

Abstract
Purpose
Sodium chloride (NaCl) has been proposed as a driving factor in autoimmune diseases through the induction of pathogenic CD4+ T helper cells that produce interleukin-17 (Th17 cells). This study investigated the effects of NaCl on inflammatory arthritis in mice and humans.
Materials and Methods
Collagen-induced arthritis (CIA) mice were fed a normal or high-salt diet ad libitum, and clinical and histologic features of arthritis were evaluated. The proportion of Th17 cells in the spleens of CIA mice fed a normal or high-salt diet was evaluated by flow cytometry, and the expression of IL-17 in joints and intestines was determined by immunohistochemical staining. We also analyzed the effect of NaCl on Th17 differentiation from peripheral blood monocytes of patients with rheumatoid arthritis (RA) and osteoarthritis (OA) and evaluated the contents of sodium and IL-17 in the synovial fluid of RA and OA patients.
Results
NaCl increased murine and human Th17 cell differentiation in a dose-dependent manner. Clinical and histological arthritis was more severe in the high-salt-fed CIA mice, compared to control CIA mice. The proportion of Th17 cells among splenocytes was higher in CIA mice fed a high-salt diet. Expression of synovial and intestinal IL-17 was also higher in high-salt-fed CIA mice. Comparison of synovial fluid between RA patients and OA patients revealed that Na+ and IL-17 were more abundant in RA synovial fluid.
Conclusion
This study suggests that NaCl can aggravate arthritis by affecting Th17 differentiation. Accordingly, limiting salt intake may be helpful for treating inflammatory arthritis, such as RA.
Keywords: Rheumatoid arthritis, salt, sodium chloride, Th17 cells, collagen-induced arthritis
INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by inflammatory polyarthritis that leads joint destruction and deformity. Although the pathogenesis of RA has not yet been elucidated, it is well known that interleukin (IL)-17-producing CD4+ T helper (Th17) cells play an important role in the development and progression of RA.1,2 IL-17 aggravates synovial inflammation and bone destruction by promoting the production of pro-inflammatory cytokines, such as IL-6, IL-1β, tumour necrosis factor-α (TNF-α), and matrix metalloproteinases. Moreover, increased expression of receptor activator of nuclear factor-κB ligand (RANKL) by IL-17 activates osteoclasts and subsequently causes bone loss. Because Th17 cells play a major role in the pathogenesis of inflammatory arthritis, efforts have been made to identify potential contributors that promote Th17 differentiation.
RA is caused by interactions among environmental factors and genetic predisposition. Because genetic susceptibility is difficult to control, efforts to identify environmental factors that promote or inhibit the development of RA are ongoing. Epidemiological studies have suggested associations between RA and environmental triggers, such as smoking and gingivitis, although the pathologic mechanisms are still unclear. Clinical trials have also been conducted on diets to examine whether dietary intake attenuates the proinflammatory response of RA.3,4,5,6 However, there is a limited amount of data on the modifiable risk factors for RA.7
Two recent studies reported that pathogenic Th17 cells can be inducted by sodium chloride (NaCl), suggesting that salt may be a risk factor for autoimmune diseases.8,9 Increased NaCl concentration enhances differentiation into Th17 cells via the p38/mitogen-activated protein kinase signaling pathway, which involves nuclear factor of activated T cells 5 (NFAT5) and serum- and glucocorticoid-inducible kinase 1 (SGK1). NFAT5 and SGK1 were originally identified as regulators that respond to cell volume and osmotic pressure. However, the activation of NFAT5 and SGK1 is also affected by inflammatory cytokines. SGK1 is expressed ubiquitously and is induced by IL-6, which is an important pro-inflammatory cytokine.10 SGK1 increases the expression of IL-23 receptor, and this increase in IL-23 receptor contributes to the stable action of IL-23 on Th17 phenotype.9 NFAT5 is highly expressed in RA synovium, and is induced by TNF-α and IL-β.11,12 In the pathogenesis of RA, NFAT5 is associated with cell survival, migration and angiogenesis.12
Based on recent studies of the proinflammatory role of salt in autoimmune diseases, we investigated the effect of a high-salt diet on inflammatory arthritis in mice with collagen-induced arthritis (CIA). Clinical and histological arthritis was evaluated in CIA mice fed a high-salt diet or normal diet, and the distribution of IL-17-producing cells in the tissues was examined in both groups. In addition, we assessed the effect of NaCl on Th17 differentiation in patients with RA and osteoarthritis (OA).
MATERIALS AND METHODS
Induction of collagen-induced arthritis and feeding of the high-salt diet
DBA/1J mice at seven weeks of age (purchased from Orient-Bio, Seongnam, Republic of Korea) were injected intradermally on the back with 100 µg of CII in complete Freund's adjuvant (Chondrex, Redmond, WA, USA). A second immunization with 100 µg of CII in incomplete Freund's adjuvant (Chondrex) was performed 3 weeks after the first immunization. To evaluate the effect of NaCl on inflammatory arthritis, CIA mice were fed a normal or a high-salt diet ad libitum (n=10 in each group). CIA mice in the high-salt group received tap water that contained 1% NaCl before the first immunization and chow that contained 4% NaCl at the first immunization. Clinical arthritis score was evaluated by two independent examiners twice a week until sacrifice on day 56. Each paw was scored on a graded scale from 0 to 4 as described previously.13 A representative arthritis score was calculated by summing the scores for the four paws. All the experimental procedures were reviewed and approved by the Animal Research Ethics Committee at the Catholic University of Korea (2014-0088-03).
Synovial fluid and serum from patients with RA and OA
Patients who satisfied the relevant classification criteria for RA14,15 or OA16 were recruited from the outpatient clinic in the Department of Rheumatology, Seoul St. Mary's Hospital, Seoul, Korea. An informed consent was obtained from all patients. Synovial fluid was obtained from arthrocentesis of swollen knee joints, which was performed for therapeutic purposes (n=17 for RA, and n=16 for OA). The Na+ concentration and leukocyte counts in the synovial fluid were measured using routine laboratory tests. Synovial IL-17 concentration was determined using a sandwich enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. Peripheral blood mononuclear cells (PBMCs) isolated from the heparinized blood of RA patients (n=3) and OA patients (n=2) were differentiated into Th17 cells under high NaCl conditions. The study was approved by the Institutional Review Board of Seoul St. Mary's Hospital (KC13TISI0240).
Murine Th17 differentiation
CD4+ T cells were isolated from spleens of control mice or CIA mice by positive selection using a magnetic sorter with microbeads (Miltenyi Biotec Inc., Bergisch Gladbach, Germany). Murine CT4+ T cells were cultured in RPMI-1640 (10% fetal bovine serum; Gibco, Carlsbad, CA, USA) and were stimulated with 2 µg/mL of plate-bound anti-CD3 (BD Biosciences, San Jose, CA, USA) for 3 days. Th17 cells were cultured with 5 µg/mL of anti-CD28 (BD Biosciences), 10 µg/mL of anti-IL-4, 10 µg/mL of anti-interferon γ (IFNγ), 50 ng/mL of IL-6, and 1 ng/mL of transforming growth factor β (TGF-β) (all from R&D Systems). NaCl was added at concentrations of 10, 20, 40, 60, or 80 mM. Because the concentration of NaCl in RPMI-1640 media is 104.4 mM, the final NaCl concentrations were 114.4, 124.4, 144.4, 164.4, or 184.4 mM during Th17 differentiation.
Human Th17 differentiation
PBMCs were collected by centrifugation of human blood at 2000 rpm at 20℃ for 30 min with Ficoll-Paque PLUS (GE Healthcare, Little Chalfont, UK). Naïve CD4+ T cells were isolated from PBMCs using a Naïve CD4+ T Cell Isolation Kit II (Miltenyi Biotec Inc.). CD4+ T cells were cultured in 96-well plates coated with 10 µg/mL anti-human CD3 (BD Biosciences). To induce Th17 differentiation, naïve CD4+ T cells were cultured with 1 µg/mL anti-CD28 (BD Biosciences), 25 ng/mL IL-23, 5 ng/mL TGF-β, 12.5 ng/mL IL-1β, and 25 ng/mL IL-6 (all cytokines from R&D Systems) for 5 days.
Flow cytometry
Murine splenocytes were incubated with allophycocyanin (APC)-conjugated anti-CD4 antibodies (BD Biosciences) and then permeabilized using a Foxp3 Staining Buffer Set (eBioscience, San Diego, CA, USA). To identify Th17 cells, murine CD4+ T cells were stained with anti-RORγt antibodies conjugated with phycoerythrin or anti-IL-17 antibodies conjugated with fluorescein isothiocyanate (both from eBioscience). For detection of IL-17, cells were incubated with 5 ng/mL of phorbol- 12-myristate-13-acetate (PMA), 500 ng/mL ionomycin, and 1 µL/mL of GolgiPlug (BD Biosciences) for 4 h prior to staining with anti-IL-17 antibodies.
Human Th17 cells were identified based on co-expression of RORγt, and IL-17. PBMCs were incubated with 50 ng/mL of PMA, 250 ng/mL of ionomycin, and 1 µL/mL of GolgiPlug for 4 h. After incubation with anti-CD4 eFluor 450 (eBioscience), cells were permeabilized using a Foxp3 Transcription Factor Staining Buffer Set (eBioscience). RORγt and IL-17 expression was detected by staining with anti-RORγt antibodies conjugated with phycoerythrin and anti-IL-17A antibodies conjugated with APC (both from eBioscience). After staining with antibodies, the cells were assessed on an LSRFortessa cell analyzer (BD Biosciences). The acquired data were analyzed using FlowJo 7.6.5 software (TreeStar Inc., Ashland, OR, USA).
Histological assessment of synovial tissues
The joint tissues from the hind paws were fixed in 4% paraformaldehyde and decalcified in 10% EDTA bone decalcifier prior to embedding in paraffin. Sections at a 5-µm thickness were stained with Hematoxylin and Eosin, Safranin O Fast Green, and Toluidine Blue. The severity of synovial inflammation and joint destruction was measured by three individual researchers in a blinded manner, as described previously.17 Arthritis scores for inflammation and destruction were determined by the severity of cellular infiltration and hyperplasia and by pannus formation and cartilage erosion, respectively.
Immunofluorescence staining
The tarsal joint, small intestine, and large intestine of the mice were fixed, transferred into 30% sucrose, and then incubated overnight at 4℃. Endogenous peroxidase activity was blocked with 3% H2O2 prepared in phosphate buffered saline (PBS). Nonspecific binding sites were blocked with 10% normal goat serum (Vector Lab, Burlingame, CA, USA) containing 1% PBA (phosphate buffered saline containing 1% bovine serum albumin). The tissue samples were incubated with rabbit polyclonal anti-IL-17 antibodies (Abcam, Cambridge, UK) diluted in 5% normal goat serum containing 1% PBA (1:500) overnight at 4℃. The sections were then incubated with Alexa Fluor 594 goat anti-rabbit IgG (H+L) antibodies (Thermo Fisher Scientific, Waltham, MA, USA) diluted in PBS (1:200) at room temperature for 40 min. Nucleus staining was performed using 4′,6-diamidino-2-phenylindole (DAPI, Roche, Basel, Switzerland). To evaluate the abundance of IL-17+ cells in the tissue samples, IL-17+ cells were manually counted by two independent investigators under a microscope at low power (×100 or ×200 magnification).
Statistical analysis
Experimental data are presented as means and standard errors. Statistical significance was determined using the Mann- Whitney U test. All p values <0.05 were considered significant. All data were analyzed using SAS software (v. 9.1; SAS Institute, Cary, NC, USA) and GraphPad Prism software (v. 5.01; GraphPad, San Diego, CA, USA).
RESULTS
NaCl increases murine Th17 differentiation
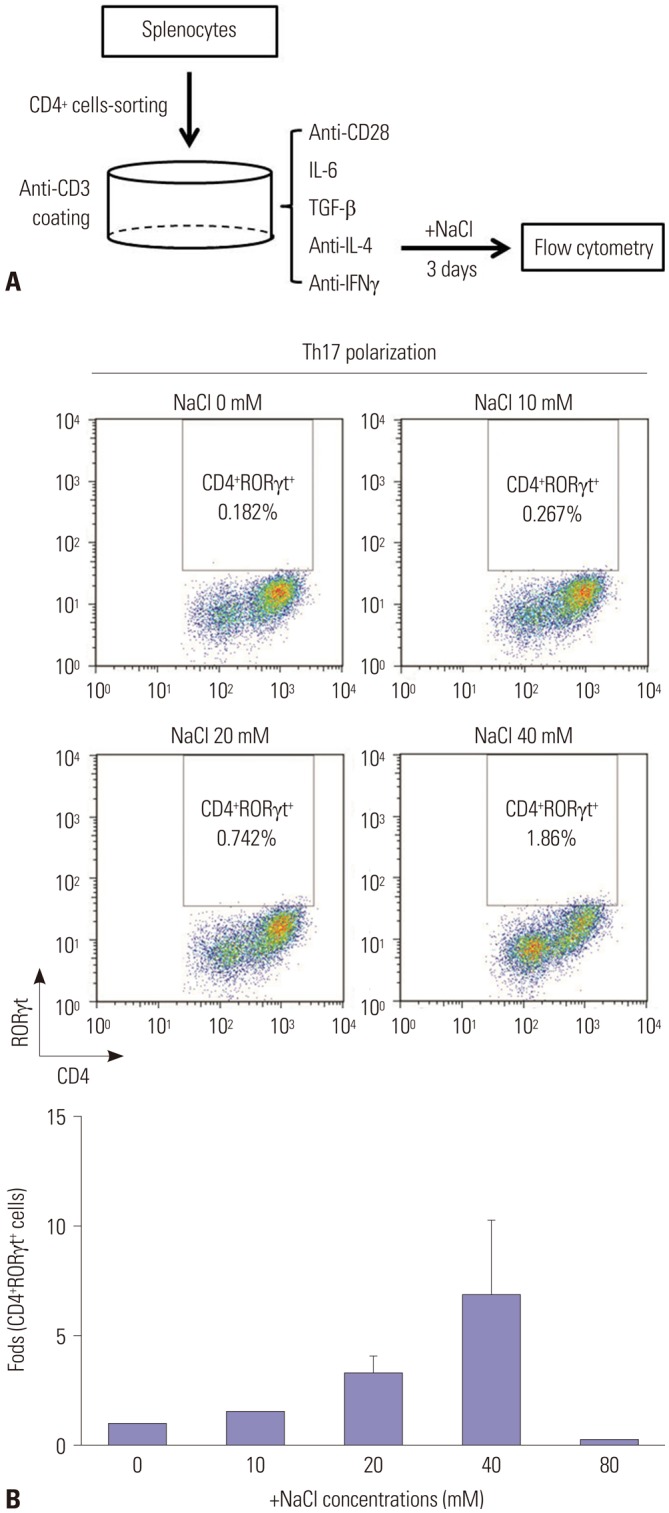
To induce Th17 differentiation, CD4+ T cells isolated from murine splenocytes were stimulated with anti-CD3 and anti-CD28 antibodies, IL-6, and TGF-β and were inhibited with anti-IL-4 and anti-IFNγ antibodies for 3 days (Fig. 1A). Murine CD4+ T cells were cultured under Th17-polarizing condition in the presence of various NaCl concentrations. NaCl up to concentration of 40 mM dose-dependently increased the proportion of CD4+RORγt+ cells (Fig. 1B). However, we observed cellular apoptosis and an abrupt decrease in the number of CD4+RORγt+ cells at a concentration of 80 mM NaCl.
Fig. 1. NaCl induces Th17 differentiation in vitro. (A) A schematic diagram for evaluating the effect of salt on Th17 differentiation. Naïve CD4+ T cells isolated from the spleens of DBA/1J mice were cultured under Th17-polarizing conditions in the absence or presence of NaCl at different concentrations (10, 20, 40, and 80 mM) for 3 days. (B) Representative flow cytometry plots of differentiated Th17 cells at different NaCl concentrations. The displayed numbers are the percentage of CD4+RORγt+-labeled cells. Fold increases were calculated as the percentage of CD4+RORγt+ Th17 cells relative to the percentage in the NaCl-deficient condition. Data are representative of three independent experiments and are expressed as mean±SEM.
High-salt diet aggravates inflammatory arthritis in mice
The high-salt diet was composed of 4% NaCl-containing chow and 1% NaCl-containing water. NaCl-enriched meals were administered to CIA mice since first immunization with CII, and the NaCl challenge was continued for the entire experimental period until day 58 (Fig. 2A). A high-salt diet exacerbates the clinical and histological features of inflammatory arthritis in CIA mice. Arthritis scores were consistently higher in the CIA mice fed a high-salt diet than in the CIA mice fed a normal diet (Fig. 2B). Joint swelling of hind paws was more severe in high-salt-fed CIA mice than in control CIA mice (Fig. 2C). Histological assessment revealed massive infiltration of inflammatory cells, cartilage destruction, and bone erosion in high-salt-fed CIA mice. Evaluation of articular cartilage by Safranin-O and Toluidine Blue staining showed more prominent thinning and erosion of cartilage in high-salt-fed CIA mice (Fig. 2D). The histological score to quantify synovial inflammation and joint destruction was significantly higher in high-salt-fed CIA mice, compared with control CIA mice (Fig. 2E).
Fig. 2. High-salt diet aggravates inflammatory arthritis in CIA mice. (A) Time schedule for salt ingestion in CIA mice. Salt-containing water and chow were started 1 week before and at the time of CII immunization, respectively. (B) Mean arthritis scores in wild-type mice (WT), CIA mice (CIA), and CIA mice fed a high-salt diet (CIA+NaCl). Arthritis of each paw was scored from 0 (no swelling) to 4 (erythema and severe swelling encompassing the ankle and foot). The arthritis score is the sum of the scores for the four paws. Data represent the mean arthritis score±SEM. *p<0.05. (C) Representative photographs of hind paws in control mice and CIA mice fed a high-salt diet. The hind paws of WT, CIA, and CIA+NaCl showed a different degree of swelling at day 58 after primary CII immunization. (D) Histological analysis of tarsal joints in control mice and CIA mice fed a high-salt diet. The tissue sections obtained at 58 days were stained with hematoxylin and eosin (H&E), Safranin O, and toluidine blue. The magnifications are indicated in the right bottom corner. (E) The mean histological score of the tarsal joints in each group. Inflammation and joint destruction were scored on a scale of 0–3 by three independent examiners. The data represent the mean histological score±SEM. ‡p<0.001.

NaCl-rich environment primes Th17 polarization in mice
Splenocytes were extracted from both high-salt-fed CIA and control CIA mice. Ex vivo analysis of splenocytes showed a higher proportion of Th17 cells expressing CD4 and RORγt in high-salt-fed CIA mice, compared with control CIA mice (fold change 1.15 vs. 1.0, p<0.05) (Fig. 3A). To evaluate the priming effect of a high-salt diet on Th17 differentiation, CD4+ T cells isolated from CIA mice in each group were cultured under Th17-polarizing condition. CD4+ T cells from high-salt-fed CIA mice were more likely to express IL-17A than those from control CIA mice (fold change 1.8 vs. 1.0, p<0.05) (Fig. 3B).
Fig. 3. NaCl-rich condition primes Th17 polarization in mice with CIA. (A) Flow cytometry analysis of naïve CD4+ T cells in spleens isolated from control CIA mice (CIA) and high-salt-fed CIA mice (CIA+NaCl). Representative plots of Th17 population among naïve CD4+ T cells in both groups are presented, and fold increases were calculated as the percentage of CD4+RORγt+ Th17 cells relative to the percentage in control CIA mice. (B) Flow cytometry analysis of naïve CD4+ T cells isolated from CIA and CIA+NaCl mice cultured under Th17-polarizing conditions. Splenocytes were isolated from CIA mice fed a normal or a high-salt diet to evaluate the priming effect of NaCl on Th17 differentiation. Purified naïve CD4+ T cells were cultured under Th17 differentiating condition for 3 days. Representative plots of differentiated CD4+IL-17A+Th17 cells from naïve CD4+ T cells isolated from both groups were presented, and fold increases were calculated as the percentage of CD4+IL-17A+ Th17 cells relative to the percentage in the control CIA group. Data are representative of three independent experiments and are expressed as mean±SEM. *p<0.05.

High-salt diet increases IL-17-producing cells in synovial and intestinal tissue in CIA mice
Because high NaCl concentrations enhanced Th17 differentiation in vitro, we performed an in vivo investigation to examine the effects of a high-salt diet on Th17 cells. Immunofluorescent staining with IL-17A showed abundant Th17 cells in the tarsal joints of CIA mice. IL-17-producing cells were identified more frequently in CIA mice fed a high-salt diet than control CIA mice (Fig. 4A). We examined the presence of IL-17+ cells in the intestinal mucosa and submucosa by immunohistochemical and immunofluorescence staining because we were also interested in whether dietary NaCl would directly affect the immune cells in the intestinal tissue. IL-17+ cells were absent in the intestine of wild-type mice, whereas CIA mice fed a normal or high-salt diet both expressed IL-17, mainly in the small intestine (Fig. 4B). IL-17-producing cells were more abundant in the intestinal submucosa of high-saltfed CIA mice, compared with control CIA mice. Although IL-17+ cell infiltration into the large intestine of CIA mice was not prominent, cellular aggregates stained with anti-IL-17 antibodies were found in the colonic submucosa of high-salt-fed CIA mice (Fig. 4C). Manual cell counting of IL-17+ cells in the synovium and gastrointestinal tract showed a significantly higher amount of IL-17+ cells in high-salt-fed CIA mice than in control CIA mice (Fig. 4D).
Fig. 4. Salt intake increases IL-17 expression in the synovium and gut of CIA mice. (A–C) Expression of IL-17A in the joint synovium, small intestine, and large intestine in WT mice and CIA mice fed a normal diet (CIA) or a high-salt diet (CIA+NaCl). Immunohistochemical staining with anti-IL-17A antibodies was performed in tissue sections of joint (A), small intestine (B), and large intestine (C) obtained from experimental animals on day 58. Representative images in each group are shown, and the area with cellular deposition was enlarged. The magnification is presented in the right bottom corner, and scale bars indicate 100 µm. (D) Quantification of IL-17+ cells in the joint and small intestine and IL-17+ spots in the large intestine observed in a low power field (LPF) (×100 or ×200 as presented in the graphs). The number of IL-17+ cells or spots was determined by three independent examiners. The data are expressed as mean±SEM. *p<0.05.

NaCl-rich environment in synovial fluid from RA patients is associated with Th17 polarization
Consistent with the results from animal experiments, human T cells isolated from patients with RA or OA showed enhanced Th17 differentiation at high NaCl concentrations (Fig. 5A). NaCl increased the expression of RORγt and IL-17, and had the greatest effect at a concentration of 40 mM. At a concentration of 60 mM NaCl, T cells exhibited apoptosis, and the expression level of RORγt and IL-17 was reduced. To confirm the effect of NaCl on Th17 differentiation of human T cells, we recruited additional patients with RA (n=3) and OA (n=2) and collected CD4+ T cells from the peripheral blood of those patients. CD4+ T cells of individual patients were cultured under Th17 polarizing condition in the absence or the presence of 40 mM NaCl. Flow cytometry showed that the expression of RORγt and IL-17 consistently increased after NaCl treatment (Fig. 5B). The CD4+ T cell response in high NaCl concentrations showed similar patterns in RA and OA patients: 1.5- to 5.0-fold increases in Th17 differentiation.
Fig. 5. Synovial fluid from RA patients contained higher levels of sodium and IL-17 compared with those from OA patients. (A) Changes in Th17 differentiation from human CD4+ T cells after treatment with various doses of NaCl. Naïve CD4+ T cells from peripheral blood were cultured under Th17-polarizing conditions with or without NaCl at different concentrations (10, 20, 40, and 60 mM) for 3 days. RORγt+IL-17A+ Th17 cells were detected by flow cytometry. Fold differences at the different concentrations were expressed relative to the NaCl-deficient condition. (B) The effect of NaCl on Th17 differentiation in human cells obtained from RA and OA patients. Representative flow cytometry plots for Th17 differentiation at NaCl 0 mM and 40 mM are shown in patients with RA (n=3) and OA (n=2). Fold increases in differentiated Th17 cells were expressed relative to the NaCl-deficient condition. The graph represents the data from individual patients. Data represent the mean of three independent experiments±SEM. *p<0.05. (C) Characteristics of synovial fluid obtained from RA patients and OA patients. Leukocyte count and sodium concentration in synovial fluid were measured by routine laboratory methods. IL-17 level in the synovial fluid was determined by ELISA. Horizontal bars for analysis of synovial fluid indicate the median of all values in a group. †p<0.01, ‡p<0.001. RA, rheumatoid arthritis; OA, osteoarthritis; WBC, white blood cell.

Because RA is characterized by inflammatory synovitis, white blood cell counts in synovial fluid from RA patients was markedly higher than in that from OA patients (median value of 16000 cells/mm3 vs. 95.5 cells/mm3, p<0.0001) (Fig. 5C). The concentration of IL-17 was also significantly higher in RA synovial fluid than in OA synovial fluid (p=0.008). Interestingly, the Na+ concentration was much higher in RA synovial fluid than in OA synovial fluid (median value of 135 mEq/L vs. 68 mEq/L, p<0.0001).
DISCUSSION
The present study showed that NaCl aggravates inflammatory arthritis in CIA mice through Th17 cell induction. Mice fed a high-salt diet showed a higher proportion of Th17 cells in the spleen and joints, compared to control CIA mice, and CD4+ T cells from high-salt-fed CIA mice were likely to differentiate into Th17 cells. High-salt diet also increased the proportion of IL-17-producing cells in the synovium and intestine of CIA mice. The synovial fluid of RA patients contained larger amounts of sodium and IL-17, suggesting an association between NaCl and Th17 cells.
Consistent with a recent report,8 we found an in vitro effect of NaCl on Th17 differentiation in a dose-dependent manner. The most potent NaCl concentration for Th17 development was 40 mM in the culture media. Interestingly, several previous studies also showed that the NaCl concentration of 40 mM had the greatest effect in modulating immune cells and that the experimental cell lines lost their viability at concentrations above 40 mM of NaCl.8,18,19 Although the tolerable concentration of NaCl in the culture condition is not clear, the threshold might be less than 60 mM of NaCl. There is one study showing the effect of high-salt diet on sodium concentration in vivo. Machnik, et al.18 reported that the skin Na+ contents in rats fed a high-salt diet and a low-salt diet were about 200 mM and 150 mM, respectively. There was no significant difference in serum Na+ level between two groups. This finding suggests that NaCl accumulates in the interstitium without a change in serum Na+ concentration. After high-salt intake, the regulatory mechanisms maintained the interstitial sodium concentration within 50 mM-difference to keep the physiological state of the body.
We also investigated the distribution of IL-17-producing cells in the gastrointestinal tract based on the assumption that the NaCl-rich dietary content would induce Th17 differentiation of the mucosal immune system. Similar to the in vitro experiments, the high-salt diet promoted intestinal IL-17 production in CIA mice. IL-17 is primarily secreted from Th17 cells, although there are a few cell populations that express IL-17 in the intestine.20 Therefore, the presence of IL-17 in the intestinal tract of CIA mice fed a high-salt diet may indicate that the salt component promotes Th17 differentiation in the intestines.
The gut is the largest organ in the immune system and is responsible for the maintenance of homeostasis between host immunity and diverse antigenic stimuli, including food and commensal microorganisms. Previous research has shown that the gut microbiota is associated with Th17 differentiation in the intestinal tissues.21,22 In this study, we did not examine changes in intestinal microflora after salt ingestion. It is unclear whether high NaCl concentration in the intestinal lumen directly induces Th17 polarization or whether salt-containing foods alter the intestinal flora and then influence T cell differentiation. Although the precise mechanism for intestinal Th17 differentiation after salt intake is difficult to define, the induction of intestinal Th17 cells by NaCl could be responsible for exacerbation of the autoimmune arthritis. There is evidence that suggests a significant association between intestinal Th17 cells and extraintestinal autoimmunity. Germ-free animals with fewer intestinal Th17 cells are resistant to the development of autoimmune arthritis and experimental autoimmune encephalomyelitis.23,24
Interestingly, the synovial Na+ concentration was significantly higher in patients with RA than in those with OA. The mean Na+ concentration in RA synovial fluid was 135 mEq/L, similar to the in vitro NaCl concentration when 40 mM NaCl was added to the culture media (104.4 mM). The highest Na+ concentration in RA synovial fluid was 157 mEq/L. In consideration of the in vitro result that T cells exhibited apoptosis at concentrations above 60 mM NaCl (≥164.4 mM in the culture media), the synovial Na+ concentration above 160 mEq/L may be a difficult condition for lymphocyte proliferation and inflammatory activation. To the best of our knowledge, this is the first report showing the Na+ concentration in RA synovial fluid. Further investigation would be required to elucidate the association between synovial Na+ concentration and inflammatory responses.
In this study, the inflammatory response of CD4+ T cells to high NaCl concentrations was not significantly different between RA patients and OA patients. However, hypernatremic synovial fluid from RA patients can provide a milieu favoring Th17 polarization. In addition to Th17 differentiation, hypertonicity is essential for lymphocyte proliferation and maturation. Physiologically, lymphoid tissue is hyperosmolar compared with serum.25 Hypertonic saline promotes T cell proliferation26 by activating the signaling pathway in T cells27 and reversing prostaglandin E-mediated T cell suppression.28 Thus, hypernatremic hypertonicity in RA synovial fluid may increase Th17 differentiation and lymphocyte proliferation, leading to exacerbation of the inflammatory environment.
Similar to previous studies, we found that a NaCl-rich environment enhances the expression of RORγt, which is a critical transcription factor for Th17 differentiation.1 Induction of RORγt in high-salt condition was consistently represented by in vitro and in vivo experiments. High-salt diet exacerbated the severity of experimental autoimmune encephalomyelitis and experimental colitis, and expression of RORγt was more prominent in mice fed a high-salt diet, compared to control mice.8,29 Microarray analysis also showed the increased expression of RORC in T cells under high-salt conditions.8,9,30 Considering the loss of sodium-induced increase of Th17 signatures, including RORγt, in SGK1-deficient T cells,9,31 SGK1 can be a link between salt and RORγt. However, it is difficult to conclude that the worsening of inflammatory arthritis by sodium intake is entirely due to Th17 polarization. Earlier studies have suggested a significant association between immunity and hypertonicity. Autoimmunity can be regulated by inhibition of angiotensin-converting enzyme,32 which is activated by hypertonicity due to salt ingestion.33 This inhibitory effect is mediated by inducing regulatory T cells and by modulating Th1 and Th17 responses.32 Hypertonic saline also stimulates innate immunity in vivo by up-regulating the expression of Toll-like receptor signaling.34,35 In addition, salt intake is associated with an increase in inflammatory cytokines. In animal models of gastric carcinogenesis, a high-salt diet increases the production of pro-inflammatory cytokines, including IL-1, IL-6, and TNF-α.36,37 In healthy adolescents, average sodium intake correlates with serum levels of TNF-α, independently of adipose tissues.38 Therefore, in addition to Th17 differentiation, various inflammatory responses may contribute to exacerbation of arthritis.
In clinical studies, an association between high-salt diet and autoimmune disease remains unclear. A recent study suggests that a high salt diet may be associated with increased disease activity in patients with multiple sclerosis.39 In Chinese patients with systemic lupus erythematosus, excessive salt intake was associated with treatment failure despite intensive glucocorticoid treatment.40 Additional epidemiologic studies and intervention studies that limit salt intake are required to clarify the causal relationship between salt intake and autoimmune diseases.
In conclusion, NaCl can aggravate arthritis by stimulating Th17 differentiation. To our knowledge, this is the first report to demonstrate a proarthritic effect of salt in an animal model. A low-salt dietary approach to alleviate the symptoms and signs of inflammatory arthritis has not yet been studied. Although diet plays an auxiliary role in primary treatment based on immunosuppressants, dietary modifications can be applied as a treatment approach for RA because there are no side effects and costs are minimal.
ACKNOWLEDGEMENTS
This work was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A092258); the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT, & Future Planning (2013R1A1A1076125); and a grant from “The Center for Evaluating Next-Generation Stem Cell-based Therapeutics,” supported by the National Institute of Food and Drug Safety Evaluation, part of the Ministry of Drug and Food Safety (14172 CENST 974).
Footnotes
This study was presented at the 34th Korean College of Rheumatology Annual Scientific Meeting (2014. 5. 16).
The authors have no potential conflicts of interest to disclose.
References
- 1.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 2.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 3.Proudman SM, James MJ, Spargo LD, Metcalf RG, Sullivan TR, Rischmueller M, et al. Fish oil in recent onset rheumatoid arthritis: a randomised, double-blind controlled trial within algorithm-based drug use. Ann Rheum Dis. 2015;74:89–95. doi: 10.1136/annrheumdis-2013-204145. [DOI] [PubMed] [Google Scholar]
- 4.Park Y, Lee A, Shim SC, Lee JH, Choe JY, Ahn H, et al. Effect of n-3 polyunsaturated fatty acid supplementation in patients with rheumatoid arthritis: a 16-week randomized, double-blind, placebo-controlled, parallel-design multicenter study in Korea. J Nutr Biochem. 2013;24:1367–1372. doi: 10.1016/j.jnutbio.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Salesi M, Farajzadegan Z. Efficacy of vitamin D in patients with active rheumatoid arthritis receiving methotrexate therapy. Rheumatol Int. 2012;32:2129–2133. doi: 10.1007/s00296-011-1944-5. [DOI] [PubMed] [Google Scholar]
- 6.McKellar G, Morrison E, McEntegart A, Hampson R, Tierney A, Mackle G, et al. A pilot study of a Mediterranean-type diet intervention in female patients with rheumatoid arthritis living in areas of social deprivation in Glasgow. Ann Rheum Dis. 2007;66:1239–1243. doi: 10.1136/ard.2006.065151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahiri M, Morgan C, Symmons DP, Bruce IN. Modifiable risk factors for RA: prevention, better than cure? Rheumatology (Oxford) 2012;51:499–512. doi: 10.1093/rheumatology/ker299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng F, Yamagiwa Y, Taffetani S, Han J, Patel T. IL-6 activates serum and glucocorticoid kinase via p38alpha mitogen-activated protein kinase pathway. Am J Physiol Cell Physiol. 2005;289:C971–C981. doi: 10.1152/ajpcell.00081.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masuda K, Masuda R, Neidhart M, Simmen BR, Michel BA, Müller-Ladner U, et al. Molecular profile of synovial fibroblasts in rheumatoid arthritis depends on the stage of proliferation. Arthritis Res. 2002;4:R8. doi: 10.1186/ar427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon HJ, You S, Yoo SA, Kim NH, Kwon HM, Yoon CH, et al. NFAT5 is a critical regulator of inflammatory arthritis. Arthritis Rheum. 2011;63:1843–1852. doi: 10.1002/art.30229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2:1269–1275. doi: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 14.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 15.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–1588. doi: 10.1136/ard.2010.138461. [DOI] [PubMed] [Google Scholar]
- 16.Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29:1039–1049. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- 17.Hückel M, Schurigt U, Wagner AH, Stöckigt R, Petrow PK, Thoss K, et al. Attenuation of murine antigen-induced arthritis by treatment with a decoy oligodeoxynucleotide inhibiting signal transducer and activator of transcription-1 (STAT-1) Arthritis Res Ther. 2006;8:R17. doi: 10.1186/ar1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 19.Müller S, Quast T, Schröder A, Hucke S, Klotz L, Jantsch J, et al. Salt-dependent chemotaxis of macrophages. PLoS One. 2013;8:e73439. doi: 10.1371/journal.pone.0073439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrido-Mesa N, Algieri F, Rodríguez Nogales A, Gálvez J. Functional plasticity of Th17 cells: implications in gastrointestinal tract function. Int Rev Immunol. 2013;32:493–510. doi: 10.3109/08830185.2013.834899. [DOI] [PubMed] [Google Scholar]
- 21.Kamada N, Núñez G. Role of the gut microbiota in the development and function of lymphoid cells. J Immunol. 2013;190:1389–1395. doi: 10.4049/jimmunol.1203100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sommer F, Bäckhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 23.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Go WY, Liu X, Roti MA, Liu F, Ho SN. NFAT5/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proc Natl Acad Sci U S A. 2004;101:10673–10678. doi: 10.1073/pnas.0403139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Junger WG, Liu FC, Loomis WH, Hoyt DB. Hypertonic saline enhances cellular immune function. Circ Shock. 1994;42:190–196. [PubMed] [Google Scholar]
- 27.Junger WG, Hoyt DB, Hamreus M, Liu FC, Herdon-Remelius C, Junger W, et al. Hypertonic saline activates protein tyrosine kinases and mitogen-activated protein kinase p38 in T-cells. J Trauma. 1997;42:437–443. doi: 10.1097/00005373-199703000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Coimbra R, Junger WG, Liu FC, Loomis WH, Hoyt DB. Hypertonic/ hyperoncotic fluids reverse prostaglandin E2 (PGE2)-induced T-cell suppression. Shock. 1995;4:45–49. doi: 10.1097/00024382-199507000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Monteleone I, Marafini I, Dinallo V, Di Fusco D, Troncone E, Zorzi F, et al. Sodium chloride-enriched diet enhanced inflammatory cytokine production and exacerbated experimental colitis in mice. J Crohns Colitis. 2017;11:237–245. doi: 10.1093/ecco-jcc/jjw139. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. 2015;125:4212–4222. doi: 10.1172/JCI81151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu C, Chen Z, Xiao S, Thalhamer T, Madi A, Han T, et al. SGK1 governs the reciprocal development of Th17 and regulatory T cells. Cell Rep. 2018;22:653–665. doi: 10.1016/j.celrep.2017.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Platten M, Youssef S, Hur EM, Ho PP, Han MH, Lanz TV, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U S A. 2009;106:14948–14953. doi: 10.1073/pnas.0903958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crestani S, Gasparotto Júnior A, Marques MC, Sullivan JC, Webb RC, da Silva-Santos JE. Enhanced angiotensin-converting enzyme activity and systemic reactivity to angiotensin II in normotensive rats exposed to a high-sodium diet. Vascul Pharmacol. 2014;60:67–74. doi: 10.1016/j.vph.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen LW, Huang HL, Lee IT, Hsu CM, Lu PJ. Hypertonic saline enhances host defense to bacterial challenge by augmenting Toll-like receptors. Crit Care Med. 2006;34:1758–1768. doi: 10.1097/01.CCM.0000218810.66485.01. [DOI] [PubMed] [Google Scholar]
- 35.Chen LW, Su MT, Chen PH, Liu WC, Hsu CM. Hypertonic saline enhances host defense and reduces apoptosis in burn mice by increasing toll-like receptors. Shock. 2011;35:59–66. doi: 10.1097/SHK.0b013e3181e86f10. [DOI] [PubMed] [Google Scholar]
- 36.Gaddy JA, Radin JN, Loh JT, Zhang F, Washington MK, Peek RM, Jr, et al. High dietary salt intake exacerbates Helicobacter pyloriinduced gastric carcinogenesis. Infect Immun. 2013;81:2258–2267. doi: 10.1128/IAI.01271-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung WK, Wu KC, Wong CY, Cheng AS, Ching AK, Chan AW, et al. Transgenic cyclooxygenase-2 expression and high salt enhanced susceptibility to chemical-induced gastric cancer development in mice. Carcinogenesis. 2008;29:1648–1654. doi: 10.1093/carcin/bgn156. [DOI] [PubMed] [Google Scholar]
- 38.Zhu H, Pollock NK, Kotak I, Gutin B, Wang X, Bhagatwala J, et al. Dietary sodium, adiposity, and inflammation in healthy adolescents. Pediatrics. 2014;133:e635–e642. doi: 10.1542/peds.2013-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farez MF, Fiol MP, Gaitán MI, Quintana FJ, Correale J. Sodium intake is associated with increased disease activity in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2015;86:26–31. doi: 10.1136/jnnp-2014-307928. [DOI] [PubMed] [Google Scholar]
- 40.Zou YF, Xu JH, Tao JH, Xu SQ, Liu S, Chen SY, et al. Impact of environmental factors on efficacy of glucocorticoids in Chinese population with systemic lupus erythematosus. Inflammation. 2013;36:1424–1430. doi: 10.1007/s10753-013-9682-3. [DOI] [PubMed] [Google Scholar]