

C. difficile is a spore-forming anaerobic bacterium which causes infection of the large intestine with high mortality rates. The C. difficile infection is difficult to prevent and treat, as the pathogen is resistant to many antimicrobial agents. Prolonged use of β-lactam antibiotics for treatment of various infectious diseases triggers the infection, as these drugs suppress the abundance of protective gut bacteria, allowing the resistant C. difficile bacteria to multiply. While resistance of C. difficile to β-lactam antibiotics plays the major role in the development of the disease, the mechanism of resistance is unknown. The significance of our research is in the discovery in C. difficile of β-lactamases, enzymes that destroy β-lactam antibiotics. These findings ultimately can help to combat deadly C. difficile infections.
KEYWORDS: Clostridium difficile, antibiotic resistance, beta-lactamases
ABSTRACT
Clostridium difficile is the causative agent of the deadly C. difficile infection. Resistance of the pathogen to β-lactam antibiotics plays a major role in the development of the disease, but the mechanism of resistance is currently unknown. We discovered that C. difficile encodes class D β-lactamases, i.e., CDDs, which are intrinsic to this species. We studied two CDD enzymes, CDD-1 and CDD-2, and showed that they display broad-spectrum, high catalytic efficiency against various β-lactam antibiotics, including penicillins and expanded-spectrum cephalosporins. We demonstrated that the cdd genes are poorly expressed under the control of their own promoters and contribute only partially to the observed resistance to β-lactams. However, when the cdd1 gene was expressed under the control of efficient promoters in the antibiotic-sensitive Clostridium cochlearium strain, it produced high-level resistance to β-lactams. Taken together, the results determined in this work demonstrate the existence in C. difficile of intrinsic class D β-lactamases which constitute a reservoir of highly potent enzymes capable of conferring broad-spectrum, clinically relevant levels of resistance to β-lactam antibiotics. This discovery is a significant contribution to elucidation of the mechanism(s) of resistance of the clinically important pathogen C. difficile to β-lactam antibiotics.
INTRODUCTION
Clostridium difficile infection is a life-threatening disease that imposes a significant burden on health care. In 2011, the estimated number of C. difficile infections in the United States exceeded 450,000, resulting in 29,300 deaths (1) and in associated excess health care costs of $4.8 billion for acute care facilities alone (2 – 4). The number of incidents of C. difficile infections in both hospitals and the outpatient setting is steadily rising, with an average annual rate of 3.3% in the United States (5 – 8). As a result, the sum of charges for C. difficile infection-associated hospitalizations increased from $20.1 billion in 2005 to $31.4 billion in 2014 (8). Antimicrobial therapy plays a crucial role in the development of C. difficile infection (9 – 11) due to the ability of many antibiotics to suppress the abundance of the gut microbiota that protects the host from invasion by pathogenic bacteria (12 – 14). Resistance of C. difficile to antibiotics is an important contributor to the emergence of the disease. C. difficile is notorious for its resistance to antimicrobial agents and is listed by the Centers for Disease Control and Prevention as the pathogen that causes the highest antibiotic resistance threat in the United States (15). While several classes of antimicrobial agents have been implicated in outbreaks of C. difficile infection, β-lactam antibiotics are recognized as major causative agents of the disease (16 – 18). β-Lactams differ significantly in their levels of activity against C. difficile (19, 20), and those levels correlate with their ability to trigger the infection. β-Lactams with low potency against C. difficile and high activity against the protective gut microbiota represent the major risk factor (21 – 24). Although resistance of C. difficile to β-lactams is recognized as a leading contributor to the development of C. difficile infection, the underlying mechanisms of resistance are currently unknown (19, 20).
RESULTS
C. difficile ATCC 43255 and C. difficile 630Δerm are resistant to β-lactam antibiotics.
While published data cumulatively indicate that C. difficile is generally resistant to many β-lactams (19, 20), there have been relatively few studies on the susceptibility of the pathogen to these antimicrobial agents. To initiate studies of the mechanism(s) of resistance of C. difficile to β-lactam antibiotics, we evaluated MIC values of various β-lactam antibiotics against two clinically important clostridial isolates, C. difficile ATCC 43255 and C. difficile 630Δerm (25 – 27), and showed that the two isolates exhibit very similar broad antibiotic resistance profiles (Table 1). They were highly resistant to the expanded-spectrum cephalosporins cefotaxime, ceftriaxone, ceftazidime, and cefepime (MICs, 64 to 256 µg/ml), the cephamycin cephalosporin cefoxitin (MIC, 128 µg/ml), and the monobactam aztreonam (MIC, 2,048 µg/ml). The MICs of the penicillins ampicillin, penicillin G, and oxacillin, of the carbapenems imipenem and meropenem, and of the cephalosporin cephalothin were significantly lower. The MICs of oxacillin, cephalothin, and meropenem were 2-to-8-fold higher for C. difficile 630Δerm.
TABLE 1.
MICs of β-lactam antibiotics against clostridial isolates
| Antibiotic | MIC (µg/ml) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
C. difficile
ATCC 43255 |
C. difficile
630Δerm |
C. cochlearium
ATCC 17787 |
|||||||
| cdd1 | cdd2 |
cdd2
intron |
Δcdd2 | Parental strain |
*cdd1 | *cdd2 | Pthl
cdd1 |
Pfdx
cdd1 |
|
| Ampicillin | 4 | 4 | 1 | 1 | 0.03 | 0.25 | 0.25 | 32 | 16 |
| Penicillin G | 4 | 4 | 1 | 1 | 0.015 | 0.25 | 0.25 | 32 | 16 |
| Oxacillin | 8 | 64 | 64 | 64 | 0.25 | 2 | 1 | 64 | 16 |
| Cephalothin | 16 | 32 | 32 | 32 | 0.12 | 0.5 | 0.25 | 32 | 16 |
| Cefotaxime | 128 | 128 | 128 | 128 | 1 | 2 | 2 | 128 | 64 |
| Ceftriaxone | 64 | 64 | 32 | 32 | 1 | 2 | 2 | 256 | 128 |
| Cefoxitin | 128 | 128 | 128 | 128 | 0.25 | 0.25 | 0.25 | 0.5 | 0.5 |
| Ceftazidime | 256 | 256 | 64 | 64 | 32 | 32 | 32 | 1,024 | 512 |
| Cefepime | 64 | 64 | 32 | 32 | 16 | 16 | 16 | 256 | 128 |
| Aztreonam | 2,048 | 2,048 | 2,048 | 2,048 | 512 | 512 | 512 | 512 | 256 |
| Imipenem | 4 | 4 | 4 | 4 | 0.03 | 0.06 | 0.06 | 0.12 | 0.12 |
| Meropenem | 2 | 4 | 4 | 4 | 0.007 | 0.015 | 0.015 | 0.03 | 0.03 |
Genomes of C. difficile ATCC 43255 and C. difficile 630Δerm encode class D β-lactamases.
Gram-positive bacteria manifest resistance to β-lactam antibiotics via two major mechanisms: production of antibiotic-degrading enzymes, namely, β-lactamases, and/or production of antibiotic-resistant targets, namely, penicillin-binding proteins (PBPs). Four molecular classes of β-lactamases (classes A through D) are widely disseminated in Gram-negative bacteria, while for a long time, only class A and B enzymes were known in Gram-positive microorganisms. Recently, we demonstrated that class D enzymes are also widely spread in the Clostridiaceae, Bacillaceae, and Eubacteriaceae families of phylum Firmicutes of Gram-positive bacteria (28).
In our effort to get insights into the mechanism of resistance of C. difficile to β-lactam antibiotics, we analyzed genomes of two strains, C. difficile ATCC 43255 and C. difficile 630Δerm, and found that each contains only one open-reading frame annotated as a putative β-lactamase gene. The enzyme from C. difficile ATCC 43255 is 312 amino acid residues long, while that from C. difficile 630Δerm is slightly shorter (Fig. 1). Both harbor conserved amino acid sequence motifs (SXXK, YGN, KTG, and WXXG) characteristic of class D β-lactamases. We named the enzyme from C. difficile ATCC 43255 CDD-1 (an abbreviation from Clostridium difficile class D β-lactamase) and named the enzyme from C. difficile 630Δerm CDD-2.
FIG 1.

Amino acid sequence alignment and genomic regions of CDD-1 and CDD-2 β-lactamases. Sequence motifs conserved in class D β-lactamases are highlighted in yellow. Putative leader sequences for the CDD-1 and CDD-2 β-lactamases are underlined.
CDD β-lactamases are intrinsic to C. difficile.
To evaluate how widely CDD-type β-lactamases are spread among C. difficile, we analyzed around 800 completed genomic sequences of C. difficile and found that the vast majority of them harbor a single chromosomal gene for the CDD-type enzyme, showing that these enzymes are intrinsic to the pathogen. To appreciate the diversity of CDD-type β-lactamases on the amino acid sequence level, we utilized BLAST software from the NCBI for the search of homologous proteins using the sequence of the mature CDD-1 enzyme as the template. Results of this study showed that class D β-lactamases from C. difficile strains isolated from different parts of the world are very closely related, with >97% of them sharing at least 95% amino acid sequence identity.
To identify the most common substitutions in CDD β-lactamases, we analyzed the reported amino acid sequences that were identical in at least five different C. difficile strains. We found 477 such strains and subdivided their enzymes into two subgroups, the CDD-1-type and CDD-2-type subgroups (Table 2). We identified the CDD-1-type enzymes in 47 strains. Of those, 41 strains encoded the CDD-1 β-lactamase, while the 6 remaining strains encoded the CDD-1 mutant (CDD-1-M1) with a single C22Y amino acid substitution. This substitution is also present in enzymes of all other 430 strains encoding the CDD-2-type β-lactamases. Among the strains harboring genes for the CDD-2-type enzymes, the vast majority (312) encoded the CDD-2 β-lactamase. All remaining strains encoded seven CDD-2-like enzymes that have from one to three amino acid substitutions compared to the amino acid sequence of the CDD-2 β-lactamase.
TABLE 2.
Most common amino acid substitutions in CDD β-lactamases
| Enzymea | Amino acid substitutions | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDD-1 (41)b | S2 | V3 | I5 | G14 | C22 | D74 | N103 | S109 | D165 | R177 | R205 | L214 |
| CDD-1-M1 (6) | Y | |||||||||||
| CDD-2 (312) | K | I | M | Y | E | D | ||||||
| CDD-2-M1 (22) | K | I | M | Y | E | D | C | |||||
| CDD-2-M2 (13) | K | I | M | Y | E | D | I | |||||
| CDD-2-M3 (11) | K | I | M | Y | E | D | Y | |||||
| CDD-2-M4 (24) | K | I | M | Y | E | Y | ||||||
| CDD-2-M5 (9) | K | I | M | Y | E | |||||||
| CDD-2-M6 (16) | K | I | M | D | Y | E | Y | |||||
| CDD-2-M7 (23) | K | I | Y | N | K | |||||||
The number of isolates with the given amino acid substitutions is indicated in parentheses. CDD-2-M1 through CDD-2-M7 are mutants of CDD-1 and CDD-2 β-lactamases.
The amino acid sequence of CDD-1, which was used as the reference sequence for comparison, is indicated in bold. The amino acid numbering indicates the position of each amino acid in the mature enzyme.
Our analysis also demonstrated that beyond the C. difficile species, the amino acid sequence similarity of CDD-1 to currently known class D enzymes is below 55%. These data indicated that CDD β-lactamases have failed to spread to other bacteria and that they represent a unique family of closely related enzymes that are native to C. difficile.
C. difficile ATCC 43255 and C. difficile 630Δerm produce active CDD β-lactamases.
To assess whether C. difficile ATCC 43255 and C. difficile 630Δerm produce active β-lactamases, we utilized a nitrocefin test. We observed a slight change in nitrocefin color in wells with both strains grown in the presence of antibiotics but not in their absence (Fig. 2A, second row). We then investigated whether the observed β-lactamase activity was due to production of the class D CDD-1 and CDD-2 enzymes or if β-lactamases of other classes are involved. Unlike serine β-lactamases of classes A and C, class D enzymes require posttranslational carboxylation of their active-site lysine for catalysis (29). This lysine may undergo decarboxylation under experimental conditions, resulting in a precipitous decrease in the catalytic activity of the enzyme. The activity can be restored by addition of a CO2 source such as sodium bicarbonate. We monitored hydrolysis of nitrocefin in the presence of 50 mM sodium bicarbonate. We observed a significant change in color when bacteria were grown in the presence of each of the β-lactams that we tested (except for cefoxitin), while the color of antibiotic-free medium changed only slightly (Fig. 2A, third row). These data indicated that both C. difficile ATCC 43255 and C. difficile 630Δerm produce an inducible class D β-lactamase.
FIG 2.
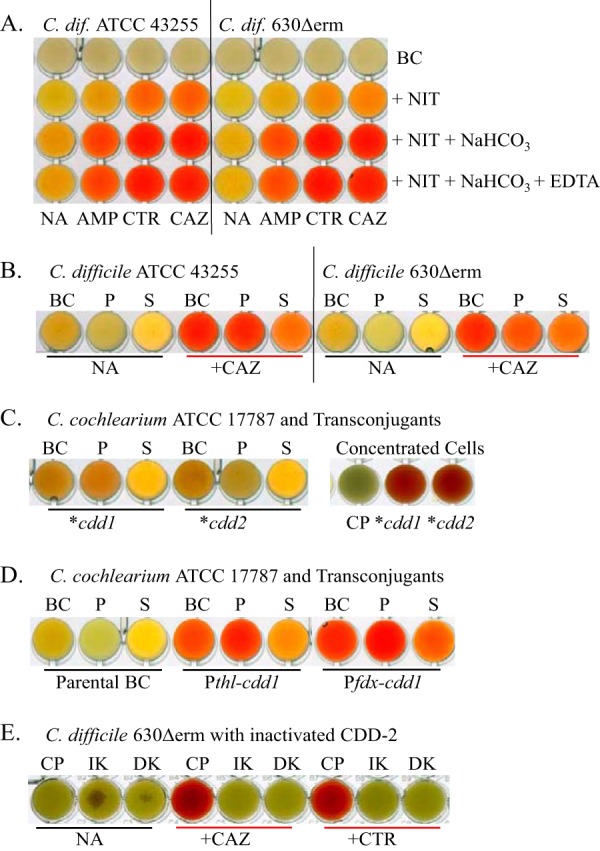
Evaluation of β-lactamase activity. (A) Induction of β-lactamase activity in C. difficile (C. dif) ATCC 43255 and C. difficile 630Δerm by three representative antibiotics. Their activity increases in the presence of NaHCO3 and is not inhibited by EDTA. (B) Distribution of β-lactamase between the bacterial cells and growth medium. (C) Expression of CDD-1 and CDD-2 enzymes from their own promoters in C. cochlearium ATCC 17787. The right panel shows 5-fold-concentrated cell pellets. (D) Expression of CDD-1 from two functional promoters in C. cochlearium ATCC 17787. (E) Loss of CDD-2 β-lactamase activity in C. difficile 630Δerm knockouts. Activity was measured in 5-fold-concentrated cell pellets. Abbreviations: BC, bacterial culture; NIT, nitrocefin; NA, no antibiotic; AMP, ampicillin; CTR, ceftriaxone; CAZ, ceftazidime; P, pellet; S, supernatant; CP, control pellet of the parental strain; IK, insertional knockout; DK, deletion knockout.
We also evaluated whether metallo β-lactamases contribute to the observed change in the nitrocefin color. The presence of EDTA, a chelating agent that strongly inhibits the activity of class B metalloenzymes, did not prevent or delay the nitrocefin reaction, which excludes the possibility of a contribution of metallo β-lactamases to nitrocefin hydrolysis (Fig. 2A, fourth row).
In Gram-positive bacteria, β-lactamases can be anchored to the bacterial cell wall and/or excreted to the milieu. We investigated whether the CDD-1 and CDD-2 enzymes are secreted or attached to the cell wall. We pelleted bacteria by centrifugation, saved the growth medium, and resuspended the pellets in a phosphate buffer. The nitrocefin color change in C. difficile ATCC 43255 was mostly associated with bacterial cell pellets, while the C. difficile 630Δerm color changes were similar in the cell pellets and supernatant (Fig. 2B). These data showed that more CDD β-lactamase is cell wall associated in C. difficile ATCC 43255 than in C. difficile 630Δerm. Cell wall association also has been demonstrated for other classes of β-lactamases from Gram-positive and Gram-negative bacteria, where the enzymes are often anchored to the membrane (30 – 33).
The CDD-1 β-lactamase displays broad-spectrum activity.
To evaluate the substrate profile of CDD enzymes, we purified the CDD-1 β-lactamase to homogeneity and undertook kinetic studies. CDD-1 exhibited high catalytic efficiency for the three penicillins (kcat/Km, 1.2 × 106 to >1.8 × 106 M−1 s−1; Table 3) and for five of the six cephalosporins (kcat/Km, 1.9 × 105 to 1.6 × 106 M−1 s−1), with the exception of cefoxitin. The catalytic efficiency determined for the monobactam aztreonam (kcat/Km, 5.0 × 105 M−1 s−1) was similar to that determined for the penicillins and cephalosporins, while that determined for the carbapenems imipenem and meropenem was 2 orders of magnitude lower. CDD-1 had high apparent affinity levels (Km, <1 to 2.7 µM) and relatively low turnover numbers (kcat, 1.8 to 3.2 s−1) for all penicillins tested. For the cephalosporins ceftriaxone, cefotaxime, and cefepime, turnover numbers were 4-to-8-fold higher and the apparent affinity decreased by up to 23-fold compared to penicillins. The steady-state kinetic parameters for aztreonam were similar to those for expanded-spectrum cephalosporins, while very low turnover rates corresponding to kcat values of 0.044 and 0.005 s−1 were observed for imipenem and meropenem, respectively. Our kinetic studies demonstrated that CDD-1 is an efficient broad-spectrum β-lactamase. We also determined the steady-state kinetic parameters for the CDD-2 enzyme with several β-lactam antibiotics (ampicillin, oxacillin, cefotaxime, and ceftriaxone) and found they were almost identical to those of CDD-1 (data not shown).
TABLE 3.
Steady-state kinetic parameters of the CDD-1 β-lactamase
| Antibiotic | kcat (s−1) | Km (µM) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| Ampicillin | 3.2 ± 0.1 | 2.7 ± 0.7 | (1.2 ± 0.3) × 106 |
| Penicillin G | 1.8 ± 0.1 | <1 | >(1.8 ± 0.1) × 106 |
| Oxacillin | 2.9 ± 0.1 | 1.6 ± 0.3 | (1.8 ± 0.3) × 106 |
| Cephalothin | 0.88 ± 0.02 | 0.8 ± 0.2 | (1.2 ± 0.2) × 106 |
| Cefoxitin | 0.009 ± 0.001 | 40 ± 3 | (2.3 ± 0.3) × 102 |
| Ceftazidime | 21 ± 1 | 110 ± 10 | (1.9 ± 0.2) × 105 |
| Cefepime | 10.5 ± 0.2 | 23 ± 1 | (4.5 ± 0.2) × 105 |
| Ceftriaxone | 12.0 ± 0.4 | 7.6 ± 0.9 | (1.6 ± 0.2) × 106 |
| Cefotaxime | 12.0 ± 0.3 | 7.3 ± 0.7 | (1.6 ± 0.2) × 106 |
| Aztreonam | 20 ± 1 | 40 ± 3 | (5.0 ± 0.5) × 105 |
| Imipenem | 0.044 ± 0.001 | 4.7 ± 0.4 | (9.4 ± 0.8) × 103 |
| Meropenem | 0.005 ± 0.001 | <4 | >(1.3 ± 0.3) × 103 |
| Nitrocefin | 40 ± 1 | 5.8 ± 0.8 | (6.9 ± 1) × 106 |
The genes for CDD-1 and CDD-2 β-lactamases express poorly under the control of their own promoters.
To test the expression levels of the cdd1 and cdd2 genes under the control of their own promoters, we analyzed the genetic composition of their upstream regions. The cdd1 gene is flanked upstream by the gene for an ATP-dependent DNA helicase (Fig. 3). There is an insertion of two genes between cdd2 and the helicase gene in C. difficile 630Δerm; thus, the β-lactamase neighbors a putative membrane protein. Hence, intergenic regions upstream of the cdd1 and cdd2 genes differ in length and nucleotide sequence. To establish whether there are functional promoters in close proximity to the cdd1 and cdd2 genes, we cloned those genes along with their 182-bp and 177-bp upstream regions, respectively, into the pMLT83151 vector, resulting in pMTL83151::*cdd1 and pMTL83151::*cdd2 constructs. As C. difficile is intrinsically resistant to expanded-spectrum cephalosporins, we expressed the CDD enzymes in a heterologous clostridial host, Clostridium cochlearium ATCC 17787, which we have shown is highly sensitive to most β-lactam antibiotics tested (Table 1) and is amenable to genetic manipulations.
FIG 3.
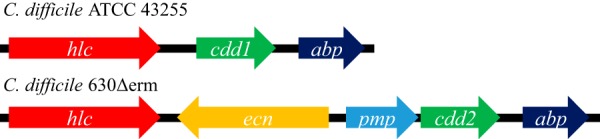
Schematic representation of regions surrounding the genes for CDD-1 and CDD-2 β-lactamases. The open reading frames are shown as arrows and intergenic regions as black lines. In strain ATCC 43255 (top), the gene for the CDD-1 β-lactamase (cdd1) is flanked by an ATP-dependent helicase (hlc) upstream and by an ABC transporter-coupled two-component system ATP-binding protein (abp) downstream. In strain 630 (bottom), two additional open reading frames are present between the gene for the CDD-2 β-lactamase (cdd2) and the hlc gene (an excinuclease ABC subunit A paralog of unknown function [ecn] and a putative membrane protein [pmp]). As a result, the intergenic region between the cdd gene and the upstream gene changed from 540 bp for cdd1 to 27 bp for cdd2.
We then introduced the pMTL83151::*cdd1 and pMTL83151::*cdd2 constructs in C. cochlearium ATCC 17787, where they were stably maintained. Expression of the CDD-1 and CDD-2 enzymes in the new host was not inducible by β-lactam antibiotics (data not shown). The change in nitrocefin color was slow with bacterial culture and was more intense with cell pellet than with the supernatant (Fig. 2C). The color change was more apparent when the cells were concentrated 5-fold. These data indicate that the CDD-1 and CDD-2 β-lactamases are poorly expressed from their own promoters in C. cochlearium ATCC 17787. The resulting low MIC values for the majority of β-lactam antibiotics tested support this assumption (Table 1). The largest effect was observed for penicillins, whose MIC values increased 8-fold to 16-fold to 0.25 to 2.0 µg/ml. The MIC values for cephalothin, cefotaxime, ceftriaxone, imipenem, and meropenem were increased 2-fold, while the MIC values of other β-lactams remained unchanged.
CDD-1 β-lactamase produces high-level resistance to β-lactam antibiotics when expressed from efficient promoters.
Next, we investigated whether CDD β-lactamases are capable of producing high-level resistance to β-lactam antibiotics when expressed from known functional clostridial promoters. To accomplish this goal, we placed the gene for the CDD-1 β-lactamase encoding the full-length enzyme under the control of the Pthl and Pfdx promoters, which are often utilized for expression of clostridial genes in various clostridial species (34, 35). The resulting constructs were cloned into shuttle vectors to produce the pMTL83122::cdd1-Pthl and pMTL83123::cdd1-Pfdx vectors, which were introduced into C. cochlearium ATCC 17787. The nitrocefin color change seen with these constructs was significantly more intense than what we observed with CDD-1 expressed under the control of its own promoter (Fig. 2D and C). As in the case of C. difficile ATCC 43255, hydrolysis of nitrocefin was more efficient in cell pellets of C. cochlearium than in the supernatants, an indication that the two bacterial species process the CDD-1 β-lactamase in the same way (Fig. 2D).
Expression of CDD-1 from the Pthl and Pfdx promoters (Pthl cdd1 and Pfdx cdd1) rendered C. cochlearium highly resistant to the majority of β-lactam antibiotics tested (Table 1). Overall, the Pthl promoter provided 2-fold-higher levels of resistance. Pthl cdd1 increased the MICs of ampicillin, penicillin G, and oxacillin 256-fold to 2,048-fold (to 32 to 64 µg/ml). It also significantly increased the MICs of the majority of cephalosporins tested. The parental C. cochlearium ATCC 17787 strain is highly sensitive to cephalosporins cephalothin, cefotaxime, ceftriaxone, and cefoxitin. Pthl cdd1 increased the MIC values of the first three of these drugs 128-fold to 256-fold. Noticeably, the MICs of cefoxitin remained practically unchanged, an indication that this antibiotic is not a substrate for the β-lactamase. The two remaining cephalosporins, ceftazidime and cefepime, had much higher MICs against the parental C. cochlearium ATCC 17787 strain. Expression of the CDD-1 β-lactamase from the Pthl promoter further increased the MICs of these antibiotics to 1,024 and 256 µg/ml, respectively. Expression of CDD-1 failed to further increase the high MICs of aztreonam. The MICs of meropenem and imipenem increased 4-fold but remained well below clinically significant levels.
Inactivation of CDD-2 β-lactamase decreases resistance of C. difficile to β-lactam antibiotics.
To further evaluate the contribution of clostridial β-lactamases to the observed resistance to β-lactam antibiotics, we attempted to inactivate the genes for the CDD-1 and CDD-2 β-lactamases. Creating gene knockouts in C. difficile is challenging, as some C. difficile strains are minimally amenable to genetic manipulations, while the majority are not (36, 37). To inactivate the cdd1 and cdd2 genes, we utilized two recently developed methodologies. One of the two, represented by the ClosTron system, utilizes a group II intron to create insertions into the gene of interest to cause its inactivation (38). The other employs a two-step allele-exchange methodology (36) to generate targeted deletions of genetic material. Our numerous attempts to knock out the cdd1 gene failed due to the extreme instability, in C. difficile ATCC 43255, of the vectors used for generation of knockouts. We succeeded, however, in creating the cdd2 gene knockout in C. difficile 630Δerm using both approaches. Inactivation of the gene abolished the β-lactamase activity as confirmed by the nitrocefin test (Fig. 2E). The MICs of β-lactams against C. difficile 630Δerm strain harboring either of the two generated cdd2 gene knockouts (cdd2 intron and Δcdd2) were identical (Table 1). We observed a 4-fold decrease in the MIC values for ampicillin, penicillin G, and ceftazidime and a 2-fold decrease for ceftriaxone and cefepime, while the MICs of other antibiotics remained unchanged. This correlates with poor expression of the gene under the control of its own promoter, which we also observed in C. cochlearium. These data demonstrated that while C. difficile 630Δerm encodes a class D β-lactamase that has high catalytic activity against various β-lactam substrates, the enzyme is poorly expressed in this strain and responsible only partially for its resistance to these antibiotics. Results of our studies also indicate that an additional, as-yet-unidentified mechanism in C. difficile 630Δerm contributes significantly to the observed high-level resistance to β-lactams.
We then aimed to reintroduce the cdd2 gene into the C. difficile 630Δerm strain harboring the Δcdd2 gene knockout and to evaluate the effect on the produced resistance. For this purpose, we used the pMTL83151::*cdd2 construct, where the gene is expressed under the control of its own promoter, and also constructed the pMTL83122::cdd2-Pthl plasmid, where the cdd2 gene is expressed under the control of the efficient Pthl promoter. We then measured the MICs of the two β-lactams, ampicillin and ceftriaxone, against the resulting transconjugants. For the transconjugant harboring the pMTL83151::*cdd2 plasmid, we observed only a 2-fold increase in the MICs for one of antibiotics, ampicillin, while for the transconjugant carrying the pMTL83122::cdd2-Pthl construct, the MIC of ampicillin increased 16-fold and that of ceftriaxone 2-fold (data not shown). These data clearly show that the cdd2 gene is expressed more efficiently under the control of the Pthl promoter than under that of its own promoter, as was also the case with the cdd1 gene in C. cochlearium. The lower magnitude of the increase in antibiotic resistance in C. difficile could have resulted from both its significant level of residual β-lactam resistance and the high degree of instability of the constructs that we observed in this host.
DISCUSSION
In our efforts to elucidate the mechanism of resistance of C. difficile to β-lactam antibiotics, we discovered that this pathogen encodes intrinsic class D β-lactamases which share a high level of identity of their amino acid sequences. Antibiotic susceptibility testing of two clostridial isolates, C. difficile ATCC 43255 and C. difficile 630Δerm, showed that they are resistant to expanded-spectrum cephalosporins and the cephamycin cefoxitin and are highly resistant to the monobactam aztreonam but are sensitive to both penicillins and carbapenems. Our kinetic studies of CDD β-lactamases demonstrated that both CDD-1 and CDD-2 possess high catalytic activity against a wide spectrum of β-lactam antibiotics. Comparison of the antibiotic resistance and catalytic profiles of CDD-1 revealed some noticeable discrepancies between them (Tables 1 and 3). While the pathogen was highly resistant to cefoxitin, CDD-1 had very low catalytic efficiency against this substrate, which indicates that the high MIC of cefoxitin likely resulted from low affinity of the drug to its target PBP. The opposite trend was observed for ampicillin, penicillin G, oxacillin, and cephalothin. Although CDD-1 has high catalytic efficiency against these substrates, C. difficile ATCC 43255 is relatively sensitive to these antibiotics. Such sensitivity could result from poor expression of the CDD-1 β-lactamase.
To validate this assumption, we knocked out the cdd2 gene and observed only moderate decreases in the MICs of β-lactams. Subsequent reintroduction of the cdd2 gene under the control of its own and the strong Pthl promoters confirmed that the β-lactamase is indeed poorly expressed under the control of its own promoter. In addition, we expressed the genes for the CDD-1 and CDD-2 β-lactamases in the highly β-lactam-sensitive C. cochlearium ATCC 17787 strain and found that both enzymes were also expressed poorly from their own promoters in this bacterium.
Our study also demonstrated that the catalytic efficiency or substrate profile or both of the CDD enzymes are unique among class D β-lactamases. Although CDD-1 has a substrate profile similar to that of the only other characterized class D β-lactamase from Gram-positive bacteria, BPU-1, it has superior (up to 200-fold-higher) catalytic efficiency against expanded-spectrum cephalosporins and aztreonam (28). While the substrate profile of CDD-1 is much broader than that of mutants of OXA-2 and OXA-10 β-lactamases from Pseudomonas aeruginosa (39, 40), it is more similar to those of some mutant class D β-lactamases from Acinetobacter baumannii (41 – 44). However, as with BPU-1, the catalytic activity of CDD-1 is much higher. Our analysis demonstrates that, among characterized class D β-lactamases, CDD-1 overall displays the broadest spectrum and the highest level of activity against expanded-spectrum cephalosporins and aztreonam. These results strongly indicate that active site of CDD-1 has a unique architecture that evolved to accommodate and efficiently hydrolyze a wide spectrum of β-lactam substrates.
Combined, our experiments showed that C. difficile strains produce intrinsic broad-spectrum CDD β-lactamases. In the two studied strains, the enzymes are poorly expressed and responsible only partially for the observed β-lactam resistance. This implies that these strains also employ an additional, non-CDD β-lactamase-mediated mechanism(s) of resistance. Importantly, our studies demonstrated that the CDD β-lactamases are very potent enzymes, and when one of them, CDD-1, was supplied with functional promoters, it produced high-level, broad-spectrum resistance to β-lactams. Further experiments are warranted to determine whether CDD β-lactamases with efficient promoters are circulating in any of the multiple clinical C. difficile isolates. In fact, acquisition of strong promoters has already been well documented with class D β-lactamase genes of Acinetobacter baumannii. Originally, these genes were poorly expressed and were of no clinical concern. However, over time, they acquired stronger promoters supplied by insertion sequences, which rendered A. baumannii highly resistant to carbapenem antibiotics. As C. difficile also harbors multiple mobile genetic elements, which constitute up to 11% of its genome (45), the acquisition of strong promoters by the cdd genes is highly likely. More-efficient promoters would provide even higher levels and broader spectra of resistance to β-lactam antibiotics. Indeed, clinical C. difficile isolates with such high levels of resistance to β-lactams have already been reported (20, 46, 47). Regardless, these intrinsic enzymes pose a serious potential threat, as they are fully capable of producing a high level of resistance to clinically important antibiotics, which would further complicate the already difficult tasks of preventing and treating C. difficile infection.
MATERIALS AND METHODS
Bacterial strains.
E. coli DH10B (New England Biolabs) was used in cloning experiments. E. coli CA434 (CHAIN Biotech, United Kingdom) was used as the donor strain for mobilization of shuttle vectors into clostridial strains. C. difficile 630Δerm (CHAIN Biotech, United Kingdom) and C. difficile ATCC 43255 are known toxigenic isolates, widely used to study various aspects of C. difficile infection. C. cochlearium ATCC 17787 was used for expression of cdd genes. All clostridial isolates were grown under anaerobic conditions at 37°C.
Antibiotic susceptibility testing.
MICs of β-lactam antibiotics against clostridial strains were determined according to Clinical and Laboratory Standards Institute guidelines (M11-A8) (48). Briefly, the broth microdilution method was used with 96-well plates. Serial dilutions of antibiotics were performed in supplemented Brucella medium, which subsequently was inoculated with the final bacterial inoculum of 106 CFU/ml. Plates were incubated for 44 to 48 h at 37°C in an anaerobic box prior to recording results. All MIC experiments were performed at least in triplicate.
Identification of β-lactamases in C. difficile.
To identify β-lactamases in C. difficile ATCC 43255 and C. difficile 630Δerm, we analyzed their genomic sequences (available at https://www.ncbi.nlm.nih.gov). Visual inspection of the amino acid sequences of these enzymes was used to identify conserved motifs of the class D β-lactamases. Around 800 completed genomic sequences of C. difficile (available at www.patricbrc.org) were analyzed to appreciate the spread and diversity of CDD enzymes.
Nitrocefin test.
To assess whether clostridia produced active β-lactamases, we utilized the chromogenic cephalosporin nitrocefin, which changes color from yellow to dark red upon hydrolysis. Overnight bacterial cultures were used for the analysis of bacterial cultures, pellets resuspended in phosphate buffer, and supernatants. Reactions performed with nitrocefin (200 µM) were monitored for up to 60 min. Sodium bicarbonate and EDTA were used at 50 mM and 10 mM, respectively, where indicated.
Cloning procedures.
For expressing the CDD-1 β-lactamase in C. cochlearium ATCC 17787, we placed the full-length cdd1 gene under the control of either the Pthl promoter or Pfdx promoter (34, 35) by cloning it between the NdeI and HindIII restriction sites of the pMTL83122 and pMTL83123 shuttle vectors (49) to generate pMTL83122::cdd1 and pMTL83123::cdd1, respectively.
To evaluate whether there are functional promoters upstream of the cdd1 and cdd2 genes, we PCR amplified DNA fragments containing the entire genes and their 182-bp and 177-bp 5′ flanking regions, respectively. We subsequently cloned the amplified fragments between the BamHI and HindIII restriction sites of the pMTL83151 vector to generate pMTL83151::*cdd1 and pMTL83151::*cdd2, respectively.
To construct the cdd1 and cdd2 gene knockouts, we utilized two alternative methodologies. The ClosTron system achieves gene inactivation by the insertion of a group II intron (38), while a two-step allele exchange technique generates targeted deletions of genetic material (36). For the insertional mutagenesis, the introns were retargeted using an online tool (www.clostron.com), custom synthesized, and inserted into the pMTL007C-E5 vector (ATUM, CA), resulting in pMTL007C-E5::Cdi-cdd1-284a and pMTL007C-E5::Cdi-cdd2-272a. For the deletional inactivation of the cdd1 and cdd2 genes, we amplified regions upstream (left arm) and downstream (right arm) of the gene fragment to be deleted. The left arm included the first 30 bp of the genes and 759 bp of their upstream region. The right arm consisted of the 511 bp of the 3′ region of the cdd genes plus 560 bp downstream. Use of this construct is expected to create deletions of 398 bp and 386 bp in the cdd1 and cdd2 genes, respectively. The arms were ligated, and the product was cloned into the PmeI restriction site of the pMTL-SC7315 vector to result in pMTL-SC7315::Δcdd1 and pMTL-SC7315::Δcdd2.
To produce a large amount of the CDD-1 β-lactamase, the gene for the mature enzyme was custom synthesized (GenScript) and cloned into the NdeI and HindIII sites of the pET-24a(+) expression vector (Novagen). The mature enzyme lacks the first 58 N-terminal amino acids, whose deletion is necessary for the optimal expression of the enzyme, as we have shown previously for the BPU-1 class D β-lactamase from another Gram-positive bacterium, Bacillus pumilus (28).
Conjugation experiments.
Various shuttle vectors were delivered to clostridial cells using an established protocol (49). Briefly, 1 ml of overnight culture of donor E. coli CA434 culture harboring a shuttle vector was pelleted and resuspended in 200 µl of the appropriate C. difficile or C. cochlearium recipient strains. The bacterial suspension was spotted onto the surface of brain heart infusion-supplemented (BHIS) agar and incubated anaerobically overnight at 37°C. Next, grown cells were resuspended in 500 µl BHIS medium and spread on selective plates to recover transconjugants. For C. cochlearium, selection was modified by using 12 µg/ml aztreonam instead of cefoxitin. After 3 days of incubation and subsequent restreaking, individual colonies were grown in selective BHIS medium overnight. DNA was isolated using a DNeasy blood & tissue kit (Qiagen) and analyzed by PCR to identify transconjugants.
Inactivation of cdd1 and cdd2 genes using ClosTron mutagenesis.
The mutagenesis procedure was performed as described earlier (37). Briefly, following introduction of pMTL007C-E5::Cdi-cdd1-284a and pMTL007C-E5::Cdi-cdd2-272a into C. difficile ATCC 43255 and C. difficile 630Δerm, respectively, the transconjugants were selected on BHIS agar supplemented with 20 µg/ml lincomycin and 250 µg/ml d-cycloserine overnight at 37°C. Next, 120 individual colonies were restreaked onto the same medium, and the resulting individual colonies were grown in BHIS agar supplemented with 20 µg/ml lincomycin for DNA isolation. Insertion of the intron into the cdd genes was verified by PCR and sequencing.
Inactivation of cdd1 and cdd2 genes by using the two-step allele exchange procedure.
The protocol was used as previously described (36). pMTL-SC7315::Δcdd1 and pMTL-SC7315::Δcdd2 were introduced into C. difficile ATCC 43255 and C. difficile 630Δerm, respectively, by conjugation. The transconjugants were restreaked onto TCC plates (BHIS agar supplemented with 15 µg/ml thiamphenicol, 10 µg/ml cefoxitine, and 250 µg/ml d-cycloserine), and the DNA from larger colonies was isolated and checked by PCR for integration of the plasmid into the chromosome. An integrant clone was then restreaked onto nonselective BHIS agar plates, and the cells were harvested in 0.5 ml phosphate-buffered saline (PBS) after 3 to 4 days of incubation and spread onto C. difficile minimal medium (CDMM) plates supplemented with 75 µg/ml 5-fluorocytosine after serial 102 to 105 dilutions. Plates with individual colonies were replica plated onto BHIS agar with and without 15 µg/ml thiamphenicol to identify clones that lost the plasmid. Deletion of the cdd genes was confirmed by PCR and sequencing.
Protein expression and purification.
For expression of the CDD-1 enzyme, BL21(DE3) cells harboring the plasmid with the cdd gene were grown at 37°C with shaking to an optical density at 600 nm (OD600) of 0.6. Expression was induced with 0.5 mM isopropyl β-d-thiogalactoside (IPTG), and the cells were grown overnight at 22°C. The cells were harvested by centrifugation, resuspended in 25 mM HEPES (pH 7.0), 1 mM EDTA, and 0.2 mM dithiothreitol (DTT), and disrupted by sonication. Subsequently, the lysate was centrifuged at 32,000 rpm at 4°C and the supernatant was loaded onto a DEAE anion exchange column (Bio-Rad) preequilibrated with the same buffer. The protein was eluted with a 0 to 250 mM NaCl gradient. The enzyme was concentrated using a Centricon Plus 70 concentrator (Millipore), and dialyzed against 25 mM HEPES (pH 7.0) using a 10-kDa-molecular-weight-cutoff (MWCO) membrane (Spectrum).
Enzyme kinetics.
All data were collected on a Cary 60 spectrophotometer (Agilent). Reaction conditions were identical to those previously reported for class D β-lactamases (50). Observed rates were calculated from the linear phase of each reaction and fitted to the Michaelis-Menten equation using GraphPad Prism 5.04 for Windows (GraphPad Software, CA) to determine the steady-state parameters kcat and Km. Standard deviations were calculated using the same software.
ACKNOWLEDGMENTS
This work was supported by grant R01AI114668 from the NIH/NIAID (to S.B.V.) and by the Pilot Project Grant Program from the Eck Institute for Global Health (to S.B.V.).
Footnotes
Citation Toth M, Stewart NK, Smith C, Vakulenko SB. 2018. Intrinsic class D β-lactamases of Clostridium difficile. mBio 9:e01803-18. https://doi.org/10.1128/mBio.01803-18.
REFERENCES
- 1.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubberke ER, Olsen MA. 2012. Burden of Clostridium difficile on the healthcare system. Clin Infect Dis 55(Suppl 2):S88–S92. doi: 10.1093/cid/cis335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, Lynfield R, Maloney M, McAllister-Hollod L, Nadle J, Ray SM, Thompson DL, Wilson LE, Fridkin SK; Emerging Infections Program Healthcare-Associated Infections and Antimicrobial Use Prevalence Survey Team. 2014. Multistate point-prevalence survey of health care-associated infections. N Engl J Med 370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller BA, Chen LF, Sexton DJ, Anderson DJ. 2011. Comparison of the burdens of hospital-onset, healthcare facility-associated Clostridium difficile infection and of healthcare-associated infection due to methicillin-resistant Staphylococcus aureus in community hospitals. Infect Control Hosp Epidemiol 32:387–390. doi: 10.1086/659156. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett JG, Gerding DN. 2008. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis 46(Suppl 1):S12–S18. doi: 10.1086/521863. [DOI] [PubMed] [Google Scholar]
- 6.Kuijper EJ, van Dissel JT, Wilcox MH. 2007. Clostridium difficile: changing epidemiology and new treatment options. Curr Opin Infect Dis 20:376–383. [DOI] [PubMed] [Google Scholar]
- 7.Lo Vecchio A, Zacur GM. 2012. Clostridium difficile infection: an update on epidemiology, risk factors, and therapeutic options. Curr Opin Gastroenterol 28:1–9. doi: 10.1097/MOG.0b013e32834bc9a9. [DOI] [PubMed] [Google Scholar]
- 8.Luo R, Barlam TF. 2018. Ten-year review of Clostridium difficile infection in acute care hospitals in the USA, 2005–2014. J Hosp Infect 98:40–43. doi: 10.1016/j.jhin.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, Monnet DL, van Dissel JT, Kuijper EJ; ECDIS Study Group. 2011. Clostridium difficile infection in Europe: a hospital-based survey. Lancet 377:63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- 10.Hensgens MP, Goorhuis A, Dekkers OM, Kuijper EJ. 2012. Time interval of increased risk for Clostridium difficile infection after exposure to antibiotics. J Antimicrob Chemother 67:742–748. doi: 10.1093/jac/dkr508. [DOI] [PubMed] [Google Scholar]
- 11.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 12.Knecht H, Neulinger SC, Heinsen FA, Knecht C, Schilhabel A, Schmitz RA, Zimmermann A, dos Santos VM, Ferrer M, Rosenstiel PC, Schreiber S, Friedrichs AK, Ott SJ. 2014. Effects of β-lactam antibiotics and fluoroquinolones on human gut microbiota in relation to Clostridium difficile associated diarrhea. PLoS One 9:e89417. doi: 10.1371/journal.pone.0089417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keeney KM, Yurist-Doutsch S, Arrieta MC, Finlay BB. 2014. Effects of antibiotics on human microbiota and subsequent disease. Annu Rev Microbiol 68:217–235. doi: 10.1146/annurev-micro-091313-103456. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Cobas AE, Moya A, Gosalbes MJ, Latorre A. 2015. Colonization resistance of the gut microbiota against Clostridium difficile. Antibiotics (Basel) 4:337–357. doi: 10.3390/antibiotics4030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CDC. 2013. Antibiotic resistance threats in the United States. www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. [PubMed]
- 16.Spencer RC. 1998. The role of antimicrobial agents in the aetiology of Clostridium difficile-associated disease. J Antimicrob Chemother 41(Suppl C):21–27. doi: 10.1093/jac/41.suppl_3.21. [DOI] [PubMed] [Google Scholar]
- 17.Gerding DN. 2004. Clindamycin, cephalosporins, fluoroquinolones, and Clostridium difficile-associated diarrhea: this is an antimicrobial resistance problem. Clin Infect Dis 38:646–648. doi: 10.1086/382084. [DOI] [PubMed] [Google Scholar]
- 18.Oldfield EC., III. 2004. Clostridium difficile-associated diarrhea: risk factors, diagnostic methods, and treatment. Rev Gastroenterol Disord 4:186–195. [PubMed] [Google Scholar]
- 19.Spigaglia P. 2016. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther Adv Infect Dis 3:23–42. doi: 10.1177/2049936115622891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilcox MH, Chalmers JD, Nord CE, Freeman J, Bouza E. 2017. Role of cephalosporins in the era of Clostridium difficile infection. J Antimicrob Chemother 72:1–18. doi: 10.1093/jac/dkw385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 22.Muto CA, Pokrywka M, Shutt K, Mendelsohn AB, Nouri K, Posey K, Roberts T, Croyle K, Krystoflak S, Patel-Brown S, Pasculle AW, Paterson DL, Saul M, Harrison LH. 2005. A large outbreak of Clostridium difficile-associated disease with an unexpected proportion of deaths and colectomies at a teaching hospital following increased fluoroquinolone use. Infect Control Hosp Epidemiol 26:273–280. doi: 10.1086/502539. [DOI] [PubMed] [Google Scholar]
- 23.Owens RC Jr, Donskey CJ, Gaynes RP, Loo VG, Muto CA. 2008. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis 46(Suppl 1):S19–S31. doi: 10.1086/521859. [DOI] [PubMed] [Google Scholar]
- 24.Bignardi GE. 1998. Risk factors for Clostridium difficile infection. J Hosp Infect 40:1–15. doi: 10.1016/S0195-6701(98)90019-6. [DOI] [PubMed] [Google Scholar]
- 25.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Δerm) and demonstration that the conjugative transposon Tn916ΔE enters the genome of this strain at multiple sites. J Med Microbiol 54:137–141. doi: 10.1099/jmm.0.45790-0. [DOI] [PubMed] [Google Scholar]
- 27.Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, Young VB. 2015. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun 83:934–941. doi: 10.1128/IAI.02768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toth M, Antunes NT, Stewart NK, Frase H, Bhattacharya M, Smith CA, Vakulenko SB. 2016. Class D β-lactamases do exist in Gram-positive bacteria. Nat Chem Biol 12:9–14. doi: 10.1038/nchembio.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc Natl Acad Sci U S A 98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez LJ, Bahr G, Nakashige TG, Nolan EM, Bonomo RA, Vila AJ. 2016. Membrane anchoring stabilizes and favors secretion of New Delhi metallo- β-lactamase. Nat Chem Biol 12:516–522. doi: 10.1038/nchembio.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen JB, Caulfield MP, Lampen JO. 1981. Lipoprotein nature of Bacillus licheniformis membrane penicillinase. Proc Natl Acad Sci U S A 78:3511–3515. doi: 10.1073/pnas.78.6.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen JB, Lampen JO. 1982. Membrane-bound penicillinases in Gram-positive bacteria. J Biol Chem 257:4490–4495. [PubMed] [Google Scholar]
- 33.Randall LB, Dobos K, Papp-Wallace KM, Bonomo RA, Schweizer HP. 2016. Membrane-bound PenA β-lactamase of Burkholderia pseudomallei. Antimicrob Agents Chemother 60:1509–1514. doi: 10.1128/AAC.02444-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Girbal L, Mortier-Barriere I, Raynaud F, Rouanet C, Croux C, Soucaille P. 2003. Development of a sensitive gene expression reporter system and an inducible promoter-repressor system for Clostridium acetobutylicum. Appl Environ Microbiol 69:4985–4988. doi: 10.1128/AEM.69.8.4985-4988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takamizawa A, Miyata S, Matsushita O, Kaji M, Taniguchi Y, Tamai E, Shimamoto S, Okabe A. 2004. High-level expression of clostridial sialidase using a ferredoxin gene promoter-based plasmid. Protein Expr Purif 36:70–75. doi: 10.1016/j.pep.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78:4683–5690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods 80:49–55. doi: 10.1016/j.mimet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Antunes NT, Fisher JF. 2014. Acquired class D β-lactamases. Antibiotics (Basel) 3:398–434. doi: 10.3390/antibiotics3030398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans BA, Amyes SG. 2014. OXA β-lactamases. Clin Microbiol Rev 27:241–263. doi: 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harper TM, June CM, Taracila MA, Bonomo RA, Powers RA, Leonard DA. 2018. Multiple substitutions lead to increased loop flexibility and expanded specificity in Acinetobacter baumannii carbapenemase OXA-239. Biochem J 475:273–288. doi: 10.1042/BCJ20170702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaitany KC, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA. 2013. Structures of the class D carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob Agents Chemother 57:4848–4855. doi: 10.1128/AAC.00762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell JM, Clasman JR, June CM, Kaitany KC, LaFleur JR, Taracila MA, Klinger NV, Bonomo RA, Wymore T, Szarecka A, Powers RA, Leonard DA. 2015. Structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbapenem-hydrolyzing class D β-lactamases from Acinetobacter baumannii. Biochemistry 54:1976–1987. doi: 10.1021/bi501547k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroder EC, Klamer ZL, Saral A, Sugg KA, June CM, Wymore T, Szarecka A, Leonard DA. 2016. Clinical variants of the native class D β-lactamase of Acinetobacter baumannii pose an emerging threat through increased hydrolytic activity against carbapenems. Antimicrob Agents Chemother 60:6155–6164. doi: 10.1128/AAC.01277-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 46.Buchler AC, Rampini SK, Stelling S, Ledergerber B, Peter S, Schweiger A, Ruef C, Zbinden R, Speck RF. 2014. Antibiotic susceptibility of Clostridium difficile is similar worldwide over two decades despite widespread use of broad-spectrum antibiotics: an analysis done at the University Hospital of Zurich. BMC Infect Dis 14:607. doi: 10.1186/s12879-014-0607-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noren T, Alriksson I, Akerlund T, Burman LG, Unemo M. 2010. In vitro susceptibility to 17 antimicrobials of clinical Clostridium difficile isolates collected in 1993–2007 in Sweden. Clin Microbiol Infect 16:1104–1110. doi: 10.1111/j.1469-0691.2009.03048.x. [DOI] [PubMed] [Google Scholar]
- 48.CLSI. 2012. Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard—8th ed. CLSI document M11-A8. Clinical and Laboratory Standards Institute, Wayne, Pa. [Google Scholar]
- 49.Heap JT, Pennington OJ, Cartman ST, Minton NP. 2009. A modular system for Clostridium shuttle plasmids. J Microbiol Methods 78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Toth M, Smith CA, Antunes NT, Stewart NK, Maltz L, Vakulenko SB. 2017. The role of conserved surface hydrophobic residues in the carbapenemase activity of the class D β-lactamases. Acta Crystallogr D Struct Biol 73:692–701. doi: 10.1107/S2059798317008671. [DOI] [PMC free article] [PubMed] [Google Scholar]