

Abstract
Background
Serum IL-22 levels are elevated in atopic dermatitis (AD), which commonly precedes asthma in the atopic march. Epicutaneous (EC) sensitization in mice results in Th2-dominated skin inflammation that mimics AD, and sensitizes the airways for antigen challenge-induced allergic inflammation characterized by the presence of both eosinophils and neutrophils. EC sensitization results in elevated serum levels of IL-22.
Objective
To determine the role of IL-22 in antigen-driven airway allergic inflammation following inhalation challenge in EC sensitized mice.
Methods
Wild type (WT) and Il22−/− mice were EC sensitized or intraperitoneally (i.p.) immunized with ovalbumin (OVA) and intranasally challenged with antigen. OVA TCR-specific T cells were Th22 polarized in vitro. Airway inflammation, mRNA levels in the lungs and airway hyperresponsiveness (AHR) were examined.
Results
EC sensitization preferentially elicited an IL-22 response compared to i.p. immunization. Intranasal challenge of mice EC-sensitized with OVA elicited in the lungs Il22 mRNA expression, IL-22 production and accumulation of CD3+CD4+IL22+ T cells that co-expressed IL-17A and TNFα. EC-sensitized Il22−/− mice exhibited diminished eosinophil and neutrophil airway infiltration, and decreased AHR following intranasal OVA challenge. Production of IL-13, IL-17A and TNFα was normal, but IFNγ production was increased in lung cells from airway-challenged EC-sensitized Il22−/− mice. Intranasal instillation of IFNγ neutralizing antibody partially reversed the defect in eosinophil recruitment. WT recipients of Th22 polarized WT, but not IL-22 deficient, TCR-OVA specific T cells, which both secrete IL-17A and TNFα, developed neutrophil-dominated airway inflammation and AHR upon intranasal OVA challenge. Intranasal instillation of IL-22 with TNFα, but not IL-17A, elicited neutrophil-dominated airway inflammation, and AHR in WT mice, suggesting that the loss of IL-22 synergy with TNFα contributed to the defective recruitment of neutrophils into the airways of Il22−/− mice. TNFα, but not IL-22 blockade at the time of antigen inhalation challenge inhibited airway inflammation in EC sensitized mice
Conclusion
EC sensitization promotes the generation of antigen-specific IL-22 producing T cells that promote airway inflammation and AHR following antigen challenge, suggesting that IL-22 plays an important role in the atopic march.
Keywords: IL-22, AD, asthma, Neutrophils
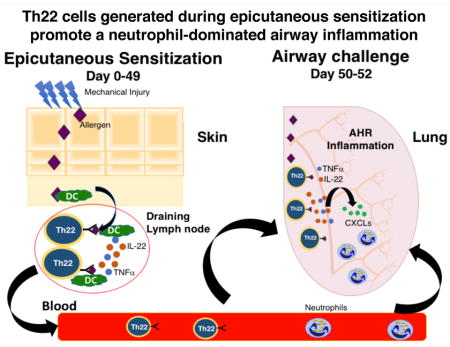
Graphical Abstract
INTRODUCTION
Asthma is a chronic inflammatory disorder of the lungs that affects up to 15% of children and teenagers worldwide1. The heterogeneity of the asthma phenotype has led to multiple classifications depending on the age of onset, severity or persistence of the disease, allergic status or response to treatment modalities. Despite decades of research, knowledge the cellular and molecular mechanisms in asthma remains incomplete, constituting a barrier for the optimal management of patients with the disease.
Atopic dermatitis (AD) often precedes asthma, a phenomenon known as atopic march2. AD is a common allergic skin inflammatory disease that often starts in infancy and affects more than 17% of children in the US. Acute AD skin lesions are characterized by epidermal and dermal thickening and by dermal infiltration of CD4+ T cells and eosinophils, predominant expression of the Th2 cytokines IL-4 and IL-13 and elevated expression of IL-22 and IL-17A3–5. The roles of Th2 cytokines and IL-17A have been extensively studied in mouse models of the atopic march6–10. However, little is known about the role of IL-22 in this disease progression.
IL-22 is a member of the IL-10 family of cytokines produced by adaptive Th17 and Th22 cells, innate lymphocytes that include γδ T cells and type 3 innate lymphoid cells (ILC3) as well as myeloid cells including neutrophils11–15. The IL-22 receptor (IL-22R) is expressed on epithelial cells, but not immune cells16, indicating an important role for IL-22 signaling in mucosal barrier function. Increased IL-22 levels have been observed in lungs, bronchoalveolar lavage fluid (BALF) and serum of patients with asthma17–19. Increased lung IL-22 levels have been observed in mouse models of asthma upon airway challenge 17, 20–22. IL-22 inhibits the expression of proinflammatory chemokines and adhesion molecules induced by IFNγ, in human bronchial epithelial cells23, suggesting that it could play a protective role in asthma. In contrast, IL-22 enhances the proliferation and migration of human airway smooth muscle cells24, 25, suggesting that it could play a pathogenic role in asthma. Genetic deletion of Il22 in mice or administration of IL-22 blocking antibody to WT mice aggravates airway inflammation and airway hyperreactivity (AHR) elicited by intranasal (i.n.) challenge of intraperitoneally (i.p.) immunized mice17, 21, 22. Reciprocally, i.n. instillation of rIL-22 before i.n. challenge of i.p. sensitized mice reduced airway inflammation and AHR17, 21, suggesting a protective role of IL-22. In contrast, Genetic deletion of Il22 in mice or administration of IL-22 blocking antibody to WT mice reduced airway inflammation in mice sensitized subcutaneously (s.c.) with OVA17. Thus, IL-22 appears to play opposing roles in antigen driven mouse models of asthma depending on the route of immunization.
Neither i.p. immunization nor s.c. immunization mimic antigen sensitization in patients. We have previously reported that mice epicutaneously (EC) sensitized by application of antigen to tape stripped skin, which mimics antigen cutaneous exposure to antigen in patients with AD, develop allergic skin inflammation with features of AD26. Antigen challenge of EC sensitized mice results in airway inflammation and AHR with features of allergic asthma26. This model mimics the atopic march in patients with AD who develop asthma driven by antigens that had been initially introduced through a disrupted skin barrier. We herein demonstrate a role for IL-22 in airway inflammation and AHR in this model, suggesting that IL-22 may play a role in asthma that develops in the atopic march in patients with AD.
METHODS
Mice and Sensitization
Il22−/− mice were described previously27. Wild type (WT) Balb/c and DO11.10 TCR transgenic mice were obtained from Charles River Laboratories and Jackson Laboratories, respectively. All mice were housed in a specific pathogen-free environment and fed an OVA-free diet. EC and IP sensitization of 6 to 10 weeks old mice was done as previously described7. All procedures were performed in accordance with the Animal Care and Use Committee of Boston Children’s Hospital.
RNA extraction and quantitative PCR analysis
RNA was extracted from whole lungs with total RNA isolation kit (Ambion). cDNA was prepared with iscript cDNA synthesis kit (Biorad). Quantitative real-time PCR was done using Taqman gene expression assays, universal PCR master mix and ABI prism 7300 sequence detection system (Applied Byosistems).
Cytokines production by spleen and lung cells
Spleen and lung single cell suspensions were cultured at 4×106/ml in the presence of OVA (200 μg/ml) for 96 hours as described previously28. Cytokines in supernatants was measured by ELISA using Ready-Set-Go! ELISA Kits (eBioscience) following the manufacturer’s instructions.
Intranasal treatments and analysis of airway inflammation and airway hyperrreactivity (AHR)
Intranasal (i.n.) challenge with OVA (50 μg) was performed daily for three days followed by analysis of airway hyperreactivity responses. Lung resistance (RL) was measured using invasive BUXCO (Buxco electronics) in response to increasing doses of methacholine administrated by nebulization to anesthetized mice 24 h after last i.n. treatment. Immediately after sacrifice, BALF was collected, total cells were counted and the number of leukocytes was determined from cytospins stained with Diff-Quik stain set (Baxter). Recombinant mouse cytokines were purchased from R&D systems. Mice were treated with mouse rIL-22 (100 or 1000 ng), rTNFα (50 ng), rIL-17A (100 ng) and rIL-13 (500, 1000or 2500 ng) alone or in combinations for 3 consecutives days. For IFNγ blocking experiments mice were treated with 50 μg of monoclonal antibody against IFNγ (Clone XMG1.2, Bioxcell), 15 μg of monoclonal antibody against IL-22 or TNF (R&D systems) or isotype control (Bioxcell or R&D systems) during intranasal OVA challenge.
Histological analysis
Lung specimens were fixed in 4% PFA and embedded in paraffin. H&E staining was performed in 5 mm sections.
Antibodies and flow cytometry
Lung cells were incubated with an anti- FcγRIII/II Ab (eBioscience) on ice for 15 min to block Fc receptors, washed and incubated with eF506 viability dye (eBioscience), APC-Cy7-conjugated anti-GR1 (RB6-8C5, eBioscience), FITC-conjugated anti-CD11b (M1/70, Biolegend), eFluor-450-anti-CD3 (17A2, eBioscience), APC-anti CD4 (GK1.5, eBioscience), APC-Cy7-conjugated anti-CD90 (53-2.1, eBioscience), PE-anti CD8 (53-6.7, eBioscience), PercPeF710-conjugated TCR γδ (eBioGL3, eBioscience) for 15 min. For ILC staining, biotin-anti-CD11b (M1/70), biotin-anti-CD11c (N418), biotin-anti-F4-80 (BM8), biotin-anti-B220 (RA3-6B2), biotin-anti-CD19 (1D3), biotin-anti-FcεR1α (MAR-1), biotin-anti-CD49b (DX5) and biotin-anti-Gr1 (RB6-8C5) from eBioscience, followed by Streptavidin-BV605 (BD) were use as lineage gate. ILCs were defined as CD45+CD3−Lin− cells. For Intracellular staining, lung cells were stimulated with PMA (50 ng/ml) and ionomycin (500 ng/ml) in presence of Golgi-stop and Golgi-plug for 4 hours in IMDM media, followed by surface staining for CD3, CD4, CD8, γδ TCR and CD90. Cells were then fixed and permeabilized using BD Cytofix/Cytoperm Kit (BD Biosciences) and stained with anti-IL-22 (Biolegend), anti-TNFα (Biolegend), anti-IL-17A (eBioscience) or anti-IFNγ (eBioscience) antibodies.. Finally, cells were washed and analyzed on LSRFortessa (BD Biosciences). Results were analyzed using Flowjo (Treestar).
Th22 in vitro polarization and adoptive transfer
Naïve CD4+ T cells from DO11.10 transgenic (Tg) mice were isolated from the spleen using Naive CD4+ T Cell Isolation Kit, mouse (Miltenyi). 1X106 naïve were cultured for 4 days in anti-CD3 Ab coated wells (2 μg/ml) with anti-CD28 Ab (2 μg/ml), IL-6 (50 ng/ml), IL-23 (50 ng/ml), anti-IL-4 Ab (10 μg/ml), anti-IFNγ Ab (10 μg/ml) and anti TGFβ Ab (10 μg/ml). Then, 2X106 Th22 in vitro-polarized cells were intravenously injected in WT mice.
Statistical analysis
Results were analyzed using 2-way ANOVA or non-parametric t tests. A p value of less than 0.05 was considered significant.
RESULTS
EC sensitization elicits a systemic IL-22 response and an antigen-specific IL-22 response in the lungs
We previously demonstrated that EC sensitization by application of antigen to tape stripped mouse skin promotes systemic type 2 and type 17 immune responses7, 29. We examined whether EC sensitization also elicits a systemic IL-22 response. Spleens cells from mice EC sensitized with OVA secreted significantly more IL-22 when cultured in vitro with OVA, compared to spleen cells from control mice EC sensitized with saline (Fig. 1A). Furthermore, as we previously described, EC sensitization with OVA, but not saline, resulted in a significant elevation of serum IL-22 levels (Fig. 1B). These findings demonstrate that EC sensitization with antigen elicits a systemic IL-22 response.
Figure 1. EC sensitization elicits a systemic IL-22 response and an antigen-specific IL-22 response in the lungs.

A-B. IL-22 secretion by OVA stimulated splenocytes (A) and IL-22 serum levels (B). C,D. Il22 mRNA expression in the lungs (C), and IL-22 secretion by OVA stimulated lung cells (D). E. Representative FACS analysis and quantitation of intracelluar expression of IL-22+ cells among CD3+CD4+ T cells and of IL-17A+ and TNFα+ cells among CD3+CD4+IL-22+ cells in the lung. Mice were EC sensitized with OVA or saline in A and B, followed by i.n. challenged with OVA in C-F. Bars represent mean±SEM (n=5–10 per group). *p<0.05.
We next investigated whether intranasal (i.n.) OVA challenge of mice EC sensitized with OVA causes Il22 mRNA expression and IL-22 production in the lungs. Following i.n. OVA challenge, Il22 mRNA levels in the lungs were significantly increased in mice EC sensitized with OVA, compared to control mice EC sensitized with saline (Fig. 1C). In addition, lungs cells from mice EC sensitized with OVA, secreted significantly higher amounts of IL-22 in response to OVA re-stimulation in vitro compared to lung cells from controls EC sensitized with saline (Fig. 1D). These results demonstrate that EC sensitization elicits an antigen-specific IL-22 response in response to following airway antigen challenge.
To identify the cellular sources of IL-22 in the lung of intranasally challenged EC sensitized mice, lung cell suspensions were analyzed by flow cytometry. FACS showed significantly increased percentages of CD3+CD4+IL-22+ cells in the lungs mice EC sensitized mice OVA, compared to mice EC sensitized mice with saline (Fig. 1E). These CD3+CD4+IL-22+ cells co-expressed the cytokines TNFα 90±5%, n= 4) and IL-17A (50±8.4% n= 4). IL-22 expression was barely detected or not detected in CD3+CD8+ cells, CD3+γδ TCR+ or CD3−Lin−CD90+ ILCs from lungs of EC sensitized mice with OVA or saline (Supplementary Figure 1). Altogether these results show that CD4+ T cells are the major source of IL-22 in the lungs of EC sensitized mice with OVA after i.n. challenge.
EC sensitization preferentially promotes an IL-22 response
IL-22 serum levels (Fig. 2A), IL-22 and TNFα secretion by splenocytes stimulated with OVA in vitro (Fig. 2B), Il22 and Tnfa mRNA expression in the lungs following OVA airway challenge (Fig. 2C) and IL-22 and TNFα secretion in response to OVA stimulation by lungs cells from airway challenged mice (Fig. 2D) were all significantly lower in mice immunized intraperitoneally (i.p.) with OVA compared to mice EC sensitized with OVA. As previously reported7, IL-17A production by OVA stimulated splenocytes was higher in EC sensitized mice than in i.p. immunized mice, while IL-4, IL-13 and INF-γ production and serum OVA specific IgE levels were comparable between the two groups (data not shown). These results show that EC sensitization preferentially promotes a systemic IL-22 response and, importantly, the production of IL-22 and TNFα in the lungs after i.n. challenge.
Figure 2. EC sensitization preferentially promotes an IL-22 response.

A-D. serum IL-22 levels (A), IL-22 secretion by OVA stimulated splenocytes (B), Il22 and Tnfa mRNA expression in the lungs (C) and IL-22 and TNFα secretion by OVA stimulated lung cells of mice EC or IP sensitized with OVA and i.n. challenged with OVA. Bars represent mean±SEM (n=3–5 per group). *p<0.05, **p<0.005 and ***p<0.001.
We had previously shown demonstrated that TGFβ, which is richly expressed in the skin, is important for inducing an IL-17A response to EC sensitization with OVA7. To examine whether TGFβ plays a role in the IL-22 response to EC sensitization we intradermally (i.d.) injected a neutralizing antibody to TGFβ 6 hrs prior to EC sensitization, then examined dendritic cells (DCs) isolated from draining lymph nodes 24 hrs after EC sensitization for their ability to drive cytokine production by naïve OVA-TCR transgenic DO11.10 T cells. TGFβ blockade had no effect on the ability of the DCs to drive IL-22 production by naïve T cells (Supplementary Figure 2). However, as we previously reported7, it significantly impaired their ability to drive IL-17A production. TGFβ blockade had no effect on the ability of DCs to drive IL-13 or INFγ production.
IL-22 is important for the development of airway inflammation and AHR following intranasal antigen challenge of EC sensitized mice
To determine the role of IL-22 in lung inflammation elicited by i.n. challenge of EC sensitized mice, we made use of Il22−/− mice. As previously reported7, 26, i.n. OVA challenge of WT mice EC sensitized with OVA elicited eosinophil and neutrophil influx into the airways, as well as AHR (Fig. 3A–C). Il22−/− mice EC sensitized with OVA accumulated significantly less total cells, eosinophils and neutrophils in BALF and demonstrated reduced peribronchial cellular infiltrates in the lungs compared to OVA sensitized WT controls (Fig. 3A,B). Importantly, following i.n. OVA challenge, Il22−/− mice EC sensitized with OVA showed a significant decrease in airway resistance in response to methacholine compared to OVA sensitized WT controls (Fig. 3C). Lungs cells from WT mice EC sensitized with OVA secreted significant more IL-13, IL-17A, IFNγ and IL-22 and TNFα in response to re-stimulation in vitro with OVA compared to mice EC sensitized with saline. Lung cells from Il22−/− mice EC sensitized with OVA and subjected to airway antigen challenge, secreted comparable amounts of IL-13, IL-17A and TNFα, but significantly more of IFNγ, in response to OVA re-stimulation in vitro compared to lung cells from OVA sensitized WT controls (Fig. 3D). The increased IFNγ production was selective to lung cells, as we previously reported normal production of IFNγ by OVA stimulated splenocytes of EC sensitized Il22−/− mice as well as normal levels of OVA specific IgG2a antibody, in addition to normal production of IL-4 and IL-13 by OVA stimulated splenocytes and normal levels of OVA specific IgG1 and IgE antibody30. The increased IFNγ production by lung cells in Il22−/− mice is consistent with previous report that IL-22 downregulates Th1 responses31. As expected, IL-22 secretion was not detected in lung cells from Il22−/− mice EC sensitized with OVA (Fig. 3D). These results demonstrate that IL-22 plays an important role for antigen driven airway inflammation and AHR in EC sensitized mice following i.n. challenge.
Figure 3. IL-22 is important for the development of airway inflammation and AHR following intranasal antigen challenge of EC sensitized mice.

A-D. Total and differential cell counts in BALF (A), H&E stained lung sections (B), and lung resistance in response to increasing doses of methacholine (C) and cytokine secretion by OVA stimulated lung cells (D) in WT and Il22−/− mice EC sensitized with OVA or saline and i.n. challenged with OVA. Bars represent mean±SEM (n=4–7 per group). *p<0.05, **p<0.05, ***p<0.001.
Increased IFNγ production underlies the decreased airway eosinophilia in inhalation challenged Il22−/− mice EC sensitized with OVA
IL-13 plays an important role in airway eosinophilia elicited by inhalation challenge in mouse models of allergic airway inflammation32. Furthermore, instillation of IL-13 in the airways elicits airway eosinophilia33. The observation that eosinophil recruitment to the antigen challenged airways was virtually absent in Il22−/− mice EC sensitized with OVA prompted us to examine whether IL-22 synergizes with IL-13 in driving airway eosinophilia. We first determined the dose of IL-13 that resulted in suboptimal airway eosinophilia (~ 70% of maximum) when instilled in the airway by intranasal inhalation (Supplementary Figure 3A). We then examined whether addition of IL-22 enhances IL-13 driven eosinophilia. Nasal instillation IL-22 by itself caused no airway eosinophilia (Supplementary Figure 3B). Co-administration of up to 1 μg of IL-22 by nasal instillation failed to enhance IL-13 driven airway eosinophilia (Supplementary Figure 3B). Thus the impaired eosinophilia in the antigen challenged airways of Il22−/− mice EC sensitized with OVA is not due to loss of a potential synergistic effect of IL-22 on IL-13 driven airway eosinophilia.
IFNγ has been reported to inhibit the ability of IL-13 to cause airway eosinophilia34. We investigated whether Il22−/− mice EC sensitized with OVA exhibit increased Ifng mRNA levels and enhanced recruitment of IFNγ producing cells in the lungs. Both CD3+CD4+ T cells and type 1 innate lymphoid cells (ILC1s) are a source of IFNγ35, Ifng expression is dependent on the transcription factor T-bet encoded by tbx2136, and both Th1 cells and ILC1s are recruited to tissues by the chemokine CCL5 which binds to the chemokine receptor CXC3 expressed on these cells35. The mRNA levels of Ifng, Tbx21, Cxcr3 and Ccl5 were significantly increased in lungs of EC OVA sensitized Il22−/− mice after i.n OVA challenge compared with their WT counterparts (Fig. 4A). In addition, CD3−Lin−CD90+IFNγ+ ILC1s, but not CD3+CD4+IFNγ+ Th1 cells or CD3+CD8+IFNγ+ cells were increased in EC sensitized Il22−/− mice after i.n OVA challenge (Fig. 4B), suggesting that IL-22 controls the recruitment of ILC1s to the lungs after intranasal challenge.
Figure 4. Increased IFNγ production underlies the decreased airway eosinophilia in inhalation challenged Il22−/− mice EC sensitized with OVA.

A, B. Ifng, Tbx21, Cxcr3 and Ccl5 mRNA levels in the lungs (A), Representative FACS analysis and quantitation of intracellular expression of IFNγ+ cells among CD3+Lin−C90+ cells, CD3+CD4+ T and CD3+CD8+ T cells in the lungs (B) of WT and Il22−/− mice EC sensitized and intranasally challenged with OVA. C, D. Effect of i.n. administration of anti-IFNγ antibody, or isotype control, on the recruitments of eosinophils (C) and neutrophils (D) to the antigen challenged airways of Il22−/− mice EC sensitized with OVA. Bars represent mean±SEM (n=5–7 per group). *p<0.05.
To examine if increased IFNγ production contributed to the impaired recruitment of eosinophils to the antigen challenged airways of Il22−/− mice EC sensitized with OVA, we treated the mice intranasally with neutralizing antibody to IFNγ during OVA airway challenge. Administration of anti-IFNγ antibody, but not isotype control, significantly increased airway eosinophilia following antigen inhalation in Il22−/− mice EC sensitized with OVA (Fig. 4C). This effect was specific to eosinophils because administration of IFNγ antibody failed to restore neutrophil recruitment to the airways in these mice (Fig. 4D). Reconstitution of airway eosinophilia in the challenged airways of EC sensitized Il22−/− mice by administration of anti-IFNγ antibody was partial as the numbers of eosinophils in the airways of these mice remained lower than in the challenged airways of EC sensitized WT mice (5.9±0.52×104 versus 2.04±0.32×105) These results suggest that the impaired eosinophilia in the antigen challenged airways of EC sensitized Il22−/− mice is in part secondary to the increased production of IFNγ in the lungs of these mice
Th22 antigen-specific CD4+ T cells drive neutrophil-dominated airway inflammation and AHR in response to intranasal antigen challenge
We next addressed the mechanisms by which lack of IL-22 impairs neutrophil recruitment into the antigen challenged airways of EC sensitized mice. We first investigated the ability of CD4+ helper T cells that produce IL-22 to elicit airway inflammation and AHR. We examined the response to i.n. antigen challenge of WT recipients of antigen specific CD4+ T cells polarized in vitro into IL-22 producing cells. Naïve splenic CD4+CD62L+ T cells from TCR-transgenic DO11.10 mice were activated with anti-CD3+anti-CD28 and IL-23 in the presence of neutralizing antibodies to IL-4, IFN-γ and TGF-β, conditions known to drive the differentiation of naïve CD4+ cells into IL-22 producing cells27, 37. T cells polarized in vitro under these conditions are thereafter designated Th22 cells. As expected, Th22 cells from DO11.10 mice, but not DO11.10/Il-22−/− mice, secreted IL-22 (Fig. 5A). The two types of cells secreted comparable levels of TNF-α and IL-13 and no detectable levels of IL-4 (Fig. 5A, and data not shown). Th22 cells from DO11.10/Il-22−/− mice secreted modrestly, but significantly, more IL-17A and IFNγ than Th22 cells from WT controls (Fig. 5A).
Figure 5. Th22 antigen-specific CD4+ T cells drive neutrophil-dominated airway inflammation and AHR in response to intranasal antigen challenge.

A. Cytokine secretion by vitro polarized Th22 cells from in WT and Il22−/− mice. B-F. Total and differential cell counts in BALF (B), frequency of neutrophils in lungs (C), H&E stained lung sections (D), chemokine mRNA levels in lungs (F), and lung resistance in response to increasing doses of methacholine (G) in WT recipients of Th22 polarized OVA-specific CD4+ T cells from DO11.10 and DO11.10/Il22−/− mice following n. challenge with OVA. Mice that received no T cells were used as controls. Bars represent mean±SEM (n=4–6 per group). *p<0.05, **p<0.05, ***p<0.001.
Following antigen inhalation challenge, recipients of Th22 polarized DO11.10 CD4+ T cells, but not DO11.10/Il-22−/− CD4+ T cells, exhibited a significant increase in the number of total cells and neutrophils in BALF and lungs (Fig. 5B, C), and increased peribronchial cellular infiltrates (Fig. 5D) compared to control mice that received no T cells. In line with the increase of neutrophils in their airways and lungs, recipients of Th22 polarized DO11.10 CD4+ T cells showed significantly increased mRNA levels of Cxcl1 and Cxcl3, but not Cxcl2, in their lungs compared to recipients of DO11.10/Il-22−/− CD4+ T cells and control mice that received no T cells (Fig. 5E). Comparable Il17a and Ifng mRNA levels were observed in airway challenged lungs of recipients of Th22 polarized cells WT and IL-22 deficient OVA-TCR transgenic T cells (Supplementary Figure 4). Moreover, intranasally challenged recipients of Th22 polarized DO11.10 CD4+ T cells demonstrated significantly increased airway resistance in response to methacholine compared to recipients of DO11.10/Il-22−/− CD4+ T cells or control mice that received no T cells (Fig. 5F). Together these results demonstrate that IL-22 produced by CD4+ T cells can drive neutrophil-dominated airway inflammation and AHR.
IL-22 synergizes with TNFα to drive neutrophil-dominated airway inflammation and AHR
We investigated whether IL-22 is sufficient to drive airway inflammation and AHR. rIL-22 was administered i.n. in two different doses (100 ng and 1000 ng) daily for three days to WT mice. Intranasal administration of rIL-22 caused no significant increase in the number of total cells or neutrophils in the BALF, no significant increase in the number of neutrophils in the lungs, no increase in peribronchial cellular infiltrates, no significant increase in mRNA levels of Cxcl1, Cxcl2 and Cxcl3 in the lungs and no significant AHR to methacholine compared to internal administration of saline (Fig. 6A–E and Supplementary Fig. 5).
Figure 6. IL-22 synergizes with TNFα to promote neutrophil airway inflammation.

A-D. Total and differential counts in BALF (A), frequency of neutrophils in lungs (B), H&E stained lung sections (C), chemokine mRNA levels in the lungs (D) and lung resistance in response to increasing doses of methacholine (E) in WT mice treated intranasally with saline, rIL-22, rTNFα or rIL-22+rTNFα. Bars represent mean±SEM (n=4–6 per group). *p<0.05, **p<0.05, ***p<0.001
Since IL-17A and TNFα were co-expressed with by CD3+CD4+IL-22+ lung cells, and IL-17A and TNFα were secreted by in vitro polarized Th22 cells which when adoptively transferred resulted in the development of neutrophil-dominated airway inflammation and AHR in response to antigen inhalation challenge. We examined whether IL-17A and TNFα might synergize with IL-22 to drive this response. We treated WT mice intranasally with rIL-22 (1000 ng) in combination with rIL-17A (100 ng) or rTNFα (50 ng). Mice treated with TNFα alone exhibited a modest, albeit significant, increase in neutrophils in BALF (Fig. 6A), no significant increase in the total lung neutrophils (Fig. 6B) minimal increase in peribronchial cellular infiltrates (Fig. 6C), no significant increase in Cxcl1, Cxcl2 and Cxcl3 mRNA levels in the lungs (Fig. 6D), and no increase in airway resistance compared to controls treated with saline (Fig. 6E). Importantly, mice treated with rIL-22+rTNFα exhibited significantly increased numbers of total cells, neutrophils and lymphocytes in BALF (Fig. 6A), increased neutrophils in the lungs (Fig. 6B), increased peribronchial cellular infiltrates (Fig. 6C), significantly increased mRNA levels of Cxcl1 and Cxcl3 in the lungs (Fig. 6D), and significantly enhanced airway resistance (Fig. 6E) compared to controls. There was no significant increase in Il13, Il33 Il25 or Il17a mRNA in the lungs of mice that received rIL-22+rTNFα (Supplementary Fig. 6). The numbers of total cells, macrophages neutrophils and lymphocytes in BALF, the numbers of neutrophils in the lungs, peribronchial cellular infiltrates, mRNA levels of Cxcl1, Cxcl2 and Cxcl3 in the lungs and airway resistance in mice treated with IL-17A alone, or in combination with IL-22 were comparable to those in control mice treated with saline (Supplementary Fig. 7). These results demonstrate that IL-22 synergizes with TNFα, but not with IL-17A, to promote neutrophil airway inflammation and suggest that IL-22 and TNFα produced by Th22 cells, synergize to drive neutrophil-dominated airway inflammation and AHR.
Blockade of TNFα, but not IL-22, during OVA intranasal challenge decreases airway inflammation in mice EC sensitized with OVA
We investigated whether blockade of IL-22 or TNFα during intranasal challenge decreased airway inflammation in EC sensitized mice. Intranasal administration of TNFα blocking antibody during OVA challenge caused a significant decrease in the number of total cells, eosinophils and neutrophils in the BALF, significant decrease in the number of neutrophils in the lungs, decrease in peribronchial cellular infiltrates and a significant decrease in mRNA levels of Cxcl1, Cxcl2 and Cxcl3 in the lungs compared to intranasal administration of isotype control antibody (Fig. 7A–D). Intranasal administration of IL-22 blocking antibody had not apparent effect on the airway inflammation induced by intranasal OVA challenge of EC sensitized mice with OVA (Fig. 7A–D). These results demonstrate that TNFα plays an important role in mediating airway inflammation in response to intranasal challenge of EC sensitized mice. In contrast, although important for the induction of airway inflammation in intranasally challenged EC sensitized mice, IL-22 plays no detectable role in the effector phase of the response to intranasal challenge (Fig. 7A–D).
Figure 7. TNFα, but not IL-22 blockade during the challenge phase inhibits airway inflammation.

A-D. Total and differential counts in BALF (A), Numbers of neutrophils in lungs (B), H&E staining of lung sections (C), chemokine mRNA levels in the lungs (D) in WT mice EC sensitized with OVA then subjected to i.n. instillation of OVA together with neutralizing anti-IL-22 IgG antibody, neutralizing anti-TNFα IgG antibody or IgG isotype controls. Bars represent mean±SEM (n=4–6 per group). *p<0.05, **p<0.05, ***p<0.001.
DISCUSSION
We demonstrate that IL-22 is produced by CD4+ T cells in the lungs of EC sensitized mice and plays a critical role in airway inflammation and AHR elicited by antigen inhalation challenge of these mice. Moreover, we show that synergistic action of IL-22 with TNFα produced by allergen-specific Th22 cells can drive neutrophil dominated airway inflammation.
Serum IL-22 levels, IL-22 production by antigen stimulated splenocytes, as well as Il22 mRNA expression and IL-22 production in the lungs after i.n. antigen challenge all increased in WT mice EC sensitized with OVA. EC sensitization preferentially elicited an IL-22 response compared to i.p. immunization. We previously showed that EC sensitization with OVA also preferentially elicits an IL-17A response compared to evidenced by increased serum IL-17A levels, IL-17A production by antigen stimulated splenocytes, as well as Il17a mRNA expression and IL-17A production in the lungs after i.n. antigen challenge7. The induction of both IL-22 and IL-17A following EC sensitization is consistent with the observation that the two cytokines are often produced by overlapping populations of Th cells that require different factors for their induction27. We previously demonstrated that IL-22 production, and to a lesser extent IL-17A production, following EC sensitization is dependent on IL-23 a cytokine critical for the induction of IL-22 producing CD4+ T helper cells 30. Whereas TGFβ is important for IL-17A induction after EC sensitization7, it had no detectable role in IL-22 induction in EC sensitized mice. We also showed that IL-23 is primarily induced in, and released by, keratinocytes following tape stripping, which is used for EC sensitization30. Il23 mRNA expression and IL-23 production are also induced in normal human skin subjected to mechanical injury by tape stripping30. Furthermore, Il23 mRNA expression is elevated in the skin of patients with AD38. Together, these findings suggest that our examination of the role of IL-22 in airway inflammation elicited by antigen inhalation in EC sensitized mice is relevant to asthma that develops in AD patients.
Our results demonstrate that IL-22 plays a pro-inflammatory role in airway inflammation in EC sensitized mice. OVA EC sensitized Il22−/− mice exhibited decreased eosinophil and neutrophil and influx in the BALF, attenuated lung inflammation and diminished AHR after i.n. challenge. In line with these results, previous reports have been shown that IL-22 deficiency or blockade by antibody reduces airway inflammation in mice sensitized subcutaneously with OVA17 or mice treated intra-tracheally with a high dose of bleomycin39. In contrast, several reports indicate a protective role of IL-22 in the lungs of i.p. sensitized mice, as i.p. sensitized Il22−/− mice exhibited enhanced airway inflammation following inhalation challenge and i.n. instillation of rIL-22. These results suggest that the route of allergen sensitization may determine whether IL-22 plays a pathogenic role or a protective role in airway inflammation elicited by antigen inhalation.
Deficiency of IL-22 increased the expression of Ifng, and its transcriptional regulator Tbx21, as well as of the chemokine Ccl5 and its receptor Cxcr3 expressed by Th1 cells and ILC1s in lungs of mice EC sensitized with OVA and challenged intranasally with antigen. It also increased, the number of IFN γ producing lung ILCs and IFNγ production by OVA stimulated lung cells in these mice. These results suggest that IL-22 inhibits Iocal IFNγ production during antigen-driven airway inflammation. IL-22 production in BALF of asthmatic patients inversely correlates with IFNγ-regulated pro-inflammatory mediators23. Furthermore, IL-22 blockade40, or selective ablation in CD4+ T cells of Ahr, a transcription factor required for Th22 polarization31, increases the number of Th1 cells in a model of collagen-induced arthritis41. IFNγ inhibits airway eosinophilia by counteracting the effects of IL-4 and IL-13 42. Importantly, IFNγ blockade partially restored in airway eosinophilia in the antigen challenged lungs of Il22−/− mice EC sensitized with OVA. This suggests that IL-22 promotes airway eosinophilia in our model by inhibiting local production of IFNγ. The partial restoration of airway eosinophilia in the antigen challenged airways of EC sensitized Il22−/− mice by IFNγ blockade suggests the presence of additional mechanisms by which IL-22 promotes eosinophil recruitment.
CD4+ T cells are the major source of IL-22 during airway inflammation36. We demonstrated that IL-22 was expressed in CD4+ T cells, but was not detectable in ILCs, CD8+ T cells or TCRγδ T cells, in the lungs of WT mice EC sensitized with OVA and challenged with antigen. DCs and NK cells have been reported as potential sources of IL-2241,42. We cannot exclude these cell types as sources of IL-22 the lungs in our model of airway inflammation. Importantly, antigen specific CD4+ T cells polarized in vitro to Th22 cells promoted neutrophil-dominated airway inflammation and AHR in WT recipients i.n. challenged with antigen. IL-22 production by the adoptively transferred Th22 cells was essential because adoptive transfer of Th22 polarized CD4+ T cells derived from Il22−/− mice failed to drive airway inflammation and AHR in WT recipients i.n. challenged with antigen.
Although IL-22 expression by Th22 polarized CD4+ T cells was essential for driving airway inflammation and AHR, IL-22 by itself was not sufficient. As previously reported39, 43, intranasal installation of rL-22 did not result in either neutrophil recruitment to the airways or AHR. The pathological or protective functions of IL-22 are often mediated by its combined action with other cytokines39, 44–48. IL-22 synergized with TNFα, a cytokine expressed by Th22 cells, in driving airway inflammation and AHR. Synergy between IL-22 and TNFα is important for the control of Chlamydia trachomatis49 and Candida albicans46 infections and is thought to be important in driving tissue inflammation in scleroderma44 and Behçet’s disease48. In contrast, i.n. installation of IL22 together with IL-17A, another cytokine expressed by Th22 cells, had no detectable effect, although IL22 and IL-17A have been reported to synergize to promote airway inflammation39, and in inducing the production of antimicrobial peptides by keratinocytes43. Given the normal production of TNFα and IL-17A by lung cells from airway challenged EC sensitized Il22−/− mice, our data suggests that loss of IL-22 synergy with TNFα contributed to the defective recruitment of neutrophils into the airways of Il22−/− mice.
Consistent with the results obtained in the s.c. immunization model17, IL-22 blockade prior to inhaled antigen challenge had no effect on airway inflammation, whereas TNFα blockade inhibited it. This suggests that while IL-22 is important in eliciting an immune response that promotes airway inflammation, it is not important in the effector phase of the response. Thus, IL-22 blockade could be beneficial during priming of the response to EC sensitization, whereas TNFα blockade could be beneficial in protecting from inhaled antigen challenge subsequent to EC sensitization.
In summary our results suggest that cutaneous sensitization via a mechanically injured skin, as occurs in AD, elicits antigen specific Th22 cells that produce IL-22 and TNFα in response to allergen exposure via the airways and thereby promote eosinophil and neutrophil-mediated lung inflammation and AHR. Our findings suggest that blockade of IL-22 may be beneficial in the prevention of allergic asthma in AD patients.
Supplementary Material
KEY MESSAGES.
Intranasal antigen challenge of epicutaneously sensitized mice elicits accumulation in the lungs of Th22 cells that co-express IL-17A and TNFa.
IL-22 is important for airway inflammation and AHR in EC sensitized mice intranasally challenged with antigen.
TNFa and IL-22 produced by Th22 cells synergize to drive neutrophil dominated airway inflammation and AHR.
Acknowledgments
Funding: This work was supported by NIH grants HHSN272201000020C and AI113294
The authors thank Dr. Hans Oettgen for critical reading of the manuscript. JMLC was supported by a postdoctoral fellowship from Consejo Nacional de Ciencia y Tecnologia (CONACYT, Mexico).
Abbreviations
- AD
atopic dermatitis
- AHR
airway hyperresponsiveness
- BALF
bronchoalveolar lavage fluid
- OVA
ovalbumin
- EC
epicutaneous
- IL
interleukin
- CXCL
Chemokine (C-X-C motif) ligand
- WT
wild type
- i.n
intranasal
- AHR
airway hyperresponsiveness
- RL
lung resistance
- i.p
intraperitoneal
Footnotes
Disclosure of potential conflict of interest: J. M. Leyva Castillo receives support from Consejo Nacional de Ciencia y Tecnologia (CONACYT, Mexico). The rest of the authors declare that they have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pearce N, Ait-Khaled N, Beasley R, Mallol J, Keil U, Mitchell E, et al. Worldwide trends in the prevalence of asthma symptoms: phase III of the International Study of Asthma and Allergies in Childhood (ISAAC) Thorax. 2007;62:758–66. doi: 10.1136/thx.2006.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol. 2003;112:S118–27. doi: 10.1016/j.jaci.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 3.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–94. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 4.Di Cesare A, Di Meglio P, Nestle FO. A role for Th17 cells in the immunopathogenesis of atopic dermatitis? J Invest Dermatol. 2008;128:2569–71. doi: 10.1038/jid.2008.283. [DOI] [PubMed] [Google Scholar]
- 5.Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–52. e2. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He R, Kim HY, Yoon J, Oyoshi MK, MacGinnitie A, Goya S, et al. Exaggerated IL-17 response to epicutaneous sensitization mediates airway inflammation in the absence of IL-4 and IL-13. J Allergy Clin Immunol. 2009;124:761–70. e1. doi: 10.1016/j.jaci.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci U S A. 2007;104:15817–22. doi: 10.1073/pnas.0706942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leyva-Castillo JM, Hener P, Jiang H, Li M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol. 2013;133:154–63. doi: 10.1038/jid.2012.239. [DOI] [PubMed] [Google Scholar]
- 9.Savinko T, Karisola P, Lehtimaki S, Lappetelainen AM, Haapakoski R, Wolff H, et al. ST2 regulates allergic airway inflammation and T-cell polarization in epicutaneously sensitized mice. J Invest Dermatol. 2013;133:2522–9. doi: 10.1038/jid.2013.195. [DOI] [PubMed] [Google Scholar]
- 10.Yu J, Oh MH, Park JU, Myers AC, Dong C, Zhu Z, et al. Epicutaneous exposure to staphylococcal superantigen enterotoxin B enhances allergic lung inflammation via an IL-17A dependent mechanism. PLoS One. 2012;7:e39032. doi: 10.1371/journal.pone.0039032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10:857–63. doi: 10.1038/ni.1767. [DOI] [PubMed] [Google Scholar]
- 12.Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–104. doi: 10.4049/jimmunol.179.12.8098. [DOI] [PubMed] [Google Scholar]
- 13.Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117–24. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem. 2000;275:31335–9. doi: 10.1074/jbc.M005304200. [DOI] [PubMed] [Google Scholar]
- 15.Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci U S A. 2013;110:12768–73. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Besnard AG, Sabat R, Dumoutier L, Renauld JC, Willart M, Lambrecht B, et al. Dual Role of IL-22 in allergic airway inflammation and its cross-talk with IL-17A. Am J Respir Crit Care Med. 2011;183:1153–63. doi: 10.1164/rccm.201008-1383OC. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Yang J, Gao YD, Guo W. Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol. 2010;151:297–307. doi: 10.1159/000250438. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Cao Y, Li K, Wang Z, Zuo P, Xiong W, et al. Increased expression of aryl hydrocarbon receptor and interleukin 22 in patients with allergic asthma. Asian Pac J Allergy Immunol. 2011;29:266–72. [PubMed] [Google Scholar]
- 20.Schnyder B, Lima C, Schnyder-Candrian S. Interleukin-22 is a negative regulator of the allergic response. Cytokine. 2010;50:220–7. doi: 10.1016/j.cyto.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Hirose K, Kawashima S, Niwa Y, Wakashin H, Iwata A, et al. IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. J Allergy Clin Immunol. 2011;128:1067–76. e1–6. doi: 10.1016/j.jaci.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 22.Taube C, Tertilt C, Gyulveszi G, Dehzad N, Kreymborg K, Schneeweiss K, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS One. 2011;6:e21799. doi: 10.1371/journal.pone.0021799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pennino D, Bhavsar PK, Effner R, Avitabile S, Venn P, Quaranta M, et al. IL-22 suppresses IFN-gamma-mediated lung inflammation in asthmatic patients. J Allergy Clin Immunol. 2013;131:562–70. doi: 10.1016/j.jaci.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 24.Chang Y, Al-Alwan L, Risse PA, Halayko AJ, Martin JG, Baglole CJ, et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. 2012;26:5152–60. doi: 10.1096/fj.12-208033. [DOI] [PubMed] [Google Scholar]
- 25.Chang Y, Al-Alwan L, Risse PA, Roussel L, Rousseau S, Halayko AJ, et al. TH17 cytokines induce human airway smooth muscle cell migration. J Allergy Clin Immunol. 2011;127:1046–53. e1–2. doi: 10.1016/j.jaci.2010.12.1117. [DOI] [PubMed] [Google Scholar]
- 26.Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. 1998;101:1614–22. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 28.Jin H, Oyoshi MK, Le Y, Bianchi T, Koduru S, Mathias CB, et al. IL-21R is essential for epicutaneous sensitization and allergic skin inflammation in humans and mice. J Clin Invest. 2009;119:47–60. doi: 10.1172/JCI32310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma W, Bryce PJ, Humbles AA, Laouini D, Yalcindag A, Alenius H, et al. CCR3 is essential for skin eosinophilia and airway hyperresponsiveness in a murine model of allergic skin inflammation. J Clin Invest. 2002;109:621–8. doi: 10.1172/JCI14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon J, Leyva-Castillo JM, Wang G, Galand C, Oyoshi MK, Kumar L, et al. IL-23 induced in keratinocytes by endogenous TLR4 ligands polarizes dendritic cells to drive IL-22 responses to skin immunization. J Exp Med. 2016;213:2147–66. doi: 10.1084/jem.20150376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basu R, O’Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37:1061–75. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ingram JL, Kraft M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J Allergy Clin Immunol. 2012;130:829–42. doi: 10.1016/j.jaci.2012.06.034. quiz 43–4. [DOI] [PubMed] [Google Scholar]
- 33.Pope SM, Brandt EB, Mishra A, Hogan SP, Zimmermann N, Matthaei KI, et al. IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J Allergy Clin Immunol. 2001;108:594–601. doi: 10.1067/mai.2001.118600. [DOI] [PubMed] [Google Scholar]
- 34.Ford JG, Rennick D, Donaldson DD, Venkayya R, McArthur C, Hansell E, et al. Il-13 and IFN-gamma: interactions in lung inflammation. J Immunol. 2001;167:1769–77. doi: 10.4049/jimmunol.167.3.1769. [DOI] [PubMed] [Google Scholar]
- 35.Bernink J, Mjosberg J, Spits H. Th1- and Th2-like subsets of innate lymphoid cells. Immunol Rev. 2013;252:133–8. doi: 10.1111/imr.12034. [DOI] [PubMed] [Google Scholar]
- 36.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 37.Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A, et al. IL-21 induces IL-22 production in CD4+ T cells. Nat Commun. 2014;5:3753. doi: 10.1038/ncomms4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Zaba LC, Cardinale I, Nograles KE, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. 2008;181:7420–7. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207:1293–305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Justa S, Zhou X, Sarkar S. Endogenous IL-22 plays a dual role in arthritis: regulation of established arthritis via IFN-gamma responses. PLoS One. 2014;9:e93279. doi: 10.1371/journal.pone.0093279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakahama T, Kimura A, Nguyen NT, Chinen I, Hanieh H, Nohara K, et al. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc Natl Acad Sci U S A. 2011;108:14222–7. doi: 10.1073/pnas.1111786108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitchell C, Provost K, Niu N, Homer R, Cohn L. IFN-gamma acts on the airway epithelium to inhibit local and systemic pathology in allergic airway disease. J Immunol. 2011;187:3815–20. doi: 10.4049/jimmunol.1100436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brembilla NC, Dufour AM, Alvarez M, Hugues S, Montanari E, Truchetet ME, et al. IL-22 capacitates dermal fibroblast responses to TNF in scleroderma. Ann Rheum Dis. 2016;75:1697–705. doi: 10.1136/annrheumdis-2015-207477. [DOI] [PubMed] [Google Scholar]
- 45.Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–85. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eyerich S, Wagener J, Wenzel V, Scarponi C, Pennino D, Albanesi C, et al. IL-22 and TNF-alpha represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur J Immunol. 2011;41:1894–901. doi: 10.1002/eji.201041197. [DOI] [PubMed] [Google Scholar]
- 47.Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–9. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- 48.Sugita S, Kawazoe Y, Imai A, Kawaguchi T, Horie S, Keino H, et al. Role of IL-22- and TNF-alpha-producing Th22 cells in uveitis patients with Behcet’s disease. J Immunol. 2013;190:5799–808. doi: 10.4049/jimmunol.1202677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao X, Zhu D, Ye J, Li X, Wang Z, Zhang L, et al. The potential protective role of the combination of IL-22 and TNF-alpha against genital tract Chlamydia trachomatis infection. Cytokine. 2015;73:66–73. doi: 10.1016/j.cyto.2015.01.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.