073/pnas.0631609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahamid J; Sharir A; Addadi L; Weiner S, Amorphous calcium phosphate is a major component of the forming fin bones of zebrafish: Indications for an amorphous precursor phase. P Natl Acad Sci USA 2008, 105 (35), 12748–12753. DOI: DOI 10.1073/pnas.0803354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahamid J; Aichmayer B; Shimoni E; Ziblat R; Li CH; Siegel S; Paris O; Fratzl P; Weiner S; Addadi L, Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. P Natl Acad Sci USA 2010, 107 (14), 6316–6321. DOI: DOI 10.1073/pnas.0914218107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahamid J; Sharir A; Gur D; Zelzer E; Addadi L; Weiner S, Bone mineralization proceeds through intracellular calcium phosphate loaded vesicles: A cryo-electron microscopy study. J. Struct. Biol 2011, 174 (3), 527–535. DOI: 10.1016/j.jsb.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Saito T; Arsenault AL; Yamauchi M; Kuboki Y; Crenshaw MA, Mineral induction by immobilized phosphoproteins. Bone 1997, 21 (4), 305–311. [DOI] [PubMed] [Google Scholar]
- 12.Jee SS; Culver L; Li Y; Douglas EP; Gower LB, Biomimetic mineralization of collagen via an enzyme-aided PILP process. J Cryst Growth 2010, 312 (8), 1249–1256. DOI: 10.1016/j.jcrysgro.2009.11.010. [DOI] [Google Scholar]
- 13.Sommerdijk NAJM; Nudelman F; Pieterse K; George A; Bomans PHH; Friedrich H; Brylka LJ; Hilbers PAJ; de With G, The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nature Materials 2010, 9 (12), 1004–1009. DOI: 10.1038/NMAT2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tay FR; Pashley DH, Guided tissue remineralisation of partially demineralised human dentine. Biomaterials 2008, 29 (8), 1127–1137. DOI: DOI 10.1016/j.biomaterials.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Hunter GK; Goldberg HA, Modulation of crystal formation by bone phosphoproteins: role of glutamic acid-rich sequences in the nucleation of hydroxyapatite by bone sialoprotein. Biochem. J 1994, 302, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.George A; Bannon L; Sabsay B; Dillon JW; Malone J; Veis A; Jenkins NA; Gilbert DJ; Copeland NG, The carboxyl-terminal domain of phosphophoryn contains unique extended triplet amino acid repeat sequences forming ordered carboxyl-phosphate interaction ridges that may be essential in the biomineralization process. J Biol Chem 1996, 271 (51), 32869–73. [DOI] [PubMed] [Google Scholar]
- 17.Gower LB, Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem Rev 2008, 108 (11), 4551–627. DOI: 10.1021/cr800443h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiner S; Sagi I; Addadi L, Structural biology. Choosing the crystallization path less traveled. Science 2005, 309 (5737), 1027–8. DOI: 309/5737/1027 [pii] 10.1126/science.1114920. [DOI] [PubMed] [Google Scholar]
- 19.Pouget EM; Bomans PH; Goos JA; Frederik PM; de With G; Sommerdijk NA, The initial stages of template-controlled CaCO3 formation revealed by cryo-TEM. Science 2009, 323 (5920), 1455–8. DOI: 323/5920/1455 [pii] 10.1126/science.1169434. [DOI] [PubMed] [Google Scholar]
- 20.Boskey AL, Amorphous calcium phosphate: the contention of bone. J Dent Res 1997, 76 (8), 1433–6. [DOI] [PubMed] [Google Scholar]
- 21.Tsuji T; Onuma K; Yamamoto A; Iijima M; Shiba K, Direct transformation from amorphous to crystalline calcium phosphate facilitated by motif-programmed artificial proteins. Proc Natl Acad Sci U S A 2008, 105 (44), 16866–70. DOI: 0804277105 [pii] 10.1073/pnas.0804277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gajjeraman S; Narayanan K; Hao J; Qin C; George A, Matrix macromolecules in hard tissues control the nucleation and hierarchical assembly of hydroxyapatite. J Biol Chem 2007, 282 (2), 1193–204. DOI: M604732200 [pii] 10.1074/jbc.M604732200. [DOI] [PubMed] [Google Scholar]
- 23.Deshpande AS; Fang PA; Simmer JP; Margolis HC; Beniash E, Amelogenin-collagen interactions regulate calcium phosphate mineralization in vitro. J Biol Chem 2010, 285 (25), 19277–87. DOI: M109.079939 [pii] 10.1074/jbc.M109.079939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He G; Ramachandran A; Dahl T; George S; Schultz D; Cookson D; Veis A; George A, Phosphorylation of phosphophoryn is crucial for its function as a mediator of biomineralization. J. Biol. Chem 2005, 280 (39), 33109–33114. DOI: 10.1074/jbc.M500159200. [DOI] [PubMed] [Google Scholar]
- 25.Deshpande AS; Fang P-A; Zhang X; Jayaraman T; Sfeir C; Beniash E, Primary Structure and Phosphorylation of Dentin Matrix Protein 1 (DMP1) and Dentin Phosphophoryn (DPP) Uniquely Determine Their Role in Biomineralization. Biomacromolecules 2011, 12 (8), 2933–2945. DOI: 10.1021/bm2005214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai L; Qi YP; Niu LN; Liu Y; Pucci CR; Looney SW; Ling JQ; Pashley DH; jyh FR, Inorganic-Organic Nanocomposite Assembly Using Collagen as Template and Sodium Tripolyphosphate as A Biomimetic Analog of Matrix Phosphoprotein. Cryst Growth Des 2011, 11 (8), 3504–3511. DOI: 10.1021/cg200663v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshpande AS; Beniash E, Bio-inspired Synthesis of Mineralized Collagen Fibrils. Cryst Growth Des 2008, 8 (8), 3084–3090. DOI: 10.1021/cg800252f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jee SS; Thula TT; Gower LB, Development of bone-like composites via the polymer-induced liquid-precursor (PILP) process. Part 1: influence of polymer molecular weight. Acta Biomater 2010, 6 (9), 3676–86. DOI: S1742–7061(10)00162–5 [pii] 10.1016/j.actbio.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 29.Li X; Chang J, Preparation of bone-like apatite-collagen nanocomposites by a biomimetic process with phosphorylated collagen. J Biomed Mater Res A 2008, 85 (2), 293–300. DOI: 10.1002/jbm.a.31397. [DOI] [PubMed] [Google Scholar]
- 30.Niu LN; Jee SE; Jiao K; Tonggu L; Li M; Wang L; Yang YD; Bian JH; Breschi L; Jang SS; Chen JH; Pashley DH; Tay FR, Collagen intrafibrillar mineralization as a result of the balance between osmotic equilibrium and electroneutrality. Nature Materials 2017, 16 (3), 370–378. DOI: 10.1038/NMAT4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li YP; Thula TT; Jee S; Perkins SL; Aparicio C; Douglas EP; Gower LB, Biomimetic Mineralization of Woven Bone-Like Nanocomposites: Role of Collagen Cross-Links. Biomacromolecules 2012, 13 (1), 49–59. DOI: 10.1021/bm201070g. [DOI] [PubMed] [Google Scholar]
- 32.Olszta MJ; Cheng XG; Jee SS; Kumar R; Kim YY; Kaufman MJ; Douglas EP; Gower LB, Bone structure and formation: A new perspective. Mat Sci Eng R 2007, 58 (3–5), 77–116. DOI: DOI 10.1016/j.mser.2007.05.001. [DOI] [Google Scholar]
- 33.Thula TT; Svedlund F; Rodriguez DE; Podschun J; Pendi L; Gower LB, Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents. Polymers (Basel) 2011, 3 (1), 10–35. DOI: 10.3390/polym3010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wingender B; Bradley P; Saxena N; Ruberti JW; Gower L, Biomimetic organization of collagen matrices to template bone-like microstructures. Matrix Biology 2016, 52–54, 384–396. DOI: 10.1016/j.matbio.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Li Y; Aparicio C, Discerning the Subfibrillar Structure of Mineralized Collagen Fibrils: A Model for the Ultrastructure of Bone. PLOS ONE 2013, 8 (9), e76782 DOI: 10.1371/journal.pone.0076782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saeki K; Chien YC; Nonomura G; Chin AF; Habelitz S; Gower LB; Marshall SJ; Marshall GW, Recovery after PILP remineralization of dentin lesions created with two cariogenic acids. Arch Oral Biol 2017, 82, 194–202. DOI: 10.1016/j.archoralbio.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nurrohman H; Saeki K; Carneiro KMM; Chien YC; Djomehri S; Ho SP; Qin C; Gower LB; Marshall SJ; Marshall GW; Habelitz S, Repair of dentin defects from DSPP knockout mice by PILP mineralization. J Mater Res 2016, 1–7. DOI: 10.1557/jmr.2015.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antebi B; Cheng X; Harris JN; Gower LB; Chen X-D; Ling J, Biomimetic Collagen-Hydroxyapatite Composite Fabricated via a Novel Perfusion-Flow Mineralization Technique. Tissue Engineering Part C-Methods 2013, 19 (7), 487–496. DOI: 10.1089/ten.tec.2012.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y; Li N; Qi YP; Dai L; Bryan TE; Mao J; Pashley DH; Tay FR, Intrafibrillar collagen mineralization produced by biomimetic hierarchical nanoapatite assembly. Adv Mater 2011, 23 (8), 975–80. DOI: 10.1002/adma.201003882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Girija EK; Yokogawa Y; Nagata F, Apatite formation on collagen fibrils in the presence of polyacrylic acid. J Mater Sci Mater Med 2004, 15 (5), 593–9. [DOI] [PubMed] [Google Scholar]
- 41.Murugan R; Ramakrishna S, Crystallographic Study of Hydroxyapatite Bioceramics Derived from Various Sources. Cryst. Growth Des. 2005, 5 (1), 111–112. DOI: 10.1021/cg034227s. [DOI] [Google Scholar]
- 42.Bigi A; Ripamonti A; Cojazzi G; Pizzuto G; Roveri N; Koch MHJ, Structural-Analysis of Turkey Tendon Collagen Upon Removal of the Inorganic Phase. International Journal of Biological Macromolecules 1991, 13 (2), 110–114. DOI: Doi 10.1016/0141-8130(91)90058-3. [DOI] [PubMed] [Google Scholar]
- 43.Lozano LF; Peña-Rico MA; Heredia A; Ocotlán-Flores J; Gómez-Cortés A; Velázquez R; Belío IA; Bucio L, Thermal analysis study of human bone. J Mater Sci 2003, 38 (23), 4777–4782. DOI: 10.1023/a:1027483220584. [DOI] [Google Scholar]
- 44.Qi YP; Li N; Niu LN; Primus CM; Ling JQ; Pashley DH; Tay FR, Remineralization of artificial dentinal caries lesions by biomimetically modified mineral trioxide aggregate. Acta Biomater 2012, 8 (2), 836–42. DOI: S1742–7061(11)00484–3 [pii] 10.1016/j.actbio.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu S; Gu L; Huang Z; Sun Q; Chen H; Ling J; Mai S, Intrafibrillar mineralization of polyacrylic acid-bound collagen fibrils using a two-dimensional collagen model and Portland cement-based resins. Eur J Oral Sci 2017, 125 (1), 72–80. DOI: 10.1111/eos.12319. [DOI] [PubMed] [Google Scholar]
- 46.Hu CM; Zilm M; Wei M, Fabrication of intrafibrillar and extrafibrillar mineralized collagen/apatite scaffolds with a hierarchical structure. J Biomed Mater Res A 2016, 104 (5), 1153–1161. DOI: 10.1002/jbm.a.35649. [DOI] [PubMed] [Google Scholar]
- 47.Li N; Niu LN; Qi YP; Yiu CKY; Ryou H; Arola DD; Chen JH; Pashley DH; Tay FR, Subtleties of biomineralisation revealed by manipulation of the eggshell membrane. Biomaterials 2011, 32 (34), 8743–8752. DOI: DOI 10.1016/j.biomaterials.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dey A; Bomans PHH; Muller FA; Will J; Frederik PM; de With G; Sommerdijk NAJM, The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nature Materials 2010, 9 (12), 1010–1014. DOI: Doi 10.1038/Nmat2900. [DOI] [PubMed] [Google Scholar]
- 49.Wang J; Chen Y; Li L; Sun J; Gu X; Xu X; Pan H; Tang R, Remineralization of dentin collagen by meta-stabilized amorphous calcium phosphate. Crystengcomm 2013, 15 (31), 6151–6158. DOI: 10.1039/C3CE40449H. [DOI] [Google Scholar]
- 50.Huang SC; Naka K; Chujo Y, Effect of molecular weights of poly(acrylic acid) on crystallization of calcium carbonate by the delayed addition method. Polym J 2008, 40 (2), 154–162. DOI: 10.1295/polymj.PJ2007162. [DOI] [Google Scholar]
- 51.Boskey AL; Villarreal-Ramirez E, Intrinsically disordered proteins and biomineralization. Matrix Biology 2016, 52–54, 43–59. DOI: 10.1016/j.matbio.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hackley VA, Colloidal Processing of Silicon Nitride with Poly(acrylic acid): I, Adsorption and Electrostatic Interactions. J Am Ceram Soc 1997, 80 (9), 2315–2325. DOI: 10.1111/j.1151-2916.1997.tb03122.x. [DOI] [Google Scholar]
- 53.Jiao K; Niu LN; Ma CF; Huang XQ; Pei DD; Luo T; Huang Q; Chen JH; Tay FR, Complementarity and Uncertainty in Intrafibrillar Mineralization of Collagen. Adv Funct Mater 2016, 26 (38), 6858–6875. DOI: 10.1002/adfm.201602207. [DOI] [Google Scholar]
- 54.Mann S, Mineralization in biological systems In Inorganic Elements in Biochemistry, Springer Berlin Heidelberg: Berlin, Heidelberg, 1983; pp 125–174. DOI: 10.1007/BFb0111320. [DOI] [Google Scholar]
- 55.Nudelman F; Bomans PHH; George A; de With G; Sommerdijk N, The role of the amorphous phase on the biomimetic mineralization of collagen. Faraday Discussions 2012, 159, 357–370. DOI: 10.1039/c2fd20062g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orgel JPRO; Irving TC; Miller A; Wess TJ, Microfibrillar structure of type I collagen in situ. P Natl Acad Sci USA 2006, 103 (24), 9001–9005. DOI: 10.1073/pnas.0502718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cantaert B; Beniash E; Meldrum FC, Nanoscale Confinement Controls the Crystallization of Calcium Phosphate: Relevance to Bone Formation. Chemistry–A European Journal 2013, 19 (44), 14918–14924. DOI: 10.1002/chem.201302835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiner S; Traub W; Wagner HD, Lamellar bone: Structure-function relations. J. Struct. Biol 1999, 126 (3), 241–255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.