

Abstract
Here, we have exploited the heightened concentration of MMP-9 to induce surface-conversional properties of nanogels with the aim of tumor-specific enhanced cellular uptake. A modular polymeric nanogel platform was designed and synthesized for facile formulation and validation of MMP-9 mediated de-PEGylation and generation of polyamine-type surface characteristics through peptide N-termini. Nanogels containing MMP-9 cleavable motifs and different poly(ethylene glycol) corona lengths (350 g/mol and 750 g/mol) were prepared and enzymatic surface conversional properties were validated by MALDI characterization of cleaved byproducts, fluorescamine assay amine quantification, and zeta potential. The nanogel with a shorter PEG length, mPEG350-NG, exhibited superior surface conversion in response to extracellular concentrations of MMP-9 compared to the longer PEG length, mPEG750-NG. Confocal microscopy images of HeLa cells incubated with both fluorescein-labeled nanogels and DiI-encapsulated nanogels demonstrated greater uptake following MMP-9 “activation” for mPEG350-NG compared to its non-treated “passive” mPEG350-NG parent, demonstrating the versatility of such systems to achieve stimuli-responsive uptake in response to cancer-relevant proteases.
Keywords: matrix metalloproteinase 9 (MMP-9), redox-responsive nanogel, active cellular uptake, stimuli-responsive, smart nanoparticle
Graphical Abstract
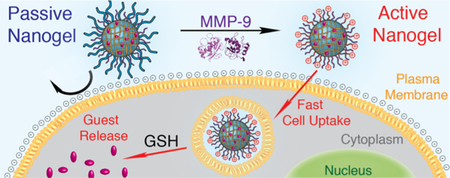
INTRODUCTION
A nanoscopic drug delivery platform that can maintain long blood circulation times without compromising selective and rapid target tissue internalization is still a big challenge in obtaining desired therapeutic profiles for clinical applications. The passive mechanism by which nanoscale systems diffuse and accumulate in tumor tissue is a result of fenestrated hypervasculature from angiogenesis and a deficient lymphatic drainage.1 Albeit somewhat controversial,2–4 this enhanced permeation and retention (EPR) effect drives the development of particles with long circulation characteristics. In the development of passive nanocarriers, poly(ethylene glycol) polymers (PEG) have been shown to achieve non-fouling characteristics for nanoparticles by reducing non-specific interactions and protein adsorption to their surfaces.5,6 The grafting, conjugation, or adsorption of PEG on the surface of macromolecules has been shown to reduce the extent of phagocytic elimination by the reticular endothelial system (RES) and achieve extended blood circulation half-lives.7–13
Passive nanogels inherently lack the ability to distinguish healthy tissues from diseased ones, and their stealth surface properties can inhibit cellular interaction resulting in reduced endosomolytic activity.14, 15 Active targeting aims to address these points through enhancement or specificity of cellular internalization. Active uptake is often pursued through one of two ways: i) targeting of unique cell-surface receptors or ii) enhanced cellular interaction with cationic materials. The former aims to improve specificity of receptor-mediated endocytosis through cancer cell surface receptors and antigens.16–20 Various systems have been explored for active targeting including the incorporation of cell-penetrating peptides,21,22 targeting peptides,23, 24 monoclonal antibodies,25–27 or small molecules.28–30 Concerns for in vivo surface decoration of such ligands is that they can limit their stealth surface, resulting in RES recognition and clearance,31 and their tumor penetration.32 Further, internalization through receptor mediated endocytosis can lead to therapeutic decomposition in the lysosome.33 The later cationic strategy is due to enhanced charge-charge interaction with the anionic phospholipid bilayer of cell membranes, resulting in adsorptive endocytosis at a higher rate than neutral or anionic formulations.34, 35 Many polycationic nanocarriers have been developed with the aim of improved cellular internalization of therapeutic contents, particularly with anionic nucleotide-based or nucleic acid therapeutics such as siRNA, mRNA, and DNA.36 Systemic administration of cationic nanomaterials however, result in indiscriminate cellular internalizations, nonspecific adsorption of serum proteins, and short circulation half-lives due to rapid RES clearance.37–39
This dilemma can be addressed by developing passive “smart” nanocarriers that become “active” in response to specific stimulus proximal to the disease site. Many externally applied stimuli have been explored (e.g. light, temperature),40 however tumor-specific endogenous stimuli can exploit inherent physiological biochemical differences in cancerous tissues.41 Such stimuli that are intrinsic to the extracellular tumor microenvironment include decreased pH and enzyme dysregulation. Nanocarriers with pH-sensitivity have been well explored because lactate production from anerobic glycolysis under hypoxic conditions in tumors causes a pH decrease (6.2–6.9).42 Nanoscopic systems that can generate positive charge or shed surface PEG in response to mildly acidic conditions have been demonstrated to facilitate phagocytosis.42–49 One disadvantage of pH dependent strategies is that inflammatory regions also have mildly acidic hypoxic environments. Further, hypoxia can be variable in incidence and severity, in some cases irregularly affecting regions distant from the nutritive vasculature where oxygen diffusion is limited and the tumor outgrows its blood supply.50, 51
The dysregulation of enzymes however, is common to many tumor types and can be highly specific to the local microenvironment.52, 53 The overexpression of matrix metalloproteinases (MMPs), zinc-dependent proteases that degrade the extracellular matrix, has been observed in various cancers and are associated with tumor invasiveness, metastasis, and angiogenesis.54 Furthermore, these tumor-associated proteases are expressed at the angiogenic invasive anterior of tumors where nanoparticles may more easily access and accumulate, supporting their candidacy as proximal activators of nanoparticles. Variable supramolecular designs in the protease-induced activation of nanoparticles have been pursued.55–65 Extracellularly overexpressed MMPs have been used to improve cell uptake through exposure of cationic polymeric block55, 56, cationic cell penetrating peptide57, 58, or through revealing underlying ligands59–61. Other approaches have used MMPs to decrease particle hydrodynamic radius62, or induce nanoparticle morphological changes63 and disassembly64, 65 to result in extracellular therapeutic release.
Here, we investigate an abridged protease activation strategy that explores the generation of peptide N-termini on the particle surface to enhance cellular uptake. This strategy can be achieved by simply installing a protease-cleavable substrate to a particle at its C-terminus that is shielded by PEG at its N-terminus (Scheme 1). Therefore, peptide hydrolysis by a relevant enzyme, here MMP-9, removes the PEG shield to reveal a polyamine-type surface from N-termini. The resultant “active” nanogel is expected to internalize more rapidly than its PEGylated parent “passive” nanogel due to charge conversion, reduced steric stabilization, and resultant enhanced membrane interactions. Such a design is versatile and flexible in that the peptide substrate can dictate the sensitivity and specificity of MMP cleavage, or be adapted to the known MMP types in a target tissue of interest. Further, the formulation is modular in nature such that the enzyme-responsive feature is post-conjugated to the pre-formed nanogel surface, allowing for facile substrate amendment and versatility in molecular design. This study focuses on proof-of-concept formulation and characterization of an MMP-9-responsive nanogel delivery platform that offers potential for tumor-specific delivery of chemotherapeutics.
Scheme 1.
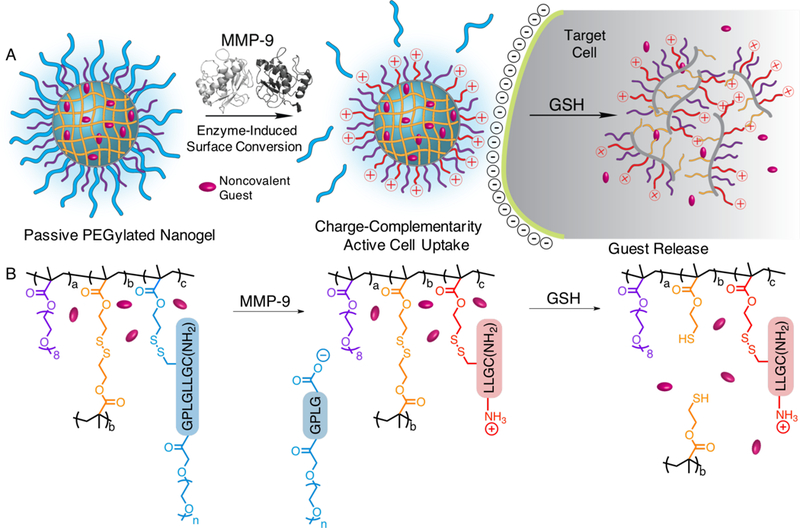
A) Schematic representation of MMP-9 responsive nanogel and resulting activated cell uptake and GSH release, and B) structural representation of polymer nanogels and stimuli-responsiveness.
EXPERIMENTAL
Materials and Methods.
Polyethylene glycol monomethyl ether (average MW 350 and 750 g/mol), 4-aminophenylmercuric acetate (APMA), methacryloyl chloride, 2-mercaptoethanol, poly(ethyleneglycol) monomethyl ether methacylate (OEGMA; MW 500 g/mol), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid, chromium trioxide, sulfuric acid, trifluoroacetic acid, triisopropylsilane, 1,2-ethanedithiol, DL-dithiothreitol (DTT), 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI), fluorescein isothiocyanate isomer 1 (FITC), cystamine dihydrochloride, trimethylamine, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), piperidine solution and fluorescamine were purchased from Sigma Aldrich and used without further purification. AIBN (2,2’-azobis(2-methylpropionitrile) was purchased from Sigma Aldrich and purified by recrystallization. Amino acids and Rink Amide AM resin (100–200 mesh) for solid phase peptide synthesis were purchased from EMD Millipore. ProMMP-9 was purchased from BioLegend Inc. Fluorogenic peptide substrate Mca-PLGL-Dpa-AR-NH2 was purchased from R&D Systems, Inc. Solvents, Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), trypsin-EDTA (0.5%, no phenol red) were purchased from Fisher Scientific. Penicillin-streptomycin-glutamine (100x) solution, PBS 7.4, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Thermo Fisher Scientific. Pyridyl disulfide ethyl methacrylate (PDSMA) was prepared as previously reported.66 Polymers were synthesized using RAFT polymerization and purified by dialysis using a membrane with 3500 MWCO. 1H-NMR spectra were recorded on a 400 MHz Bruker NMR spectrometer using the residual proton resonance of the deuterated solvent as the internal standard. Polymer molecular weights were estimated by gel permeation chromatography (GPC, Waters) using THF as eluent at a flow rate of 1 mL/min by a refractive index detector compared to poly(methyl methacrylate) (PMMA) standard. UV-visible absorption spectra were recorded on a PerkinElmer Lambda 35 UV/Vis Spectrophotometer. High performance liquid chromatography was conducted using a Shimadzu Prominence Modular HPLC.
Formulation of Nanogels:
Deionized water was added to the polymer (10 mg/mL) and made into a particle suspension using repeated chill and sonicate cycles until the solution appeared dissolved. Particles of about 20 nm were achieved by chemically crosslinking this equilibrium assembly of the polymer at 25 °C in 25 mM Na2SO4 using a calculated amount of DTT for 1 hour as reducing agent as previously reported.67, 68 Crosslinking was determined to be 25 mole percent PDS by calculating the amount of byproduct, 2-pyridinethione, using its molar extinction coefficient (8.08 × 103 M−1 cm−1 at 343 nm)69 using UV-Vis spectroscopy. Dynamic light scattering (DLS) experiments to obtain particle size were performed by using a digital correlator and goniometer with a light source operating at 514 nm using a Malvern Nanozetasizer-ZS. Final nanogel size measurements were obtained at 25 °C at a correlation time of 30 seconds. Dust was removed by filtering the solution through 0.45 μm polycarbonate filter. For each sample, 3 readings were recorded averaging 10 runs for the same sample.
PEG-Peptide conjugation to nanogels:
The substrates mPEG(350)-GPLG↓LLGC(NH2) (53.3 mg, 54.6 µmol), or mPEG(750)-GPLG↓LLGC(NH2) (72.7 mg, 54.6 µmol) were each dissolved in methanol (100 µL) and added separately to 5 mL of aqueous crosslinked nanogel (Bare NG) solutions (10 mg/mL, 109.3 µmol PDS) and stirred open to the atmosphere for 12 hours to allow the methanol to evaporate. The peptide LLGC(NH2) (3.97 mg, 9.84 µmol) was added to 1 mL of aqueous crosslinked nanogel solution (10 mg/mL, 21.9 µmol PDS) and stirred for 12 hours. The conjugation extent for each mPEG350-NG, mPEG350-NG, and LLGC-NG, was quantified by UV-Vis spectroscopy by calculating the amount of generated byproduct 2-pyridinethione. Unconjugated peptide and 2-pyridinethione byproduct was removed by dialysis against DI water in a 100 KDa MWCO cellulose dialysis membrane for 48 hours. DLS of peptide-conjugated nanogels were obtained in phosphate buffer pH 7.4 at a concentration of 2.5 mg/mL.
PEG-Peptide Cleavage by LC-MS:
MMP-9 cleavable substrates mPEG(350)-GPLG↓LLGC(NH2) (1 mg, 0.1 mmol) and mPEG(750)-GPLG↓LLGC(NH2) (1.3 mg, 0.1 mmol) were incubated with activated MMP-9 (1 µg, 10.9 nM) in TTC buffer 7.4 (1 mL) at 37 °C in 5% CO2 with shaking for 24 hours. The mixture was analyzed by LCMS. Chromatographic separation was conducted by a Thermo Scientific (Waltham, MA) Ultimate 3000 HPLC with a Thermo Acclaim PepMap RSLC C18 column (300 µm x 15 cm, 2 µm particle size). MMP-9 incubated substrates were eluted using a gradient of acetonitrile containing 0.1% formic acid that increased from 5% to 50% for 45 min at a flow rate of 4 µl/min. Substrate products were measured immediately by mass spectrometry. Mass analysis was carried out on a Bruker HCTultra (Billerica, MA) ion trap mass spectrometer equipped with an electrospray ionization source. Typically, the electrospray needle voltage was kept at ~4 kV, and the capillary temperature was set at 220 °C.
Nanogel Activation with MMP-9:
Nanogels (2.5 mg/mL polymer) decorated with MMP-9 cleavable substrates (3 mM) in TTC buffer pH 7.4 was incubated with activated MMP-9 at final enzyme concentrations of 0.54, 5.4, and 54 nM (0.05, 0.5, and 5 µg/mL MMP-9, respectively) at 37 °C in 5% CO2 with shaking. At determined time points the enzyme activity was quenched with 1% (v/v) acetic acid to pH 3.5, then cleaved peptide was isolated from the mixture for MALDI characterization using a regenerated cellulose Amicon Ultra centrifugal filter with 3KDa MWCO. Activated nanogel was purified by dialysis against DI water in a 100 KDa MWCO cellulose dialysis membrane for 24 hours.
MALDI Characterization:
Matrix solutions were prepared dissolving α-cyano-4-hydroxycinnamic acid (CHCA) (10 mg/mL) in a solution containing ACN (50 μL), H2O (47.5 μL) and TFA (2.5 μL). The matrix solution was then mixed with the centrifuge filtered nanogel solution that had been incubated with MMP-9. A Bruker Autoflex III time-of-flight mass spectrometer was used for the MALDI-MS analysis of all samples. Acquisition of all mass spectra was done in reflectron mode with an accelerating voltage of 19 kV. Each spectrum is the average of 500 laser shots at 50% laser power.
Fluorescamine Assay:
The percentage of cleavage on the particle was determined by quantifying the N-terminal amines using fluorescamine Assay. Formulated product of cleavage nanogel (LLGC-NG) was used to generate a concentration dependent fluorescence calibration curve. Various concentrations of LLGC-NG (7.4 µg/mL-0.74 mg/mL) were prepared in PBS buffer pH 7.4. MMP-9 incubated and purified nanogels mPEG350-NG and mPEG750-NG were diluted in PBS buffer pH 7.4 (to 0.74 mg/mL) so that their maximum expected amine content would be within the LLGC-NG calibration curve. Non-activated mPEG350-NG and mPEG350-NG (0.74 mg/mL) were prepared as negative controls. In a 96 well (flat-bottomed) plate PBS buffer pH 7.4 (150 µL) and sample solutions (20 uL) were added to each well. Then, fluorescamine solution in DMSO (12 uL, 0.68 mg/mL) was added to each well. The fluorescence was obtained using a Molecular Devices Spectramax M5 plate reader (excitation: 390 nm; emission 465 nm). Average fluorescence values and deviation were obtained from replicate readings (n=3). Concentration of LLGC-NG versus fluorescence was used to generate an amine calibration curve, and the fluorescence of known test samples were used to determine amine content.
Cell Culture:
Human cervical carcinoma (HeLa) cell line was grown in DMEM supplemented with 10% (FBS), 1% L-glutamine, and 1% antibiotic-antimycotic (comprised of 100 units/mL penicillin and 100 µg/mL streptomycin). All cells were grown at 5% CO2 and 37 °C. Digestion of cells for culture was done according to HeLa and 293A protocols from ATCC.
Confocal Microscopy Cellular Uptake:
HeLa cells were seeded at 30–50% confluency (~10,400 cells/cm2) in 4-chamber 35 mm glass bottom dishes and incubated overnight at 37 °C overnight in 5% CO2 before performing uptake. Culture media was removed and cells washed with PBS one time before adding new culture media containing fluorescein-labeled nanogels diluted to 0.1 mg/mL in DMEM (10×, diluted to 1× with PBS). Samples were incubated for 2 hours. Nuclear staining (NucBlue, 80 µL per mL media) was added in the final 30–60 min of incubation. Media was removed from cells and washed 2–4 times with PBS, then live cell imaging buffer was added for confocal imaging. Assessment of fluorescein-conjugated nanogel intracellular uptake was recorded using 488 nm laser and nuclear stain was detecting using a 405 nm wavelength laser. Assessment of DiI-loaded nanogel intracellular uptake was recorded using 540 nm laser and nuclear stain was detecting using a 405 nm wavelength laser. Confocal microscopy was performed on a Nikon Yokogawa spinning disk confocal microscope equipped with 40× or 100× oil objectives and an Andor EMCCD camera.
Flow Cytometry:
HeLa cells were seeded at 100,000 cells/mL in a glass bottom dish and maintained at 37 °C overnight in 5% CO2 before performing uptake. Culture media was removed and cells washed with PBS one time before adding new culture media containing fluorescein-labeled nanogels diluted to 0.05 mg/mL in DMEM (10×, diluted to 1× with PBS). Samples were incubated for 2 hours, then washed with PBS, trypsinized and collected by centrifugation. The cells were re-suspended in 200 µL PBS buffer and stored at 4 °C. A minimum of 100,000 cells were analyzed for each sample using a BD LSRFortessa.
Release Studies:
The release of encapsulated model guest DiI was monitored by fluorescence spectroscopy using a Molecular Devices Spectramax M5 plate reader. DiI (0.35 mg) was dissolved in acetone (100 µL) then added to a 350 µL solution of 10 mg/mL aggregate of polymer p(OEGMA-co-PDSMA) and stirred was for 8 hours at room temperature, open to the atmosphere to allow for acetone to evaporate. Aggregates were crosslinked then post-functionalized with substrates as previously described. Nanogels were purified by dialysis using 100 KDa MWCO membrane and any non-encapsulated DiI was removed by filtration through a 0.45 µm filter (encapsulation efficiency 50%). APMA activated MMP-9 was purified by centrifuge dialysis (10 KDa MWCO) with TNC buffer. In a 96 well (flat-bottomed) plate TNC buffer pH 7.4 (90 µL) containing relevant stimuli (GSH, ProMMP-9, or MMP-9) and concentrations were added to each well. Then, nanogel solutions (10 µL, 17 µg/mL) of either Bare NG, mPEG350-NG, and mPEG750-NG were added to each well. The fluorescence was obtained for each well (excitation: 520 nm; emission 565 nm), and the average fluorescence values and deviation were obtained from replicate readings (n=3) over 48 hours.
RESULTS AND DISCUSSION
Enzyme-Responsive Substrate Synthesis and Proteolysis Validation
Key elements of the suggested nanocarrier are the polymeric nanogel responsible for guest incorporation and intracellular guest release, along with the post-modifiable enzyme responsive substrate that will facilitate cellular internalization. The matrix metalloproteinase-9 responsive peptide-poly(ethylene glycol) monomethyl ether (Mw 350 and 750 g/mol) substrates, mPEG(350)-GPLG↓LLGC(NH2) and mPEG(750)-GPLG↓LLGC(NH2), were inspired from collagenous peptide sequences that were shown to be hydrolyzed by type IV collagenases, or MMP-9 between the Gly-Leu bond.70, 71 Of those investigated, the sequence AcPLG↓LLG-OC2H5 was found to be the best substrate for MMP-9, with a similar turnover number (kcat) to denatured collagen and a specific activity of 80 µmol/mg/h.70 A minimum of six amino acid residues and peptide charge neutrality was found to exhibit more rapid gelatinolytic activity than peptides comprised of less amino acid residues or charged residues. From N-to C-terminus, the substrate for this study was thus designed to contain PEG, an additional glycine residue, the validated PLG↓LLG sequence, and an amide-neutralized cysteine. Two PEG lengths were explored with the expectation that a shorter length may have greater enzyme accessibility, however if proteolysis is sufficient, longer lengths may engender greater difference in cellular interaction from its “passive” to “active” state. The MMP-9 cleavable PEG-peptide conjugates mPEG(350)-GPLG↓LLGC(NH2) and mPEG(750)-GPLG↓LLGC(NH2) were made using solid phase peptide synthesis, purified by high performance liquid chromatography, and characterized using electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (Figure S6–S8).
Prior to nanogel conjugation, proteolytic susceptibility of substrates by MMP-9 was evaluated. Synthesized substrates mPEG(350)-GPLG↓LLGC(NH2) and mPEG(750)-GPLG↓LLGC(NH2) were incubated with MMP-9 and characterized by liquid chromatography mass spectrometry (LCMS). For both substrates, chromatographic separation of unreacted substrate from digested products was achieved and products were identified (Figure S11). For mPEG(350)-GPLG↓LLGC(NH2) the unreacted substrate eluted at 36–39 min and has a distribution of peaks separated by 44 Da, the mass of one ethylene glycol unit, that is centered at m/z 1020 ([M+H]+, M=1019 g/mol) (Figure 1). The expected product LLGC(NH2) eluted at 15–16 minutes with m/z 404 ([M+H]+, M=403 g/mol), and mPEG(350)-GPLG eluted at 25–31 minutes with a distribution of peaks separated by 44 Da centered at m/z 635 ([M+H]+, M=634 g/mol). The 44 Da mass difference between each peak indicates the repeating ethylene glycol units, which further validate the structure of the cleaved product. Some disulfide LLGC dimer was also observed, which likely occurred during peptide incubation. Likewise, for mPEG(750)-GPLG↓LLGC(NH2), the unreacted substrate eluted at 39–40 min with m/z 1328 ([M+H]+, M=1327 g/mol). The expected product LLGC(NH2) eluted at 15–17 minutes with m/z 404, and product mPEG(750)-GPLG eluted at 25–35 minutes with m/z 987 ([M+H]+, M=986 g/mol). The absence of other products supports the specificity of MMP-9 cleavage between the Gly-Leu bond.
Figure 1.

LC-MS ESI characterization of A) mPEG(350)-GPLGLLGC(NH2) 1) starting material and MMP-9 cleavage products 2) mPEG(350)-GPLG and 3) LLGC(NH2). ESI characterization of B) mPEG(750)-GPLGLLGC(NH2) 1) starting material and MMP-9 cleavage products 2) mPEG(750)-GPLG and 3) LLGC(NH2).
Matrix Metalloproteinase-Responsive Nanogel Synthesis and Characterization
To afford nanoparticles with MMP-9 responsive characteristics, we utilized the previously reported self-crosslinking disulfide-based nanogel scaffold.67 The polymeric nanogels are achieved using amphiphilic random copolymers that self-assemble into nanoscale aggregates with disulfide functionality that allows for both controlled guest entrapment and stimuli-responsive release. Redox-responsive guest release from nanoassemblies are of significant interest due to their sensitivity to concentrations of glutathione (GSH) in blood plasma (10 μM) compared to the cytosol (10 mM).67, 72, 73 While stable encapsulation is observed at extracellular GSH concentrations, these nanogels release their therapeutic cargo following cellular internalization and exposure to cytosolic concentrations of GSH. In this work, we expanded this platform to include a second stimuli-responsive feature at the particle surface, which can modify its cell interaction and facilitate cellular uptake.
The p(OEGMA-co-PDSMA) random copolymer used for nanogel formation was synthesized by a reversible addition–fragmentation chain transfer (RAFT) polymerization of monomers poly(ethylene glycol) methyl ether methacrylate (OEGMA) and pyridyldisulfide ethyl methacrylate (PDSMA). After polymerization, the polymer was purified by dialysis against dichloromethane to remove unreacted monomers, then characterized by NMR and GPC (Figure S1, S2). We used the characteristic resonances of the 2-pyridylthio moieties of PDSMA (δHa = 8.45, δHb = 7.66, and δHc = 7.10 ppm) and methoxy moieties of OEGMA (δHd = 3.37 ppm) to calculate the relative ratios of the monomer incorporated in the polymer as 7:3 PDS to OEG. The GPC obtained number-average molecular weight (Mn) and dispersity (Đ = Mw/Mn) of the copolymer was 31.9 kDa and 1.4, respectively.
The nanogel was then formulated by dispersing the p(OEGMA-co-PDSMA) random copolymer in an aqueous phase in the presence of Na2SO4 (25 mM), a Hofmeister salt that we have previously demonstrated to finely control particle size due to the lower critical solution temperature (LCST) and relative hydration or dehydration behavior of PEG.74 The ~20 nm aggregates were then locked in conformation through intra-aggregate disulfide crosslink formation (Figure 2). The covalent crosslinking is achieved via the addition of a sub-stoichiometric amount of reducing agent, DL-dithiothreitol (DTT), which generates the corresponding quantity of free thiols on PDS moieties within the aggregate interior, which then react with remaining activated PDS moieties in the polymer chain. For these nanogels, we used 12.5 mol% DTT, relative to PDS in the aggregates, to result in 25% crosslinked nanogels. The crosslinking for resultant Bare-NG was quantified by the reaction byproduct 2-pyridinethione using its characteristic absorption peak at 343 nm (Figure S12).
Figure 2.
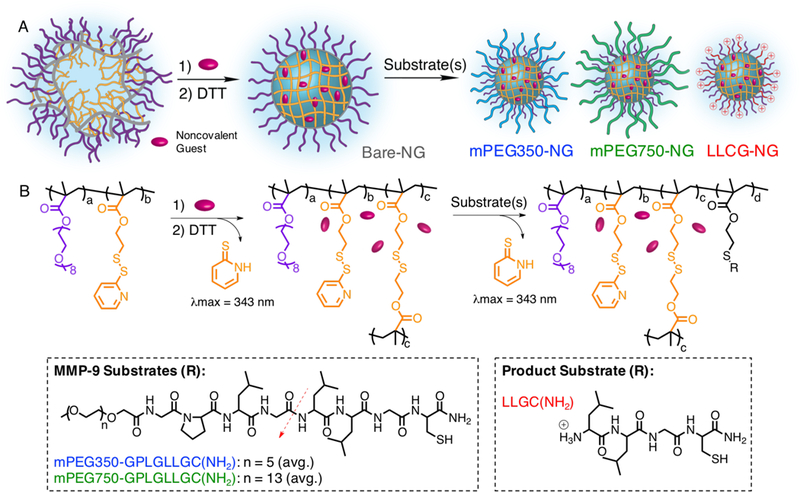
A) Schematic representation of polymeric nanogel formation, and B) structural representation of polymer nanogels synthesis with structure of substrates.
Next, nanogels were decorated with either mPEG(350)-GPLG↓LLGC(NH2), mPEG(750)-GPLG↓LLGC(NH2), or positive control LLGC(NH2) through a thiol-disulfide exchange reaction between the cysteine thiols of the peptide and PDS groups of the nanogel (Figure 2). Peptides dissolved in methanol were added to the aqueous crosslinked nanogels and post modification was quantified by absorbance using the reaction byproduct 2-pyridinethione (Figure S12). The resulting nanogels denoted mPEG350-NG, mPEG750-NG, and LLGC-NG contained ~30% peptide functionalization respective to PDS, for each mPEG(350)-GPLG↓LLGC(NH2), mPEG(750)-GPLG↓LLGC(NH2), and LLGC(NH2), respectively (Table 1). Conjugation efficiency for LLGC(NH2) was 73%, while the conjugation efficiencies for mPEG(350)-GPLG↓LLGC(NH2) and mPEG(750)-GPLG↓LLGC(NH2) were 64% and 56%, respectively. Nanogels were purified by dialysis and MALDI characterization of nanogel confirmed removal of free peptide (Figure 3). Post-modification of the peptides to the nanogels did not appreciably change the particle sizes, or cause any observable precipitation or aggregation as characterized by DLS (Figure 3). Due to the known lower critical solution temperature (LCST) characteristics of PEG, the temperature responsiveness of these nanogels were characterized by DLS. Size transitions were observed well above 37 °C, suggesting nanoparticle integrity at biologically-relevant conditions (Figure S13).
Table 1.
Characterization of substrate-functionalized nanogels.
| Sample | Crosslink (% PDS) | Substrate Identity | Substrate (% PDS) | Size (nm) |
|---|---|---|---|---|
| Bare NG | 25 | -- | N/A | 16 |
| mPEG350-NG | 25 | mPEG350-GPLGLLGC(NH2) | 32 | 18 |
| mPEG750-NG | 25 | mPEG750-GPLGLLGC(NH2) | 28 | 18 |
| LLGC-NG | 25 | LLGC(NH2) | 33 | 16 |
Figure 3.
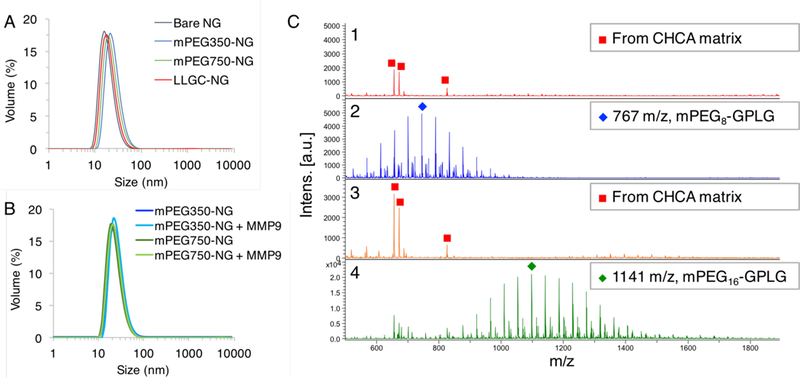
DLS sizes by volume of A) Bare NG and substrate-modified nanogels and B) MMP-9 responsive NGs mPEG350-NG and mPEG750-NG before and after MMP (54 nM) treatment. C) MALDI characterization of substrate containing nanogels 1) mPEG350-NG, 2) mPEG350-NG pretreated with MMP-9 (54 nM), 3) mPEG750-NG, 4) mPE7350-NG pretreated with MMP-9 (54 nM).
Surface-Conversional Validation and Quantification
The proteolytic capability of MMP-9 to cleave peptide from the nanogel surface was confirmed using MALDI-MS. On a substrate decorated particle surface, we expect MMP-9 proteolysis to generate free mPEG-GPLG in solution while LLGC(NH2) remains conjugated to the particle by the C-terminal cysteine. The nanogels were incubated with active MMP-9 at 37 °C for 24 hours in PBS pH 7.4. Following this incubation, any free cleaved peptide was isolated for analysis using 3 kDa centrifuge filtration. The centrifuged solutions were then mixed with matrix solutions containing CHCA and characterized by MALDI-MS. As a control, nanogels not incubated with MMP-9 were characterized to demonstrate that masses corresponding to free substrates were not observed prior to proteolysis, while their cleavage products were evident following MMP-9 treatment (Figure 3). MALDI analysis of mPEG350-NG incubated with MMP-9 generated a distribution of peaks separated by 44 Da that is centered at m/z 767, which correlates to [M+H]+ of product mPEG8-GPLG. MMP-9 mediated mPEG750-NG proteolysis also exhibited a distribution of peaks separated by 44 Da with a distribution maxima of m/z 1,141, which correlates to [M+Na]+ of product mPEG16-GPLG. For both nanogels, these proteolysis products have a slightly higher PEG distribution shift than observed on free substrate. This slight change can be attributed to either preferential conjugation of longer PEG species to the particle due to enhanced aqueous solubility, or higher MALDI ionization efficiency of the longer PEG species. Additionally, the lower m/z range indicates no other side reactions are occurring with the particles (Figure S14).
We then investigated the generation free amines on the nanogel surfaces by MMP-9 proteolysis and aimed to quantify the reaction using a fluorescamine assay. In this assay, non-fluorescent fluorescamine (4-phenylspiro[furan-2(3H), 1’-phthalan]-3,3’-dione) reacts directly with primary amines to form a fluorescent product (390 nm excitation, 465 nm emission).75 Previous reports have observed near complete reaction between lysines and N-terminus of peptides with fluorescamine.75–77 Because fluorescence is proportional to the quantity of free amines present, this assay is often used to detect and quantify proteins with high sensitivity. The fluorescamine assay has been frequently used to monitor supramolecular processes as well, such as characterization of poly(L-lysine)-DNA complexes that are post-modified a multivalent polymer.78 Likewise, we expect the N-termini generated by MMP-9 proteolysis on a nanogel surface to be quantifiable using the fluorescamine assay.
Here we used the positive control nanogel, modified with 33% LLGC(NH2), to demonstrate the reaction between fluorescamine and a modeled MMP-9 created N-terminus. Moreover, the fluorescence generated is proportional to the concentration of N-termini present from the functionalized peptide. Because the fluorescamine reaction is highly efficient, the fluorescence from this sample can be assumed to represent a nanogel with 100% cleavage. A concentration dependent calibration curve was generated using 3 molar excess of fluorescamine in DMSO with various concentrations of LLGC-NG (mg/mL) PBS buffer pH 7.4 and monitoring the fluorescence intensity (Figure 4). The fluorescamine assay confirmed that the fluorescence intensity was linearly proportional to the concentration of LLGC-NG, and appropriate for quantification of N-termini in solution.
Figure 4.
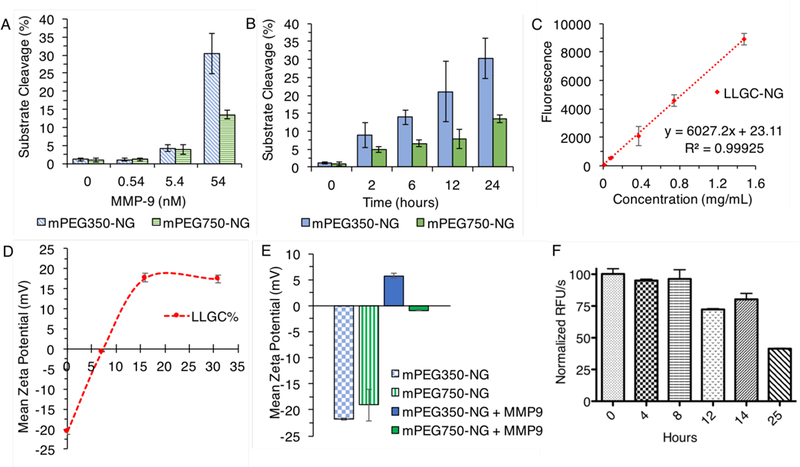
Fluorescamine assay results for MMP-9 substrate cleave percent by A) concentration and B) time for nanogels (avg. ± std, n=3 repeat), and C) fluorescence calibration of LLGC-NG (avg. ± std, n=4 measurements). D) Mean zeta potential of nanogels conjugated with increasing LLGC% (avg. ± std, n=3 measurements). E) Mean zeta potential of mPEG350-NG and mPEG750-NG before and after MMP-9 treatment (avg. ± std, n=3 measurements). F) Confirmation of MMP-9 activity over the 24-hour experiment, expressed as normalized RFU/s with 8 hours ProMMP-9 activation with APMA as time 0 of nanogel incubation (avg. ± std, n=2 repeat).
Nanogels were incubated with low nanomolar concentrations of MMP-9 (0.54–5.4 nM) to evaluate responsivity to levels reported in plasma (50–500 ng/mL) for various cancers.79–81 There have been significant efforts to determine plasma or serum concentrations of MMP-9 to discern cancer progression and prognosis significance. Local tumor concentrations are expectedly higher, and cell culture of different cancer lines report MMP-9 excretions in the high nanomolar to low micromolar range.64, 82 This is heterogeneous across different tumor types83, so we assigned an MMP-9 concentration of 54 nM (5 µg/mL) as a conservative estimate to evaluate responsivity at tumor-relevant extracellular matrix concentrations. To understand concentration affects, mPEG350-NG and mPEG750-NG nanogels were incubated with 0.54, 5.4, and 54 nM MMP-9, and at determined time points quenched, purified by 100 kDa dialysis, then reacted with fluorescamine for quantification. Obtained fluorescence intensities were calculated against the linear fit of LLGC-NG to determine percentage of N-termini, i.e. cleavage, generated by MMP-9.
Nanogels incubated without MMP-9 showed negligible fluorescence, confirming the expected absence of amines on the starting material (Figure 4). At 0.54 nM MMP-9, negligible (~1%) cleavage was observed for both mPEG350-NG and mPEG750-NG after a 24-hour incubation, which were similar in value to those obtained for 0 nM MMP-9. At moderate concentrations (5.4 nM), as expected low cleavage values were still obtained, with ~4% for both nanogels. Under extracellular relevant concentrations of MMP-9 (54 nM), 30±6% of the substrate was cleaved from the mPEG350-NG nanogel surface after 24 hours. For mPEG750-NG, however, approximately half the amount of cleavage was obtained, with 14±1% cleaved at 24 hours. To evaluate this result, time dependent cleavage of 54 nM MMP-9 was obtained. This analysis showed that much higher cleavage occurred with mPEG350-NG compared to mPEG750-NG, however was progressive over the full 24 hours for both systems. By 24 hours, the activity of MMP-9 had severely depleted. This might explain the linear cleavage observed for both nanogels, as while substrate accessibility may improve with time, and thus proteolysis increase, the enzyme activity is congruently decreasing with time. This hypothesis is consistent with observed differences between nanogels of different PEG lengths, as it is expected that the longer PEG lengths can cause more steric hindrance and restrict the extent of protease-mediated substrate cleavage.
We were also interested in evaluating this surface conversional property by examining nanocarrier zeta potential change. Substrate-decorated nanogels mPEG350-NG and mPEG750-NG gave zeta potentials of −22 mV and −19 mV, respectively (Figure 4). Although the negative zeta potential of anticipated neutral PEG surface seems unexpected, several reports indicate PEG-decorated nanostructures do have negative zeta potentials.43, 84, 85 The cause of this, however, is thus far unclear. Nanogels were then decorated with increasing amounts of amine terminus containing peptide LLGC(NH2) to test the veracity of the amine terminus to inflict a positive zeta potential on these nanogels. Indeed, conjugation of LLGC(NH2) developed a positive charge, with zeta potential becoming more neutral at −1 mV for 7% conjugation and +18 mV for 16% conjugation, which did not become more positive with higher peptide conjugation. This guided our rationale that even small amounts of this substrate could impact the surface charge of these nanogels. The zeta potentials of mPEG350-NG did indeed become positive after incubation with MMP-9, with a mean zeta potential of +6 mV after 24 hours of treatment. This suggests a significant shift in surface character from the initial value of −22 mV. Measurably positive surface charge was not achieved with mPEG750-NG, with maximum value of −1 mV after 24 hour MMP-9 incubation, a consistent finding with the lower cleavage extent observed by fluorescamine assay on this particle. This suggests that the 14% cleavage and the resultant amine character is insufficient to convert the surface charge from a negatively PEGylated particle to positive. These results further support that accessibility of the protease to the substrate limited the surface-conversion capability.
Guest Encapsulation and Stimuli-Responsive Release
For this nanocarrier, guest encapsulation under both stimuli GSH and MMP-9 was evaluated. To test the structural stability and guest release in response to stimuli, nanogels encapsulated with a model hydrophobic guest 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) were prepared for mPEG350-NG, mPEG750-NG, and Bare NG to serve as a MMP-9 non-responsive control (Figure S15). Due to the hydrophobicity of DiI, we expect any dye release from the nanogels interior to result in precipitation and resulting fluorescence decrease. Nanogels were prepared in TNC buffer, pH 7.4, and the fluorescence of DiI was monitored over time in response to GSH, MMP-9, or ProMMP-9 to serve as non-catalytic protein control (Figure S16) at various concentrations.
The trends in guest release were similar for Bare-NG, mPEG350-NG, and mPEG750-NG (Figure 5). In all cases, the nanogel control, which is exposed to no stimulus, exhibited minor release over 48 hours, with 4±3, 4±5, and 8±4% for Bare-NG, mPEG350-NG, and mPEG750-NG, respectively (Table S1). This release is likely due to loss of any adsorbed or poorly encapsulated DiI from buffer dilution. In response to extracellular concentrations of reduced GSH (10 µM), MMP-9 (54 nM), and GSH (10 µM) with MMP-9 (54 nM) release percentages were also minor, ranging from 6–12% at 48 hours (Figure 5). Inactive proenzyme ProMMP-9 and serum concentrations of MMP-9 also exhibited little release, irrespective of concentration (Figure S16). Aside from enzymatic guest stability, these results also suggest the disulfides in the particle are not very responsive to these low extracellular reducing concentrations, which is especially important because the enzyme-responsive substrate is conjugated to the particle through thiol-disulfide exchange. However, with cytosolic redox conditions, as expected, the disulfide-based nanogel crosslinks were responsive to reduced glutathione. Over a 48-hour period, we observed 51±3, 42±1, and 48±1% release of encapsulated DiI in the presence of 10 mM GSH at pH 7.4, for Bare-NG, mPEG350-NG, and mPEG750-NG, respectively (Figure 5). Further, DLS characterization of the nanogels was executed under severe dilution conditions (1000-fold) and we observed maintained particle integrity of MMP-9 (54 nM) treated nanogels, while those treated with 10 mM GSH lost its aggregation capability. These results suggest that these particles have unique and environment-specific dual-stimuli responsivity of both surface activation by MMP-9 and guest release by GSH.
Figure 5.
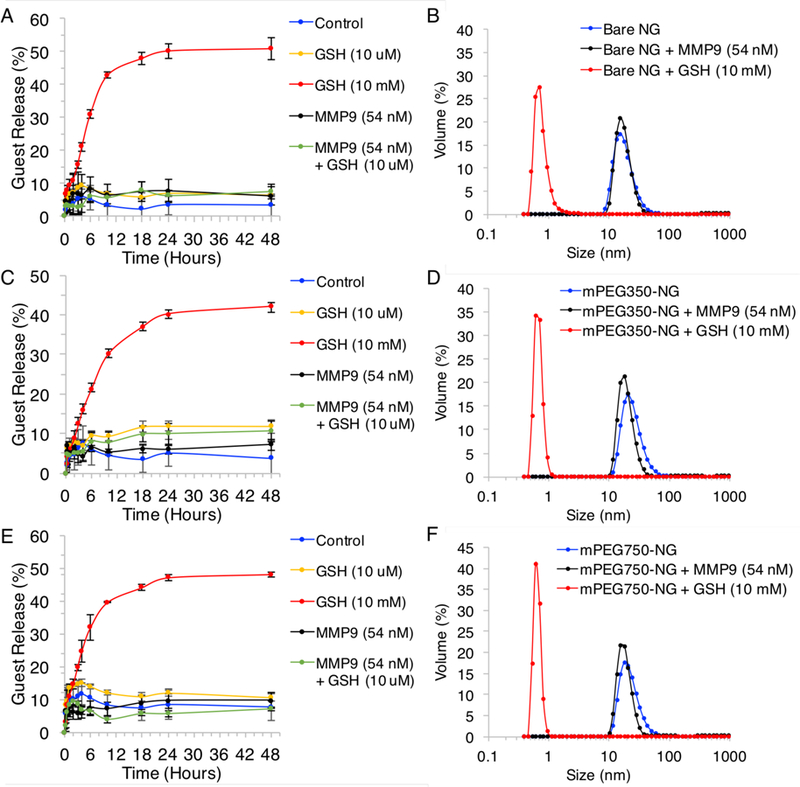
Guest release based on normalized fluorescence for nanogels A) Bare over 48 hours in response to GSH (10 mM and 10 µM), MMP-9 (54 nM), and MMP-9 (54 nM) + GSH (10 µm) compared to no stimulus control, and B) corresponding DLS of nanogel sizes upon exposure to MMP-9 (54 nM) and GSH (10 mM) and serial dilution. C) Guest release and D) DLS plots of mPEG350-NG upon exposure to stimuli, and E) guest release and F) DLS plots of mPEG750-NG upon exposure to stimuli.
Cellular Uptake Analysis
Confocal microscopy was used to evaluate whether the nanogels were taken up by human HeLa cells at an enhanced rate following MMP-9 treatment. The HeLa cell line has very weak expression of MMP-9,86 making it an appropriate choice for non-activated nanogel controls. Cytotoxicity of these nanogels was evaluated for the HeLa cell line using the MTT cytotoxicity assay, demonstrating >70% viability even up to 1 mg/mL (Figure S17). To monitor the in vitro cellular uptake, nanogels were first covalently modified with 5% thiol-modified fluorescein label. Negative “passive” controls mPEG350-NG(F) (fluorescein) and mPEG750-NG(F), amine-decorated positive control LLGC-NG(F), and test samples MMP-treated mPEG350-NG(F) Active and mPEG750-NG(F) Active were all prepared with fluorescein tag and were the same size by DLS (Figure S18).
Nanogels were incubated with HeLa cells for 2 hours before confocal imaging, and NG uptake was observed using laser excitation at 488 nm. Consistent with expectations, from microscopy images “passive” PEG-decorated mPEG350-NG(F) showed much lower cellular uptake than amine-decorated LLGC-NG(F) (Figure 6). This result supported our anticipation that the poly-amine character from N-termini could be sufficient in modifying the adsorption of nanogels on cellular membranes. Indeed, once treated with MMP-9, mPEG350-NG(F) Active exhibited uptake much more reminiscent of positive control LLGC-NG(F). Therefore, the extent of enzymatic proteolysis achieved on this nanogel was sufficient to achieve a surface-conversion and alter its cellular uptake. The results are likely a combination of PEG reduction of endosomolytic activity and improved charge-charge adsorption of polyvalent amines on the phospholipid bilayer of cells. Microscopy z-stack images at 100× objective supported these results (Figure S19). Nanogel mPEG750-NG(F), however, did not exhibit an observable difference in uptake following MMP-9 treatment, which shows that the extent of cleavage that occurs on these particles is crucial in obtaining this improved cell-membrane interaction. These results were supported quantitatively by flow cytometry, which showed highest fluorescence intensities of FITC in HeLa cells with positive control LLGC-NG, and a greater fluorescence intensity for mPEG350-NG Active compared to mPEG350-NG (Figure 6). Consistent with microscopy observations, mPEG750-NG did not show uptake enhancement following MMP activation, with similar median FITC fluorescence intensities. It is possible that either higher protease concentrations or longer incubation times could improve the extent of MMP-9 surface charge turnover for longer PEG nanogel, but these results suggest a fine balance is required between substrate PEG shielding and enzyme accessibility to achieve the “passive” to “active” conversion.
Figure 6.
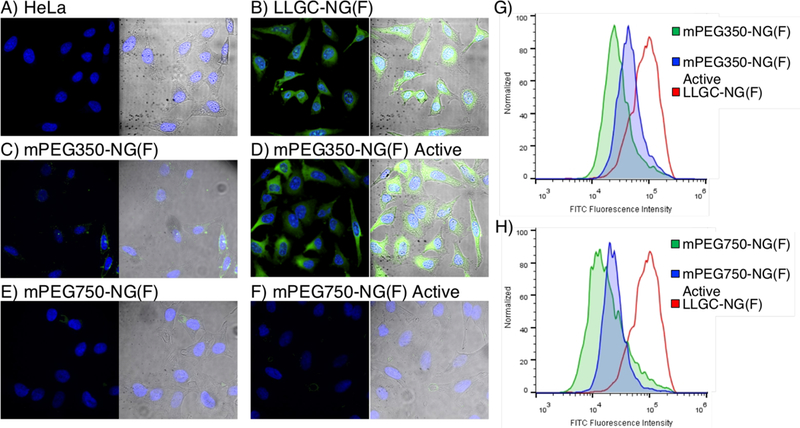
Confocal microscopy images using a 40× objective of composite (left) with nucleus (blue 405 nm) and fluorescein-labeled NG (green, 488 nm), and composite with brightfield (right) image overlays of HeLa cells after 2-hour incubation with A) no sample, and fluorescein-labeled nanogels B) LLGC-NG(F) positive control, C) mPEG350-NG(F), D) mPEG350-NG(F) Active, E) mPEG750-NG(F), F) mPEG750-NG(F) Active. Flow cytometric histogram profiles of FITC fluorescence intensity for HeLa cells incubated with LLGC-NG(F) and G) mPEG350-NG(F) and mPEG350-NG(F) Active, and H) mPEG750-NG(F) and mPEG750-NG(F) Active.
We then investigated the internalization behavior of DiI-encapsulated mPEG350-NG before and after MMP treatment to determine the capability to deliver a hydrophobic guest. DiI loaded mPEG350-NG was activated with MMP-9, then purified by 100 kDa dialysis and filtration, resulting in some loss of DiI (~18%, calculated by absorbance spectra at λmax = 558 nm) (Figure 7). Nanogels were incubated with HeLa cells for 2 hours at the same nanogel concentration (0.1 mg/mL) so that comparative uptake could be evaluated despite the DiI loss. Uptake was observed by confocal microscopy using laser excitation at 540 nm. Even with less fluorophore guest, significantly different cellular uptake was observed for mPEG350-NG(DiI) and mPEG350-NG(DiI) active (Figure 7). These results suggest that the higher internalization is due to the greater uptake of MMP-9 activated nanogels, consistent with observations with fluorescein-labeled nanogels. The enhanced delivery of a hydrophobic guest illustrates the potential for the protease-activation strategy to improve the internalization of therapeutic cargo in MMP-rich environments.
Figure 7.
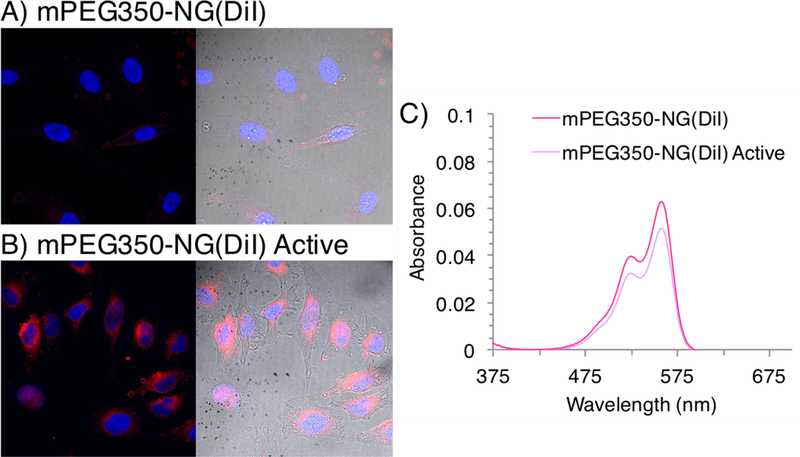
Confocal microscopy images using a 40× objective of composite (left) with nucleus (blue 405 nm) and DiI-loaded NG (red, 540 nm), and composite with brightfield (right) image overlays of HeLa cells after 2-hour incubation with A) mPEG350-NG(DiI) and B) mPEG350-NG(DiI) Active. C) UV-visible absorbance spectra of DiI encapsulation before and after MMP-9 activation. Dye release calculated from decrease in absorbance at λmax 558 nm.
CONCLUSION
In summary, we report on a nanogel capable of converting its surface properties from a passive PEGylated particle to an active amine decorated particle in the presence of MMP-9, a protease frequently observed in the tumor extracellular environment. We have shown that: (i) the incorporation of MMP-9 substrate on the nanogel surface can lead to a MMP-9 dependent surface conversion from PEG to N-termini, (ii) this conversion is dependent on time and the concentration of MMP-9 present, and circulation concentrations of MMP-9 were inadequate while tumor extracellular concentrations of MMP-9 were adequate in achieving surface conversion, (iii) the surface conversion process can be used to enhance nanocarrier cellular uptake specifically under desired MMP-9 upregulated environments, and (iv) this surface proteolysis does not cause guest release or nanogel disassembly, however intracellular concentrations of reducing glutathione results in guest release and nanogel disassembly.
Considering the molecular design, we expect that the sensitivity and specificity of peptidase response can be modified based on the identity of the proteolysis-susceptible peptide incorporated. Because this strategy of N-terminus generation can be achieved by simply installing the substrate to the particle at its C-terminus, it allows for flexible sequence selection and adaptation to the appropriate upregulated MMPs or other proteases in the target tissue. The modular formulation achieved by post-assembly modification with substrate using thiol-disulfide exchange allows for facile incorporation of a variety of responsive sequences, opening a variety of avenues for the design of these protease-responsive surface conversional nanogels.
Supplementary Material
ACKNOWLEDGEMENT
We thank the National Institutes of Health (CA-169140), the U.S. Army Research office (W911NF-15-1-0568), the Technology Fund from the UMass President’s Office for partial support of this work. We also thank NIH for training grants to M. R. Gordon (T32 GM008515) and to F. Anson and K. Singh (T32 GM108556). Microscopy data was gathered in the Light Microscopy Facility and Nikon Center of Excellence at the Institute for Applied Life Sciences, UMass Amherst with support from the Massachusetts Life Sciences Center.
REFERENCES
- 1).Liu DL; Chang X; Dong CM Reduction-and thermo-sensitive star polypeptide micelles and hydrogels for on-demand drug delivery. Chem. Commun 2013, 49 (12), 1229–1231. [DOI] [PubMed] [Google Scholar]
- 2).Bae YH; Park K Targeted drug delivery to tumors: myths, reality and possibility. J. Controlled Release 2011, 153, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Kobayashi H; Watanabe R; Choyke PL Improving Conventional Enhanced Permeability and Retention (EPR) Effects; What Is the Appropriate Target? Theranostics 2014, 4, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Jain RK; Stylianopoulos T Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol 2010, 7, 653–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Molineux G PEGylation: engineering improved pharmaceuticals for enhanced therapy. Cancer Treat Rev 2002, 28 (Suppl. A), 13–16. [DOI] [PubMed] [Google Scholar]
- 6).Moribe K; Maruyama K Reviews on PEG coated liposomal drug carriers. Drug Delivery Syst 2001, 16 (3), 165–171. [Google Scholar]
- 7).Romberg B; Hennink WE; Storm G Sheddable coatings for long-circulating nanoparticles. Pharm. Res 2008, 25, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Moghimi SM; Hunter AC; Murray JC Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev 2001, 53, 283–318. [PubMed] [Google Scholar]
- 9).Vittaz M; Bazile D; Spenlehauer G; Verrecchia T; Veillard M; Puisieux F; Labarre D Effect of PEO surface density on long-circulating PLA-PEO nanoparticles which are very low complement activators. Biomaterials 1996, 17, 1575–1581 [DOI] [PubMed] [Google Scholar]
- 10).Owens DE III; Peppas NA Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm 2006, 307, 93–102. [DOI] [PubMed] [Google Scholar]
- 11).Verbaan FJ; Oussoren C; Snel CJ; Crommelin DJ; Hennink WE; Storm G Steric stabilization of poly(2-(dimethylamino)ethyl methacrylate)-based polyplexes mediates prolonged circulation and tumor targeting in mice. J. Gene Med 2004, 6, 64–75. [DOI] [PubMed] [Google Scholar]
- 12).Vader P; van der Aa LJ; Engbersen JF; Storm G; Schiffelers RM Physicochemical and Biological Evaluation of siRNA Polyplexes Based on PEGylated Poly(amido amine)s. Pharm. Res 2012, 29, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Crownover E; Duvall CL; Convertine A; Hoffman AS; Stayton PS RAFT-synthesized graft copolymers that enhance pH-dependent membrane destabilization and protein circulation times. J. Controlled Release 2011, 155, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Chaudhari KR; Ukawala M; Manjappa AS; Kumar A; Mundada PK; Mishra AK Mathur R; Monkkonen J; Murthy RS Opsonization, biodistribution, cellular uptake and apoptosis study of PEGylated PBCA nanoparticle as potential drug delivery carrier. Pharm. Res 2012, 29, 53–68. [DOI] [PubMed] [Google Scholar]
- 15).Li Y; Kröger M; Liu WK; Endocytosis of PEGylated nanoparticles accompanied by structural and free energy changes of the grafted polyethylene glycol. Biomaterials, 2014, 35, 8467–8478. [DOI] [PubMed] [Google Scholar]
- 16).Matsumura Y; Maeda H A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- 17).Guo M; Que C; Wang C; Liu X; Yan H; Liu K Multifunctional superparamagnetic nanocarriers with folate-mediated and pH-responsive targeting properties for anticancer drug delivery. Biomaterials 2011, 32, 185–194. [DOI] [PubMed] [Google Scholar]
- 18).Narayanan S; Binulal NS; Mony U; Manzoor K; Nair S; Menon D Folate targeted polymeric ‘green’ nanotherapy for cancer. Nanotechnology 2010, 21, 285107. [DOI] [PubMed] [Google Scholar]
- 19).Zhang Q; Liu F; Nguyen KT; Ma X; Wang X; Xing B; Zhao Y Multifunctional Mesoporous Silica Nanoparticles for Cancer-Targeted and Controlled Drug Delivery. Adv. Funct. Mater 2012, 22, 5144–5156. [Google Scholar]
- 20).Davis ME; Zuckerman JE; Choi CH; Seligson D; Tolcher A; Alabi CA; Yen Y; Heidel JD; Ribas A Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Sarko D; Beijer B; Boy RG; Nothelfer E-M; Leotta K; Eisenhut M; Altmann A; Haberkorn U; Mier W The Pharmacokinetics of Cell-Penetrating Peptides. Mol. Pharmaceutics 2010, 7, 2224–2231. [DOI] [PubMed] [Google Scholar]
- 22).Jin E; Zhang B; Sun X; Zhou Z; Ma X; Sun Q; Tang J; Shen YQ; Kirk EV; Murdoch WJ; Radosz M Acid-Active Cell-Penetrating Peptides for in Vivo Tumor-Targeted Drug Delivery. J. Am. Chem. Soc 2013, 135, 933–940. [DOI] [PubMed] [Google Scholar]
- 23).Ruoslahti E RGD and Other Recognition Sequences for Integrins. Annu. Rev. Cell. Dev. Biol 1996, 12, 697–715. [DOI] [PubMed] [Google Scholar]
- 24).Aktan B; Chambre L; Sanyal R; Sanyal A “Clickable” Nanogels via Thermally Driven Self-Assembly of Polymers: Facile Access to Targeted Imaging Platforms using Thiol-Maleimide Conjugation. Biomacromolecules 2007, 18, 490–497. [DOI] [PubMed] [Google Scholar]
- 25).Friden PM; Walus LR; Musso GF; Taylor MA; Malfroy B; Starzyk RM Anti-Transferrin Receptor Antibody and Antibody Drug Conjugates Cross the Blood Brain Barrier. Proc. Natl. Acad. Sci. U.S.A 1991, 88, 4771–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Iakoubov LZ; Torchilin VP A Novel Class of Antitumor Antibodies: Nucleosome-Restricted Antinuclear Autoantibodies (ANA) from Healthy Aged Nonautoimmune Mice. Oncol. Res 1997, 9, 439–446. [PubMed] [Google Scholar]
- 27).Lukyanov AN; Elbayoumi TA; Chakilam AR; Torchilin VP Tumor-Targeted Liposomes: Doxorubicin-Loaded Long-Circulating Liposomes Modified with Anti-Cancer Antibody. J. Controlled Release 2004, 100, 135–144. [DOI] [PubMed] [Google Scholar]
- 28).Gabizon A; Horowitz AT; Goren D; Tzemach D; Mandelbaum-Shavit F; Qazen MM; Zalipsky S Targeting Folate Receptor with Folate Linked to Extremities of Poly(ethylene glycol)-Grafted Liposomes: In Vitro Studies. Bioconjugate Chem 1999, 10, 289–298. [DOI] [PubMed] [Google Scholar]
- 29).Zhu L; Ye Z; Cheng K; Miller DD; Mahato RI Site-Specific Delivery of Oligonucleotides to Hepatocytes after Systemic Administration. Bioconjugate Chem 2008, 19, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Zhu L; Mahato RI Targeted Delivery of siRNA to Hepatocytes and Hepatic Stellate Cells by Bioconjugation. Bioconjugate Chem 2010, 21, 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Phillips MA; Gran ML; Peppas NA Targeted Nanodelivery of Drugs and Diagnostics. Nano Today, 2010, 5(2),143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Lee H; Fonge H; Hoang B; Reilly RM; Allen C The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles. Mol. Pharm 2010, 7, 1195–1208. [DOI] [PubMed] [Google Scholar]
- 33).Muro S New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2010, 2, 189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Mutsaers SE; Papadimitriou JM Surface charge of macrophages and their interaction with charged particles. J. Leukocyte Biol 1988, 44, 17–26. [DOI] [PubMed] [Google Scholar]
- 35).Richard JP; Melikov K; Vives E; Ramos C; Verbeure B; Gait MJ; Chernomordik LV; Lebleu B Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem 2003, 278, 585–590. [DOI] [PubMed] [Google Scholar]
- 36).Ulbrich K; Subr V Polymeric anticancer drugs with pH-controlled activation. Adv. Drug Delivery Rev 2004, 56, 1023–1050. [DOI] [PubMed] [Google Scholar]
- 37).Lee HJ; Pardridge WM Monoclonal Antibody Radiopharmaceuticals: Cationization, Pegylation, Radiometal Chelation, Pharmacokinetics, and Tumor Imaging. Bioconjugate Chem 2003, 14, 546–553. [DOI] [PubMed] [Google Scholar]
- 38).Li W; Nakayama M; Akimoto J; Okano T Effect of block compositions of amphiphilic block copolymers on the physicochemical properties of polymeric micelles. Polymer 2011, 52, 3783–3790. [Google Scholar]
- 39).Dobrovolskaia MA; Patri AK; Simak J; Hall JB; Semberova J; De Paoli Lacerda SH; McNeil SE Nanoparticle size and surface charge determine effects of PAMAM dendrimers on human platelets in vitro. Mol. Pharm 2012, 9, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Ganta S; Devalapally H; Shahiwala A; Amiji M A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Controlled Release 2008, 126 (3), 187–204. [DOI] [PubMed] [Google Scholar]
- 41).Fleige E; Quadir MA; Haag R Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Delivery Rev 2012, 64 (9), 866–884. [DOI] [PubMed] [Google Scholar]
- 42).Kato Y; Ozawa S; Miyamoto C; Maehata Y; Suzuki A; Maeda T; Baba Y Acidic extracellular microenvironment and cancer, Cancer Cell Int 2013, 13, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Li L; Raghupathi K; Yuan C; Thayumanavan S Surface charge generation in nanogels for activated cellular uptake at tumor-relevant pH. Chem. Sci 2013, 4, 3654–3360. [Google Scholar]
- 44).Gu J; Cheng WP; Liu J; Lo SY; Smith D; Qu X; Yang Z pH-Triggered Reversible “Stealth” Polycationic Micelles, Biomacromolecules, 2008, 9, 255–262. [DOI] [PubMed] [Google Scholar]
- 45).Gao Y; Yang C; Liu X; Ma R; Kong D; Shi L A Multifunctional Nanocarrier Based on Nanogated Mesoporous Silica for Enhanced Tumor-Specific Uptake and Intracellular Delivery. Macromol. Biosci 2011, 12, 251–259. [DOI] [PubMed] [Google Scholar]
- 46).Ding CX; Gu JX; Qu XZ; Yang ZZ Preparation of Multifunctional Drug Carrier for Tumor-Specific Uptake and Enhanced Intracellular Delivery through the Conjugation of Weak Acid Labile Linker. Bioconjugate Chem 2009, 20, 1163–1170. [DOI] [PubMed] [Google Scholar]
- 47).Ding D; Kwok RTK; Yuan Y; Feng G; Tang BZ; Liu B A Fluorescent Light-up Nanoparticle Probe with Aggregation-induced Emission Characteristics and Tumor-acidity Responsiveness for Targeted Imaging and Selective Suppression of Cancer Cells. Mater. Horiz 2015, 2, 100–105. [Google Scholar]
- 48).Sun C-Y; Shen S; Xu C-F; Li H-J; Liu Y; Cao Z-T; Yang X-Z; Xia J-X; Wang J Tumor Acidity-Sensitive Polymeric Vector for Active Targeted siRNA Delivery. J. Am. Chem. Soc 2015, 137, 15217–15224. [DOI] [PubMed] [Google Scholar]
- 49).Mizuhara T; Saha K; Moyano DF; Kim CS; Yan B; Kim Y-K; Rotello VM Acylsulfonamide-Functionalized Zwitterionic Gold Nanoparticles for Enhanced Cellular Uptake at Tumor pH. Angew. Chem., Int. Ed 2015, 54, 6567–6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Wilson WR; Hay MP Targeting hypoxia in cancer therapy. Nature Reviews Cancer, 2011, 11, 393–410 [DOI] [PubMed] [Google Scholar]
- 51).Brown JM Tumor hypoxia in cancer therapy. Methods Enzymol 2007, 435, 297–321. [DOI] [PubMed] [Google Scholar]
- 52).Choi KY; Swierczewska M; Lee S; Chen XY Protease-Activated Drug Development. Theranostics 2012, 2, 156–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).de la Rica R; Aili D; Stevens MM Enzyme-responsive nanoparticles for drug release and diagnostics. Adv. Drug Delivery Rev 2012, 64, 967–978. [DOI] [PubMed] [Google Scholar]
- 54).Coleman JD; Thompson JT; Smith RW 3rd; Prokopczyk B; Vanden HJP Role of Peroxisome Proliferator-Activated Receptor β/δ and B-Cell Lymphoma-6 in Regulation of Genes Involved in Metastasis and Migration in Pancreatic Cancer Cells. PPAR Res 2013, 2013, 121956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55).Li H; Yu SS; Miteva M; Nelson CE; Werfel T; Giorgio TD; Duvall CL Matrix Metalloproteinase Responsive, Proximity-Activated Polymeric Nanoparticles for siRNA Delivery. Adv. Funct. Mater 2013, 23, 3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56).Li J; Ge Z; Liu S PEG-sheddable polyplex micelles as smart gene carriers based onMMP-cleavable peptide-linked block copolymers, Chem. Commun, 2013, 49, 6974–6976. [DOI] [PubMed] [Google Scholar]
- 57).Crisp JL; Savariar EN; Glasgow HL; Ellies LG; Whitney MA; Tsien RY Dual targeting of integrin αvβ3 and matrix metalloproteinase-2 for optical imaging of tumors and chemotherapeutic delivery. Molecular Cancer Therapeutics 2014, 13, 1514–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58).Harris TJ; von Maltzahn G; Lord ME; Park JH; Agrawal A; Min DH; Sailor MJ; Bhatia SN Protease-Triggered Unveiling of Bioactive Nanoparticles. Small 2008, 4, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59).Zhu L; Kate P; Torchilin VP Matrix Metalloprotease 2-Responsive Multifunctional Liposomal Nanocarrier for Enhanced Tumor Targeting. ACS Nano, 2012, 6(4), 3491–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Fay F; Hansen L; Hectors SJCG; Sanchez-Gaytan BL; Zhao Y; Tang J; Munitz J; Alaarg A; Braza MS; Gianella A; Aaronson SA; Reiner T; Kjems J; Langer FJ Hoeben HM, Janssen C; Calcagno GJ; Strijkers ZA; Fayad C; Peŕez-Medina R; Mulder WJM Investigating the Cellular Specificity in Tumors of a Surface-Converting Nanoparticle by Multimodal Imaging. Bioconjugate Chem 2017, 28, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61).Zhang J; Yuan ZF; Wang Y; Chen WH; Luo GF; Cheng SX; Zhuo RX; Zhang XZ Multifunctional Envelope-Type Mesoporous Silica Nanoparticles for Tumor-Triggered Targeting Drug Delivery. J. Am. Chem. Soc 2013, 135, 5068–5073. [DOI] [PubMed] [Google Scholar]
- 62).Yu SS; Lau CM; Thomas SN; Jerome WG; Maron DJ; Dickerson JH; Hubbell JA; Giorgio TD Size-and charge-dependent non-specific uptake of PEGylated nanoparticles by macrophages. Int. J. Nanomed 2012, 7, 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Callmann CE; Barback CV; Thompson MP; Hall DJ; Mattrey RF; Gianneschi MC Therapeutic Enzyme-Responsive Nanoparticles for Targeted Delivery and Accumulation in Tumors. Adv. Mater 2015, 27, 4611–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64).Kulkarni PS; Haldar MK; Nahire RR; Katti P; Ambre AH; Muhonen WW; Shabb JB; Padi SKR; Singh RK; Borowicz PP; Shrivastava DK; Katti KS; Reindl K; Guo B; Mallik S MMP-‐9 Responsive PEG Cleavable Nanovesicles for Efficient Delivery of Chemotherapeutics to Pancreatic Cancer. Mol. Pharmaceutics 2014, 11, 2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65).Liu Y; Ding X; Li J; Luo Z; Hu Y; Liu J; Dai L; Zhou J; Hou C; Cai K Enzyme responsive drug delivery system based on mesoporous silica nanoparticles for tumor therapy in vivo. Nanotechnology 2015, 26, 145102–145116. [DOI] [PubMed] [Google Scholar]
- 66).Ghosh S; Basu S; Thayumanavan S Simultaneous and Reversible Functionalization of Copolymers for Biological Applications. Macromolecules 2006, 39, 5595–5597. [Google Scholar]
- 67).Ryu J-H; Chacko RT; Jiwpanich S; Bickerton S; Babu RP; Thayumanavan S Self-cross-linked polymer nanogels: a versatile nanoscopic drug delivery platform. J. Am. Chem. Soc 2010, 132, 17227–17235. [DOI] [PubMed] [Google Scholar]
- 68).Ryu J; Jiwpanich S; Chacko R; Bickerton S; Thayumanavan S Surface functionalizable polymer nanogels with facile hydrophobic guest encapsulation capabilities. J. Am. Chem. Soc 2010, 132, 8246–8247. [DOI] [PubMed] [Google Scholar]
- 69).Kavimandan NJ; Losi E; Wilson JJ; Brodbelt JS; Peppas NA; Synthesis and Characterization of Insulin-Transferrin Conjugates. Bioconjugate Chem 2006, 17, 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70).Seltzer JL; Weingarten H; Akers KT; Eschbach ML; Grant GA; Eisen AZ Cleavage Specificity of Type IV Collagenase (Gelatinase) from Human Skin. Use of synthetic peptides as model substrates. The Journal of Biological Chemistry 1989, 264(33), 19583–19586. [PubMed] [Google Scholar]
- 71).Collier IE; Wilhelm SM; Eisen AZ; Marmer BL; Grant GA; Seltzer JL; Kronberger A; He C; Bauer EA; Goldberg GI The Structure OF THE Human Skin Fibroblast Collagenase Gene. J. Biol. Chem 1988, 263, 6579–6587. [PubMed] [Google Scholar]
- 72).Saito G; Swanson JA; Lee KD Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv. Drug Delivery Rev 2003, 55, 199–215. [DOI] [PubMed] [Google Scholar]
- 73).Meister A; Anderson ME Glutathione. Ann. Rev. Biochem 1983, 52, 711–760. [DOI] [PubMed] [Google Scholar]
- 74).Li L; Ryu JH; Thayumanavan S Effect of Hofmeister Ions on the Size and Encapsulation Stability of Polymer Nanogels. Langmuir, 2013, 29 (1), 50–55. [DOI] [PubMed] [Google Scholar]
- 75).Udenfriend S; Stein S; Böhlen P; Dairman W; Leimgruber W; Weigele M Fluorescamine: A Reagent for Assay of Amino Acids, Peptides, Proteins, and Primary Amines in the Picomole range. Science, 1972, 178(4063), 871–872 [DOI] [PubMed] [Google Scholar]
- 76).Gore M Spectrophotometry and Spectrofluorimetry: A Practical Approach Ed. Oxford University Press, Incorporated; (New York, NY: 2000) p. 63. [Google Scholar]
- 77).Böhlen P; Stein S; Dairman W; Udenfriend S Fluorometric assay of proteins in the nanogram range. Arch. Biochem. Biophys 1973, 155, 213–220. [DOI] [PubMed] [Google Scholar]
- 78).Read ML; Etrych T; Ulbrich K; Seymoura LW Characterisation of the binding interaction between poly(L-lysine) and DNA using the fluorescamine assay in the preparation of nonviral gene delivery vectors. FEBS Lett 1999, 461, 96–100. [DOI] [PubMed] [Google Scholar]
- 79).Tutton MG; George ML; Eccles SA; Burton S; Swift RI; Abulafi AM Use of Plasma MMP-2 and MMP-9 Levels as a Surrogate for Tumor Expression in Colorectal Cancer Patients. Int. J. Cancer, 2003,107, 541–550. [DOI] [PubMed] [Google Scholar]
- 80).İnce AT; Yıldız K; Gangarapu V; Kayar Y; Baysal B; Karatepe O; Kemik AS; Şentürk H Serum and biliary MMP-9 and TIMP-1 concentrations in the diagnosis of cholangiocarcinoma. Int. J. Clin. Exp. Med 2015, 8(2), 2734–2740. [PMC free article] [PubMed] [Google Scholar]
- 81).Jumper C; Cobos E; Lox C Determination of the serum matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) in patients with either advanced small-cell lung cancer or non-small-cell lung cancer prior to treatment. Respiratory Medicine 2004, 98, 173–177. [DOI] [PubMed] [Google Scholar]
- 82).Hyuga S; Nishikawa Y; Sakata K; Tanaka H; Yamagata S; Sugita K; Saga S; Matsuyama M; Shimizu S Autocrine Factor Enhancing the Secretion of Mr 95,000 Gelatinase (Matrix Metalloproteinase 9) in Serum-free Medium Conditioned with Murine Metastatic Colon Carcinoma Cells. Cancer Research, 1994, 54, 3611–3616. [PubMed] [Google Scholar]
- 83).Sun Q; Ojha T; Kiessling F; Lammers T; Shi Y Enhancing Tumor Penetration of Nanomedicines. Biomacromolecules 2017, 18, 1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84).Radovic-Moreno AF; Lu TK; Puscasu VA; Yoon CJ; Langer R; Farokhzad OC Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics. ACS Nano 2012, 6, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85).Bahadur KCR; Xu P Multicompartment Intracellular Self-Expanding Nanogel for Targeted Delivery of Drug Cocktail. Adv. Mater 2012, 24, 6479–6483. [DOI] [PubMed] [Google Scholar]
- 86).Schröpfer A; Kammerer U; Kapp M; Dietl J; Feix S; Anacker J Expression pattern of matrix metalloproteinases in human gynecological cancer cell lines. BMC Cancer 2010, 10, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.