

Abstract
Sickle cell disease is a common, life-threatening genetic disorder that is best managed when diagnosed early by newborn screening. However, sickle cell disease is most prevalent in low-resource regions of the world where newborn screening is rare and diagnosis at the point-of-care is challenging. In many such regions, the majority of affected children die, undiagnosed, before the age of five years. A rapid and affordable point-of-care test for sickle cell disease is needed. The diagnostic accuracy of HemoTypeSC, a point-of-care immunoassay, for sickle cell disease was evaluated in individuals who had sickle cell disease, hemoglobin C disease, the related carrier (trait) states, or a normal hemoglobin phenotype. Children and adults participated in low-, medium- and high-resource environments [Ghana (n=383), Martinique (n=46), and USA (n=158)]. Paired blood specimens were obtained for HemoTypeSC and a reference diagnostic assay. HemoTypeSC testing was performed at the site of blood collection, and the reference test was performed in a laboratory at each site. In 587 participants, across all study sites, HemoTypeSC had an overall sensitivity of 99.5% and specificity of 99.9% across all hemoglobin phenotypes. The test had 100% sensitivity and specificity for sickle cell anemia. Sensitivity and specificity for detection of normal and trait states were >99%. HemoTypeSC is an inexpensive (<$2 per test), accurate, and rapid point-of-care test that can be used in resource-limited regions with a high prevalence of sickle cell disease to provide timely diagnosis and support newborn screening programs.
Keywords: sickle cell disease, hemoglobin, point-of-care, rapid test, HemoTypeSC, diagnostic
INTRODUCTION
Sickle cell disease (SCD) is a group of genetic blood disorders caused by sickle hemoglobin [HbS; HBB:c.20A>T(p.E7V)] and characterized by acute and chronic multiorgan damage and dysfunction due to vaso-occlusion and hemolysis. Manifestations of SCD include painful episodes, cardiopulmonary disease, stroke, nephropathy, susceptibility to invasive bacterial infections, and early mortality.1,2 In many high-resource regions, universal newborn screening programs coupled with prophylactic interventions have dramatically reduced the mortality and morbidity of SCD during the first 20 years of life.3–8 However, in sub-Saharan Africa and central India, where more than 90% of annual SCD births occur, newborn screening programs have not been implemented universally, if at all, due in large part to the cost and logistical burden of laboratory diagnostic tests.9 Up to 90% of children with SCD in sub-Saharan Africa are thought to die before the age of 5 years, undiagnosed,10 making SCD one of the leading causes of childhood mortality in the region.11–13 Individuals with SCD in these regions are commonly identified only after hospitalization for severe pain or other overt or life-threatening manifestation of the disease. Its effects on mortality and quality of life and its economic burden on regional healthcare systems have led SCD to be declared both a disease of public concern by the United Nations General Assembly14 and a priority non-communicable disease by World Health Organization.15
Early diagnosis and intervention programs for SCD are projected to be cost-effective in sub-Saharan Africa and India,16,17 and the World Health Organization estimates that such programs would prevent 70% of existing SCD mortality.18 However, the main barriers to implementing newborn screening programs at scale include the cost of diagnostic methods, lack of adequately distributed laboratory infrastructure, and lack of adequate, sustained funding. Standard clinical laboratory methods to identify Hb variants include gel-based or capillary electrophoresis, isoelectric focusing (IEF), and high-performance liquid chromatography (HPLC). These methods require about 1 mL of whole blood, uninterrupted electrical supply, dedicated operating personnel, and necessitate the transport of blood samples from the POC to possibly distant testing facilities.19 Furthermore, these methods require the re-contacting of affected newborns’ families in order to deliver testing results – sometimes weeks or months after sample collection. It is clear that a rapid, inexpensive, and highly-accurate POC solution for SCD diagnosis is urgently needed.
Several rapid diagnostic methods have been described for SCD. The sickle cell solubility test (Sickledex®)20 can rapidly identify the presence of HbS in a blood sample, but it does not distinguish between sickle cell trait and SCD. Furthermore, this test is not reliable when HbS levels are below 15–20%, so it is not suited for newborn screening. A variation of the sickle cell solubility test has been reported to distinguish HbA/S and HbS/S from HbA/A phenotypes through visualization of differential diffusion patterns on filter paper in the presence of a solubility buffer.21 However, these diffusion patterns may be difficult to distinguish, and a computer and scanner are required for optimal visualization. Recently, a lateral flow immunoassay called Sickle SCAN® has been described, which is capable of distinguishing HbA-, HbS-, and HbC-related phenotypes in a large drop of blood (5 μL).22 Sickle SCAN® has a very high limit of detection for HbA (40%), and inconsistent visualization of HbA test results has been reported,23 likely related to its design as a sandwich-type immunoassay utilizing polyclonal antibodies. These designs are susceptible to the prozone effect,24 where high concentrations of a particular hemoglobin can yield false negative results. The accuracy of Sickle SCAN® in laboratory and high-resource environments is reported to be >95%,22,23 but a recent study in Tanzania illustrated that its specificity in the field was only 91%25 – indicating a high chance of false positive results. Furthermore, the Sickle SCAN® test comprises a cassette and a separate buffer-filled assay vial, which is a complexity that may lead to higher cost. Finally, a device called HemeChip has been recently reported to function as a smaller version of a cellulose acetate electrophoresis machine.26 While this device has the ability to provide quantitative measurements of HbA, HbS, and HbF in a sample of blood, HemeChip requires a continuous power supply and an initial investment to purchase the machine.
HemoTypeSC™ is a competitive lateral-flow immunoassay that uses monoclonal antibodies to detect hemoglobins A, S, and C in a 1.5-μL sample of whole blood (a prototype version of the test has been described27). The competitive, rather than sandwich, format lends itself to high sensitivity for detection of HbA, HbS, and HbC and consistent performance even with samples containing high Hb concentrations. The accuracy of the prototype test in controlled laboratory environments was shown to be 100% for all relevant hemoglobin phenotypes.27 The very simple design of this test leads to an end-user cost of less than $2 per test. HemoTypeSC requires no instrumentation, electricity, or refrigeration, and the results are determined by visual inspection in about 10 minutes. Here, we describe a global, multicenter evaluation of the diagnostic performance of HemoTypeSC for detecting SCD, HbC disease, and the related carrier states (HbS trait and HbC trait) using whole blood at the POC in limited-resource environments. The majority of samples were collected and tested in Ghana, a location selected for its high prevalence of both the HbS and the HbC variants. This report describes the most comprehensive global study reported to date for the evaluation of a POC test for SCD.
METHODS
Study Design and Oversight
We conducted a blinded, multicenter, prospective diagnostic accuracy study. The investigational assay was HemoTypeSC, and the reference (“gold-standard”) tests were agarose gel electrophoresis, IEF, and capillary zone electrophoresis. Three different study centers were selected in order to evaluate the test’s performance in low-, medium-, and high-resource environments compared to local reference methods. The study was designed, implemented, and supervised by Operation International Kids (a USA-based, non-profit humanitarian organization) together with investigators from Institut National de la Santé et de la Recherche Médicale (INSERM) and Cincinnati Children’s Hospital Medical Center. The HemoTypeSC test system was designed by Silver Lake Research Corporation and provided without charge for this study. All authors reviewed and analyzed the data and attest to its accuracy and completeness as well as the fidelity of adherence to the study protocol.
Study Populations
At Holy Family Hospital in Techiman, Ghana (low-resource), newborns, children, and adult family members were enrolled from the ambulatory pediatric clinic and neonatal intensive care unit, and also adult hospital staff. At the study center in Martinique (medium-resource), coordinated by INSERM investigators, predominantly newborn children were enrolled from the maternity ward. At Cincinnati Children’s Hospital Medical Center in the USA (high-resource), de-identified blood samples from standard care clinical testing from neonates, children and some adults were tested. Specimens from the USA center were either archived (refrigerated for <1 year) or freshly-drawn whole blood samples. None of the participants in Ghana or Martinique had prior Hb phenotype testing at the time of enrollment. The ethics boards at each site approved the study. All participants in Ghana and Martinique provided informed consent. A waiver of informed consent was granted by the Institutional Review Board for the use of anonymous, clinical samples in Cincinnati. This study is registered at ClinicalTrials.gov (NCT03619798).
Study Procedure
From each participant, a 1.5-μL sample of blood was first collected for the HemoTypeSC test using the provided blood collection device from a heel-prick (newborns, infants, and small children) or venipuncture (older children and adults). A larger blood sample was obtained in a collection tube for confirmatory analysis. In Ghana and Martinique, HemoTypeSC testing was performed immediately upon sample collection from each participant, while at the USA study center, blood was transported to a diagnostic laboratory where aliquots were used for HemoTypeSC testing. After transport of tube-collected specimens to the laboratories at each study center, one of three confirmatory (reference) methods was used: agarose gel electrophoresis in Ghana, IEF in Martinique, and capillary zone electrophoresis in the USA. For specimens that produced discordant (different, not matching) results between the HemoTypeSC and the reference test, another “gold-standard” reference test (HPLC) was used to adjudicate the Hb phenotype. HPLC testing of discordant samples was also conducted at the testing center at which the original samples were tested.
HemoTypeSC Test Procedure
The HemoTypeSC test kit (included components, testing procedure, and interpretation guidelines) is illustrated in Supp. Fig 1. Operators were trained to use the device by reading the test instruction sheet (Supp. Fig. 1) that describes the following five steps. First, using the included dropper pipette, six drops of water were dispensed into a test tube. Bottled drinking water was used at the Ghana and Martinique study sites; tap water was used at the US site. Second, a 1.5 μL blood droplet was collected with the included blood collection device. Third, the blood collection device was inserted into the water within the test tube and swirled to ensure blood transfer from the absorbent pad of the blood collection device into the water. Fourth, the test strip was inserted into the water-blood mixture within the test tube. Finally, the result on the test strip was interpreted visually after 10 minutes.
Confirmatory Laboratory Procedures
In Ghana and Martinique, agarose gel electrophoresis and IEF were performed according to standard clinical laboratory protocols. In the USA, screening capillary zone electrophoresis was performed on Capillarys™ 2 Flex Piercing System (Sebia SA, France), and variant Hbs were then confirmed by acid gel electrophoresis (Sebia Hydragel Acid Hemoglobin) and IEF. High-pressure liquid chromatography (HPLC) was performed on a VARIANT II Instrumentation System (Bio-Rad, USA) for discordant samples only.
Analysis
The primary objective was to determine the sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) of HemoTypeSC, compared to confirmatory or reference tests, in detecting normal Hb (HbA) and common pathologic Hbs (HbS and HbC), in trait and disease states, in whole blood specimens. The disease states of interest included SCD, comprising sickle cell anemia (HbSS), sickle-β0-thalassemia (indistinguishable from HbSS by HemoTypeSC), and sickle-hemoglobin C disease (HbSC), as well as HbC disease (HbCC). The initial test population included all specimens for which there was sufficient blood for reference testing. Following Hb screening with both the investigational and reference methods, the main analysis population was assembled, which included all specimens that produced clear, interpretable results from both testing methods. Specimens from the main analysis population that produced discordant test results between HemoTypeSC and the reference method were subjected to HPLC testing for adjudication of results, where the HPLC result was considered the true phenotype.
RESULTS
Study Participants
A total of 608 participants registered for the study, including 402 from Ghana (low-resource), 46 from Martinique (medium-resource), and 160 from the USA (high-resource; Fig. 1). Eighteen participants from the Ghana study center and two participants from the USA study center provided insufficient blood for testing by a confirmatory method and were excluded prior to analysis. Thus, 588 participants were included in the study. One specimen from the Ghana study group subsequently produced indeterminate confirmatory method results and was ruled ineligible for data analysis. Therefore, 587 participants comprised the main analysis population (Fig. 1). Both sexes and a wide range of ages (newborn to age 30 years) were represented (Table 1). At the low- and medium-resource study centers in Ghana and Martinique, 16.6% (71/429) of participants were under 1 month of age, when the predominant Hb is fetal hemoglobin (HbF).
Figure 1. Participant Enrollment and Construction of Main Analysis Population.
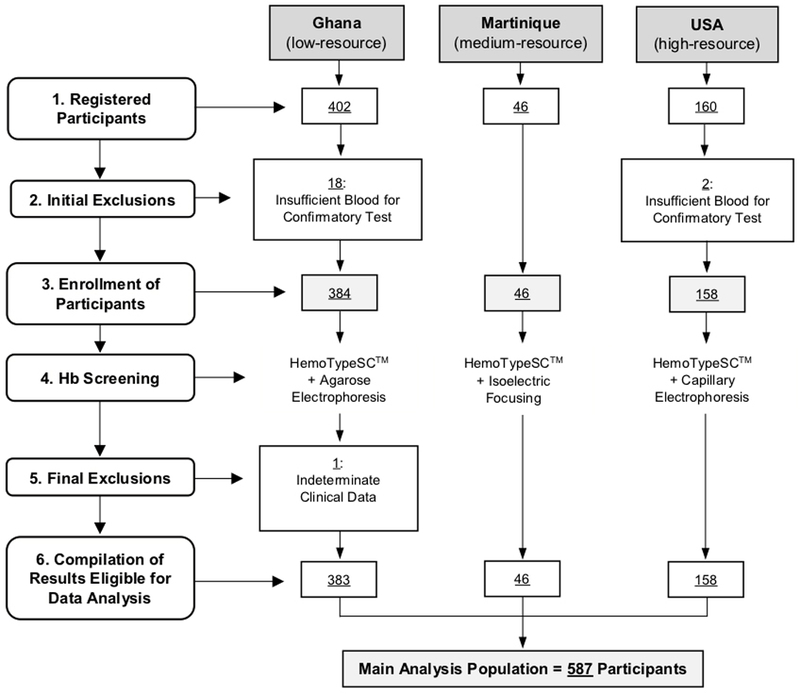
Newborns, children, and adults were registered for the study from low- (Ghana), medium- (Martinique), and high-resource testing environments. A total of 20 participants (mostly very small newborns) were initially excluded from the study due to the lack of adequate collected blood volume for confirmatory testing. Ultimately, 588 individuals were enrolled in the study, and Hb screening was conducted by comparing HemoTypeSC to three separate, clinically established reference methods (agarose gel electrophoresis, IEF, and capillary zone electrophoresis). Following a final exclusion of one specimen producing indeterminate reference method results, a total of 383 results from Ghana, 46 results from Martinique, and 158 results from the USA (total N=587) comprised the main analysis population.
Table 1.
Demographic Attributes of Participants
| Attributea | Ghana | Martinique | USA | Total | |
|---|---|---|---|---|---|
| Main Analysis Population:b | 383 | 46 | 158 | 587 | |
| Sex: | Male | 143 (37.3) | 24 (52.2) | 77 (48.7) | 244 (41.6) |
| Female | 240 (62.7) | 22 (47.8) | 81 (51.3) | 343 (58.4) | |
| Age: | ≤1 Week | 18 (4.7) | 39 (84.8) | 0 (0.0) | 57 (9.7) |
| 1 Week-1 Month | 14 (3.7) | 0 (0.0) | 17 (10.8) | 31 (5.3) | |
| 1 Month-1 Year | 40 (10.4) | 4 (8.7) | 30 (19.0) | 74 (12.6) | |
| 1–5 Years | 81 (21.1) | 3 (6.5) | 20 (12.7) | 104 (17.7) | |
| >5 Years | 230 (60.1) | 0 (0.0) | 91 (57.6) | 321 (54.7) | |
Data presented as number (% of total).
Participants who were ultimately eligible for data analysis following exclusions due to insufficient blood for confirmatory testing or indeterminate clinical results.
Analysis by Hb Variant
The HemoTypeSC test combines independent competitive immunoassays for each of three Hb variants (HbA, HbS, and HbC). Therefore, the sensitivity and specificity of HemoTypeSC were first calculated for the detection of each particular Hb (Table 2). For HbA, HemoTypeSC sensitivity and specificity were both 100% (503 HbA-positive results from 503 samples containing HbA, and 83 HbA-negative results from 83 samples not containing HbA). Overall, sensitivity and specificity for detection of HbS and HbC were similar at >99% (Table 2).
Table 2.
HemoTypeSCTM Detection Analysis by Hemoglobin Varianta
| Location | Hb | Sensitivityb | Specificityc | PPVd | NPVe | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP/(TP+FN) | % | TN/(FP+TN) | % | TP/(TP+FP) | % | TN/(TN+FN) | % | ||
| Ghana | A | 368/368 | 100.0 | 14/14 | 100.0 | 368/368 | 100.0 | 14/14 | 100.0 |
| S | 49/50 | 98.0 | 331/332 | 99.7 | 49/50 | 98.0 | 331/332 | 99.7 | |
| C | 67/68 | 98.5 | 313/314 | 99.7 | 67/68 | 98.5 | 313/314 | 99.7 | |
| Martinique | A | 45/45 | 100.0 | 1/1 | 100.0 | 45/45 | 100.0 | 1/1 | 100.0 |
| S | 8/8 | 100.0 | 38/38 | 100.0 | 8/8 | 100.0 | 38/38 | 100.0 | |
| C | 5/5 | 100.0 | 41/41 | 100.0 | 5/5 | 100.0 | 41/41 | 100.0 | |
| USA | A | 90/90 | 100.0 | 68/68 | 100.0 | 90/90 | 100.0 | 68/68 | 100.0 |
| S | 95/95 | 100.0 | 63/63 | 100.0 | 95/95 | 100.0 | 63/63 | 100.0 | |
| C | 48/48 | 100.0 | 110/110 | 100.0 | 48/48 | 100.0 | 110/110 | 100.0 | |
| Total | A | 503/503 | 100.0 | 83/83 | 100.0 | 503/503 | 100.0 | 83/83 | 100.0 |
| S | 152/153 | 99.3 | 432/433 | 99.8 | 152/153 | 99.3 | 432/433 | 99.8 | |
| C | 120/121 | 99.2 | 464/465 | 99.8 | 120/121 | 99.2 | 464/465 | 99.8 | |
TP = true positives, TN = true negatives, FP = false positives, FN = false negatives. Values presented represent the HemoTypeSC test results compared to the reference method, following ultimate adjudication of discordant results.
Any specimen that was either heterozygous or homozygous for each particular Hb variant was considered a true positive for that variant.
Any specimen that lacked each particular Hb variant was considered a true negative for that variant, regardless of other Hb variants present.
Positive predictive value.
Negative predictive value.
Analysis by Hb Phenotype (Normal, Disease, or Carrier States)
HemoTypeSC was next compared to reference methods for determination of clinical accuracy in detecting Hb phenotypes AA (normal), AS (HbS trait), AC (HbC trait), SS (sickle cell anemia or sickle-β0-thalassemia), SC (sickle-HbC disease) and CC (Hb C disease; Table 3). The overall accuracy of HemoTypeSC across all Hb phenotypes in the main analysis population was 99.5% (584/587 Hb phenotypes correctly identified). The three specimens incorrectly identified by HemoTypeSC (described below) were all from the Ghana study center. HemoTypeSC correctly identified every HbSS and HbCC phenotype with no false positive results, exhibiting sensitivity and specificity of 100% across all study centers (52/52 Hb phenotypes correctly identified). For normal Hb phenotype AA and trait Hb phenotypes AS and AC, HemoTypeSC exhibited overall sensitivities and specificities of >99% (499/501 Hb phenotypes correctly identified). The test also correctly identified two specimens from recently-transfused HbSC patients at the USA center, with a resulting Hb phenotype of HbASC. For the HbSC disease phenotype, due to one false negative resulting from an indeterminate HemoTypeSC readout, sensitivity was 96.9%, with a specificity and positive predictive value of 100% and negative predictive value of 99.8% (31/32 correctly identified). For all disease phenotypes (HbSS, HbCC, and HbSC), HemoTypeSC sensitivity was 98.8% (83/84 phenotypes correctly identified), with a specificity of 100%.
Table 3.
HemoTypeSCTM Detection Analysis by Phenotypea
| Location | Hb | Sensitivity | Specificity | PPVb | NPVc | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP/(TP+FN) | % | TN/(FP+TN) | % | TP/(TP+FP) | % | TN/(TN+FN) | % | ||
| Ghana | AA | 272/274 | 99.3 | 109/109 | 100.0 | 272/272 | 100.0 | 109/111 | 98.2 |
| AS | 39/39 | 100.0 | 343/344 | 99.7 | 39/40 | 97.5 | 343/343 | 100.0 | |
| AC | 55/55 | 100.0 | 327/328 | 99.7 | 55/56 | 98.2 | 327/327 | 100.0 | |
| SS | 2/2 | 100.0 | 381/381 | 100.0 | 2/2 | 100.0 | 381/381 | 100.0 | |
| SC | 8/9 | 88.9 | 374/374 | 100.0 | 8/8 | 100.0 | 374/375 | 99.7 | |
| CC | 4/4 | 100.0 | 379/379 | 100.0 | 4/4 | 100.0 | 379/379 | 100.0 | |
| Martinique | AA | 34/34 | 100.0 | 12/12 | 100.0 | 34/34 | 100.0 | 12/12 | 100.0 |
| AS | 7/7 | 100.0 | 39/39 | 100.0 | 7/7 | 100.0 | 39/39 | 100.0 | |
| AC | 4/4 | 100.0 | 42/42 | 100.0 | 4/4 | 100.0 | 42/42 | 100.0 | |
| SS | − | − | − | − | − | − | − | − | |
| SC | 1/1 | 100.0 | 45/45 | 100.0 | 1/1 | 100.0 | 45/45 | 100.0 | |
| CC | − | − | − | − | − | − | − | − | |
| USA | AA | 39/39 | 100.0 | 119/119 | 100.0 | 39/39 | 100.0 | 119/119 | 100.0 |
| AS | 35/35 | 100.0 | 123/123 | 100.0 | 35/35 | 100.0 | 123/123 | 100.0 | |
| AC | 14/14 | 100.0 | 144/144 | 100.0 | 14/14 | 100.0 | 144/144 | 100.0 | |
| SS | 36/36 | 100.0 | 122/122 | 100.0 | 36/36 | 100.0 | 122/122 | 100.0 | |
| SC | 22/22 | 100.0 | 136/136 | 100.0 | 22/22 | 100.0 | 136/136 | 100.0 | |
| CC | 10/10 | 100.0 | 148/148 | 100.0 | 10/10 | 100.0 | 148/148 | 100.0 | |
| ASCe | 2/2 | 100.0 | 156/156 | 100.0 | 2/2 | 100.0 | 156/156 | 100.0 | |
| Total | AA | 345/347 | 99.4 | 240/240 | 100.0 | 345/345 | 100.0 | 240/242 | 99.2 |
| AS | 81/81 | 100.0 | 505/506 | 99.8 | 81/82 | 98.8 | 505/505 | 100.0 | |
| AC | 73/73 | 100.0 | 513/514 | 99.8 | 73/74 | 98.7 | 513/513 | 100.0 | |
| SS | 38/38 | 100.0 | 503/503 | 100.0 | 38/38 | 100.0 | 503/503 | 100.0 | |
| SC | 31/32 | 96.9 | 555/555 | 100.0 | 31/31 | 100.0 | 555/556 | 99.8 | |
| CC | 14/14 | 100.0 | 527/527 | 100.0 | 14/14 | 100.0 | 527/527 | 100.0 | |
| ASCe | 2/2 | 100.0 | 585/585 | 100.0 | 2/2 | 100.0 | 585/585 | 100.0 | |
| OVERALL | 584/587 | 99.5 | 3,428/3,430 | 99.9 | 584/586 | 99.7 | 3,428/3,431 | 99.9 | |
TP = true positives, TN = true negatives, FP = false positives, FN = false negatives. Values presented represent the HemoTypeSC test results compared to the reference method, following ultimate adjudication of discordant results.
Positive predictive value.
Negative predictive value.
Discordant Results
Four of 587 total specimens (<1%) yielded discordant results between HemoTypeSC and the reference method (Supp. Table 1). The first discordant specimen yielded a HemoTypeSC/agarose electrophoresis phenotype discrepancy of HbAC/HbAA. HPLC confirmed the HbAC HemoTypeSC result. Therefore, this result was included in the detection analysis by Hb variant (Table 2) as a true positive for both HbA and HbC and in the detection analysis by phenotype (Table 3) as a true positive for HbAC. The second discordant specimen also yielded a HemoTypeSC/agarose electrophoresis phenotype discrepancy of HbAC/HbAA, and HPLC confirmed the HbAA reference method result. This result was therefore included in the detection analysis by Hb variant as a false positive for HbC and in the detection analysis by phenotype as a false positive for HbAC/false negative for HbAA. The third discordant specimen yielded a HemoTypeSC/agarose electrophoresis phenotype discrepancy of HbAS/HbAA. The patient who donated this specimen was unable to be re-contacted and was thus lost to follow-up. Since secondary confirmatory testing by HPLC was not possible in this case, the result was accepted as ultimately discordant. This result was included in the detection analysis by Hb variant as a false positive for HbS and in the detection analysis by phenotype as a false positive for HbAS/false negative for HbAA. The fourth discordant specimen yielded an indeterminate HemoTypeSC result and an agarose electrophoresis result of HbSC. Since the investigational test result was not known, confirmatory HPLC testing was not conducted, and the agarose electrophoresis result was accepted as the correct result. This result was included in the detection analysis by Hb variant as a false negative for both HbS and HbC and in the detection analysis by phenotype as a false negative for HbSC. In summary, of the four initially discordant results, HemoTypeSC was adjudicated to be correct for one result and incorrect for one result, with the two remaining results accepted as discordant due to a lack of HemoTypeSC or HPLC confirmatory data.
Samples with Insufficient Blood Sample Volume
There were 18 participants from Ghana who were registered for the study but unable to provide sufficient blood by venipuncture for confirmatory testing by agarose gel electrophoresis. All of these specimens came from premature newborns, from whom only a few drops of blood could be feasibly collected (dried blood spot testing was not a standard practice at this testing center). HemoTypeSC testing could, however, still be conducted on these samples, and results included 13 HbAA, two HbAS, two HbAC, and one HbSC. Because confirmatory testing was not conducted, these participants were excluded from the main analysis population.
DISCUSSION
In this international, multi-center study, HemoTypeSC exhibited >99% sensitivity and specificity for detection of each of three Hb types, HbA, HbS, and HbC, and >99% sensitivity and specificity for each relevant Hb phenotype (normal, carrier, and disease states) except HbSC (for which sensitivity was >96%). HemoTypeSC gave discordant results compared to a reference test in only three of 587 samples. Only one discordant result was confirmed by HPLC to be an inaccuracy of the HemoTypeSC; one other discordant result was resolved in favor of HemoTypeSC, indicating an error of the reference method. All the discordant results originated from the Ghana study center, but differences in overall accuracy across the three centers were not statistically significant (data not shown). No other rapid, POC test for the detection of SCD and related Hb variants has been reported to have such high accuracy in published reports, especially in low-resource settings. It is notable that HemoTypeSC was both highly sensitive and highly specific for the detection of SCD. As the prevalence of SCD is expected to be <5% even in the most affected areas, any test with a lower specificity would yield a much higher rate of false positive results.
The previously-described prototype version of HemoTypeSC demonstrated 100% accuracy in a laboratory setting, including specimens that contained >80% HbF.27 Equivalent results were obtained in this study, as HemoTypeSC had 100% accuracy the subset of newborns (N=57; about 10% of the study population) tested in low- and medium-resource study centers. The elevated HbF levels of newborns may be expected to decrease the accuracy of some laboratory methods for Hb typing.28,29 However, HemoTypeSC is not susceptible to interference by Hb F, so it can be used in individuals of any age, including newborns.
This study demonstrates the advantages of HemoTypeSC when only a small volume of blood can be obtained for testing. For example, specimens from 18 premature, low birth weight newborns in Ghana for whom adequate blood could not be feasibly collected by venipuncture for agarose gel electrophoresis were able to be tested using HemoTypeSC, for which only 1.5 μL of blood is needed (e.g., from a heel-stick). Even if consistent newborn screening for SCD were currently performed at this clinic (it is not), these particular infants would have been sent home shortly after birth without Hb phenotype information, including one who had sickle-HbC disease. This highlights the immediacy of results from POC testing, which can direct early preventive therapies for newborns with SCD. Rapid POC diagnosis can also direct the care of acutely ill or symptomatic patients (e.g., evaluation of a patient with fever and anemia of unknown cause).
It is important to note that HemoTypeSC provides a qualitative result (the presence or absence of particular Hbs), so certain Hb phenotypes are not discernible. Sickle-β+-thalassemia, a form of SCD, is distinguished from HbS trait by Hb quantitation. In sickle-β+-thalassemia, HbS is present in greater abundance than HbA, and vice-versa for HbS trait. HemoTypeSC will provide a result of sickle cell trait (HbA and HbS both present) in both cases. Consequently, because the level of HbA is abnormally low yet still detectable by HemoTypeSC (HbA >2.5%), some cases of sickle-β+-thalassemia may be misclassified unless follow-up testing is performed. Likewise, HemoTypeSC cannot differentiate sickle-β0-thalassemia from HbSS, although this distinction is not clinically important, because both forms of SCD are managed similarly. Additionally, because the device detects HbA through sensing a single amino acid on the β-globin chain, other clinically significant Hbs that share this identical amino acid, such as HbD, HbE, and HbO, will be identified as HbA. Therefore, users of the test in regions where these Hbs are most common, such as the Middle East and Southeast Asia, should expect that certain compound heterozygous forms of SCD will be classified as HbS trait. Other previously-published POC Hb screening devices20–22,26 share these limitations.
Once patients leave a hospital or clinic, especially in rural areas, it may be difficult to re-establish contact. In many rural regions of Africa and India, Hb screening is conducted primarily at central hospitals or laboratories. Consequently, diagnostic specimens are shipped away from the POC for analysis, which adds expense, logistical difficulties, and delays in test results. Rapid POC screening is a promising solution to these difficulties. HemoTypeSC requires no instrumentation, electricity, or refrigeration. Operator training is minimal (see Methods), and the results are determined by visual inspection in about 10 minutes. HemoTypeSC can enable affordable, rapid, and accurate diagnostic testing and newborn screening programs for SCD at the POC in low-resource regions with a high prevalence of SCD.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Christopher Akanbobnaab and Joycelyne Nugent in Ghana; Lisiane Kéclard in Guadeloupe; Mary Reynaud and Jennifer Korpik in Cincinnati; and Robert DiNello, Kin Lung Siu, Jerry Lin, and Kwamina Bentsi-Barnes at Silver Lake Research Corporation.
FINANCIAL SUPPORT
Research funding was provided by National Heart, Lung and Blood Institute of the National Institutes of Health under Award Number R44HL123670. HemoTypeSC test kits were donated by Silver Lake Research Corporation, Azusa, CA, USA.
(Funded by the National Heart, Lung and Blood Institute; ClinicalTrials.gov number, NCT03619798.)
MG and ES are employees of Silver Lake Research Corporation. CTQ has received research and consultancy funding from Silver Lake Research Corporation through a grant from NIH-NHLBI to Silver Lake Research Corporation (R43HL123670).
Footnotes
CONFLICTS OF INTEREST
The other authors report no potential conflicts of interest.
REFERENCES
- 1.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44. [DOI] [PubMed] [Google Scholar]
- 2.McCavit TL. Sickle cell disease. Pediatr Rev 2012;33:195–204; quiz 5–6. [DOI] [PubMed] [Google Scholar]
- 3.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica 2007;92:905–12. [DOI] [PubMed] [Google Scholar]
- 4.Wong TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014;124:3850–7; quiz 4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le PQ, Gulbis B, Dedeken L, et al. Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr Blood Cancer 2015;62:1956–61. [DOI] [PubMed] [Google Scholar]
- 6.Gardner K, Douiri A, Drasar E, et al. Survival in adults with sickle cell disease in a high-income setting. Blood 2016;128:1436–8. [DOI] [PubMed] [Google Scholar]
- 7.Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease. Blood 2004;103:4023–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115:3447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aygun B, Odame I. A global perspective on sickle cell disease. Pediatr Blood Cancer 2012;59:386–90. [DOI] [PubMed] [Google Scholar]
- 10.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med 2011;41:S398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 2013;381:142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun 2010;1:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010;115:4331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.United Nations General Assembly. 63/237. Recognition of sickle-cell anaemia as a public health problem. Resolution Adopted by the General Assembly 2009. [Google Scholar]
- 15.World Health Organization; Uniting against NCDs: the time to act is now. The Brazzaville declaration on non-communicable diseases prevention and control in the WHO African region Geneva, Switzerland: World Health Organization, Regional Office for Africa; 2011. [Google Scholar]
- 16.McGann PT, Grosse SD, Santos B, et al. A Cost-Effectiveness Analysis of a Pilot Neonatal Screening Program for Sickle Cell Anemia in the Republic of Angola. J Pediatr 2015;167:1314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patra PK, Khodiar PK, Hambleton IR, Serjeant GR. The Chhattisgarh state screening programme for the sickle cell gene: a cost-effective approach to a public health problem. J Community Genet 2015;6:361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.World Health Organization. Sickle Cell Disease: A Strategy For The WHO Africa Region. Report of the Regional Director 2010;Document number AFR/RC60/8. [Google Scholar]
- 19.Ryan K, Bain BJ, Worthington D, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. Br J Haematol 2010;149:35–49. [DOI] [PubMed] [Google Scholar]
- 20.Canning DM, Huntsman RG. An assessment of Sickledex as an alternative to the sickling test. J Clin Pathol 1970;23:736–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X, Kanter J, Piety NZ, Benton MS, Vignes SM, Shevkoplyas SS. A simple, rapid, low-cost diagnostic test for sickle cell disease. Lab Chip 2013;13:1464–7. [DOI] [PubMed] [Google Scholar]
- 22.Kanter J, Telen MJ, Hoppe C, Roberts CL, Kim JS, Yang X. Validation of a novel point of care testing device for sickle cell disease. BMC Med 2015;13:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGann PT, Schaefer BA, Paniagua M, Howard TA, Ware RE. Characteristics of a rapid, point-of-care lateral flow immunoassay for the diagnosis of sickle cell disease. Am J Hematol 2016;91:205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillet P, Mori M, Van Esbroeck M, Van den Ende J, Jacobs J. Assessment of the prozone effect in malaria rapid diagnostic tests. Malar J 2009;8:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smart LR, Ambrose EE, Raphael KC, et al. Simultaneous point-of-care detection of anemia and sickle cell disease in Tanzania: the RAPID study. Ann Hematol 2018;97:239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alapan Y, Kim C, Adhikari A, et al. Sickle cell disease biochip: a functional red blood cell adhesion assay for monitoring sickle cell disease. Transl Res 2016;173:74–91 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quinn CT, Paniagua MC, DiNello RK, Panchal A, Geisberg M. A rapid, inexpensive and disposable point-of-care blood test for sickle cell disease using novel, highly specific monoclonal antibodies. Br J Haematol 2016;175:724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohlfing CL, Connolly SM, England JD, et al. The effect of elevated fetal hemoglobin on hemoglobin A1c results: five common hemoglobin A1c methods compared with the IFCC reference method. Am J Clin Pathol 2008;129:811–4. [DOI] [PubMed] [Google Scholar]
- 29.Little RR, Roberts WL. A review of variant hemoglobins interfering with hemoglobin A1c measurement. J Diabetes Sci Technol 2009;3:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.