

Abstract
Protocols for preparing and culturing primary keratinocytes from newborn and adult mouse epidermis have evolved over the last 35 years. The present protocol is now routinely applied to mice of various genetic backgrounds for in vitro studies of signaling pathways in differentiation and in cell transformation, and for assessing the in vivo phenotype of altered keratinocytes in grafts of cells on immunodeficient mice. Crucial in the development and application of the procedure was the observation that keratinocytes proliferate in media of low calcium concentration, but rapidly commit to differentiation at calcium concentrations greater than 0.07 mM after the initial attachment period. Preparing primary keratinocytes from 10 newborn mice requires from 2 to 3 hours of hands-on time. Related procedures are also provided: preparing immature hair follicle buds, developing dermal hair follicles and fibroblasts from newborn mice, preparing primary keratinocytes from adult mice, and grafting cell mixtures on athymic nude mice.
Introduction
The mammalian skin is a complex organ where controlled spacial and temporal growth and differentiation are vital for survival of the organism. Furthermore, skin appendages have evolved for specialized functions that distinguish mammals from other land vertebrates. The ease of manipulating differentiation of keratinocytes from mouse skin in culture by controlling the calcium concentration in the medium1–3 has greatly advanced the understanding of the controls on epidermal proliferation and differentiation. Further, this model system has provided a means to dissect the cellular and molecular basis for epidermal pathology such as neoplastic transformation induced by chemical initiation and promotion, by introduction of oncogenes or other disease related genetic changes4, and the consequences of pharmacological exposures5–7. Remarkably, the discovery that calcium concentration is a major determinant of the differentiation state of mammalian keratinocytes in vitro parallels the finding that a similar calcium gradient is maintained across the epidermis in the living skin8,9.
While other techniques have been reported that support the growth and maintenance of mouse keratinocytes for limited periods of time10–12, the reduced calcium model has proven to be the most effective for probing the pathways that regulate basal keratinocyte physiology. The steps of this protocol for preparing and culturing primary keratinocytes from newborn mice are easily mastered. The procedure has been performed on numerous strains of mice, including genetically altered mice with defects that limit survival after birth. Depending on mouse strain, epidermal cells from one newborn to 2-day old mouse yield 3 to 4 nearly confluent 60 mm culture dishes of keratinocytes by 3 days after plating. In brief, the dermis easily separates from the epidermis at the basement membrane after floating skin on cold trypsin overnight (16-18 hours). All cell fractions are prepared with cold medium containing 8% fetal bovine serum and 1.3 mM calcium (HiCa medium) and are kept on ice between manipulations. The epidermis is minced with scissors in cold medium, the resulting suspension is triturated by pipetting to dissociate cell clumps, and stratum corneum sheets are removed by filtration of the suspension through a 100 μm mesh filter. The resulting cell suspension containing basal and suprabasal single cells and cell clumps as well as immature hair follicle buds is plated on tissue culture plastic and incubated overnight either in 0.2 mM calcium medium for more rapid attachment or in LoCa medium. By 8 to 18 hours later, basal cells and hair follicle buds have attached and spread and show typical epithelioid morphology. Hair follicle buds contribute 50-60% of the attached population. Loosely attached or floating rounded cells and cell aggregates, representing suprabasal and cornifying cells, are removed by washing with PBS before feeding the attached cells with maintenance medium containing 0.05 mM calcium and 8% Chelex-treated fetal bovine serum (LoCa medium). Medium is changed every 2 days.
The biology of basal keratinocytes in vivo is most closely reflected by that of primary keratinocytes during the first week to 10 days of culture in LoCa medium. By 4 days after plating most of the initially attaching cells that were already committed to differentiation have detached so that the remaining attached cells show typical cobblestone morphology1, are proliferating, and are negative for suprabasal differentiation markers3. By 10 days in culture, heterogeneity in morphology begins to become obvious, suggesting the onset of senescence. Therefore most experiments are performed between 4 and 7 days of culture when results are generally most reproducible from experiment to experiment. The useful culture life varies between mouse strains with BALB/c keratinocytes having the lowest initial plating efficiency and the highest propensity to senesce compared to FVB/N, CD-1, C57BL/6, or SENCAR. Cell proliferation under low calcium conditions can be prolonged by the use of fibroblast-conditioned medium or by addition of growth factors (KGF or EGF). Limited attempts in this laboratory to derive stable clonally-derived cell lines in fibroblast conditioned medium or growth factor-supplemented medium gave rise to independent cell lines that often differed markedly from each other, suggesting a degree of genetic instability during prolonged culture13. This is borne out by the finding that keratinocyte cell lines readily become near-tetraploid even at early passage in growth factor-supplemented medium.14–16
An integral part of the protocol is the procedure for removing calcium from fetal bovine serum by exposure to a chelating resin on a scale that is appropriate for the needs of the laboratory. Prior to purchase of serum, it is necessary to obtain several different serum lots from different sources, which, after treating with the chelating resin, are tested for support of growth of healthy keratinocytes over a 2 to 3 week period.
A commercially available potential alternative to LoCa medium is a serum-free keratinocytes medium available from Lonza (Cat.# CC-3108, KGM-2 w/o Ca++, “BulletKit”) consisting of a calcium-free basal medium plus a kit containing growth factors, cytokines, and supplements consisting of epinephrine, transferrin, bovine pituitary extract, EGF, insulin, hydrocortisone, and antibiotic in individual vials. Optimum concentrations of calcium and of the other factors for growing murine keratinocytes would need to be determined. This medium has not been tested in this laboratory as an alternative to the LoCa medium. A related medium, KBM-2, containing 0.15 mM calcium (Lonza Cat# CC-3103) without additional growth factors was used by Castilho et al.17 for short term in vitro studies on primary keratinocytes from newborn mice prepared according to this protocol.
If the aim is to prepare and culture primary epidermal cells from newborn mice for short term studies, only steps 1-40 of this protocol need to be considered. However, in a laboratory devoted to various aspects of skin research, there are likely to be needs for subpopulations of cells from the epidermis and dermis not only from newborn mice, but also from adult mice. For this reason, this protocol also includes complementary procedures for preparing primary keratinocytes from dorsal skin and from tail skin of adult mice4 (Box 1 and Box 2), and procedures for preparing several subfractions of dermal and epidermal cells from newborn mice that may be useful in the context of this protocol. These are single dermal cells, primarily fibroblasts (steps 41-58) and intermediate size developing hair follicles (steps 59-64) both obtained from the dermis after digestion with collagenase, and immature hair follicle buds from the epidermal cell fraction as crude hair follicle buds suitable for grafting18 (steps 65-73), and as more purified hair follicle buds suitable for collagen matrix co-cultures (steps 74-81) to study the in vitro consequences of signaling between mesenchymal cells and hair follicles19,20. Freshly prepared dermal cells are grafted in combination with hair follicles to reconstitute haired skin which depends on the dermal papilla cells contained in the dermal cell preparation18. One week cultured dermal cells are essentially devoid of functional dermal papilla cells and are used in combination with variously modified test keratinocytes as source of mesenchymal cells to reconstitute a transgenic or a hybrid skin or tumors in grafts on athymic nude mice in order to determine the in vivo phenotype of the test keratinocytes21,22. The steps of the grafting procedure are included in Box 3.
Box 1: Preparation of primary keratinocytes from adult dorsal skin of mice in the resting phase of the hair cycle4 TIMING up to 5 hrs for 5 mice with interruptions.
Separation of epidermis from dermis of adult mouse skin is only possible if the area to be excised is in the resting phase of the hair cycle (telogen), which is usually the case for back skin between age of 6 to 12 weeks after birth. Telogen must be verified in white mice by clipping the hair on the back skin and observing no hair growth for 3 days. In mice with pigmented hair, the skin after clipping the hair will appear to be pink during the resting phase of the hair cycle, and various shades of grey to black during the growing phase.
Additional Supplies and Reagents
Autoclaved porcelain spot plate
Several autoclaved 12.5 cm diameter Whatman #1 filters
Filter sterilized solution of 0.025 M NH4OH / 0.5% (vol/vol) of TritonX100 in PBS
Filter sterilized 1 mM EDTA in PBS
Procedure
Euthanize mice by deep CO2 narcosis and treat the shaved area of the dorsal skin with depilatory cream (Nair) for about 3 min.
Remove excess Nair with cotton gauze and remove remaining Nair under running water while rubbing the area gently with gauze.
Wash entire mouse with Betadine, de-ionized water and 70% ethanol as for newborn mice.
Remove Naired back skin area with sterile instruments and place epidermis side down on a sterile surface such as the rough underside of an autoclaved porcelain spot plate.
Using a scalpel blade or single edged razor blade, carefully scrape off the hypodermis while holding an edge of the skin in place with the edge of a Scoopula type sterile spatula. Skin should be stretched flat at this point.
Apply a piece of autoclaved Whatman #1 filter paper, somewhat larger than the piece of skin, to the now exposed surface of the dermis.
Cut the skin including the attached paper into 1 cm wide strips using sterile scissors and place, with the paper still attached, dermis side down on the surface of recently thawed cold 0.25% trypsin overnight at 4°C or on 1% trypsin in PBS for 1h at 37°C.
Peel off epidermis and/or scrape off epidermal cells with the rounded edge of curved forceps into HiCa medium and proceed as for newborn epidermis (steps 26 – 34). Depending on the size of skin excised, the yield is from 5 to 10 million cells per mouse.
For culture, plate cells in LoCa medium on Fibronectin/Collagen–coated dishes or on extracellular matrix deposited by newborn mouse keratinocytes on plastic culture dishes. Coating culture dishes with the Fibronectin/Collagen mixture is achieved by wetting the culture surface with an excess of the solution, removing the excess with the same pipette, and incubating the coated dishes for 30 min in the incubator. Extracellular matrix coated dishes covered with PBS can be prepared at any time and stored at 4°C until needed: rinse confluent monolayers of newborn mouse keratinocytes (4-6 days after plating) with PBS and incubate at room temperature (20 to 25 °C) with a solution of 0.025 M NH4OH/0.5% Triton ×100 (vol/vol) in PBS for 1 to 5 min while monitoring the dissolution of the soluble portion of the cells under an inverted phase microscope. Remove insoluble cell debris by several washes with 1 mM EDTA in PBS, followed by several washes with PBS.
Box 2: Preparation of primary keratinocytes from adult mouse tail skin4 TIMING up to 5 hrs with interruptions.
Keratinocytes can be isolated from tail skin at any time, regardless of age of mice, because the hair density on tails is too low to interfere with separation of dermis from epidermis.
Remove tails from euthanized mice that had been washed with Betadine and alcohol (see Box 1)
Pull skin from tail starting at the base toward the tip.
Cut the tube of skin into 3 pieces, discarding the tip.
Open each tube with a longitudinal cut and flatten the skin dermis side down on the culture surface of a culture dish. Subsequent steps are the same as for newborn mouse skin, steps 16-34. The yield of keratinocytes is from 5-10 million cells per tail.
Plate the keratinocytes on fibronectin/collagen coated dishes or extracellular matrix coated dishes as for keratinocytes from adult telogen back skin (Box 1).
Preparation of mouse fibroblasts from newborn mouse dermises TIMING 3 hrs for 5 dermises
-
41.
Rinse dermises (from Step 24) with fibroblast medium (Fb medium).
-
42.
Transfer rinsed dermises to a suitable container (e.g. 125 ml or 250 ml sterile Erlenmeyer flask) and add 2 ml 0.35 % collagenese per dermis. ! CAUTION For adequate agitation ( in step 43), the height of the liquid level in the flask should be no more and 1 cm .
-
43.
Incubate dermises with agitation at 37°C in a shaking incubator or water bath set at low speed for 30 min.
-
44.
Add 12.5 μl DNASE per dermis and continue incubation for 10 min.
-
45.
Dilute dermal digest with Fb medium to 100 ml for 5 dermises and filter through 100μm mesh Nytex cloth placed into a funnel (removes undigested dermal fragments and very large hair follicles).
-
46.
Centrifuge the filtrate at 150g for 5 min – save the supernate and pellet.
-
47.
Centrifuge the 150g supernate from step 46 at 450g for 5 min to sediment dermal cells that did not sediment at 150g due to the viscosity of the dermal digest; save the pellet containing fibroblasts and discard the supernate.
-
48.
Resuspend the 150g pellet from step 46 with Fb medium and centrifuge at 20g for 3 min (save the pellet for purifying intermediate size dermal hair follicles, step 59ff, if desired); centrifuge the 20g supernate at 230g for 5 min, save pellet which contains the bulk of the dermal fibroblasts.
-
49.
Combine the resuspended 450g (step 47) and 230g (step 48) pellets and centrifuge at 230g for 5 min. Wash the resulting fibroblast pellet two more times with Fb medium by resuspension and centrifuging at 230g for 5 min.
-
50.
Resuspend the final fibroblast pellet in 10ml Fb medium/dermis equivalent and filter through fine (20 micron mesh) Nytex filter to remove any remaining hair follicles.
-
51.
Determine cell number per ml by counting a suitable aliquot. One newborn mouse dermis typically yields 30 to 50 million cells.
Culturing primary fibroblasts TIMING up to 3 hrs for 5 dermis equivalents depending on intended use
-
52.
Plate fresh dermal cells at 1.5 to 2 mouse equivalents of dermal cells per T150 or T175 in Fb medium and incubate at 36°C for 45 to 60 min to allow attachment of fibroblast-like cells . The dermal cell preparation contains primarily fibroblasts, but also other dermal cells, such as dermal papilla cells, as well as some epithelial cells detaching from developing hair follicles and sebaceous gland structures, and precursors to other cells such as adipocytes, melanocytes, endothelial cells, and nerve cells. Figure 2 shows the morphology of cells in the dermal cell preparation within 5 min after plating in Fb medium (Fig. 2e) and 3 days later (Fig. 2f). Rapidly attaching cells are obvious in Figure 2e, and two strings of elongated cells, probably representing other types of cells, are obvious in the lower right quadrant of Figure 2f. Depending on intended use, dermal cells can be used either uncultured, cultured overnight or cultured longer, and passaged. (See box 3, on grafting cells to athymic nude mice.) The following steps markedly reduce keratinocyte contamination in overnight cultured fresh dermal cells while retaining functional dermal papilla cells (needed for hair follicle reconstitution). This precaution to markedly reduce, if not eliminate the presence of keratinocytes in the dermal cell preparation, may be important when combining keratinocytes from one genetic background with fibroblasts of different genetic backgrounds to reconstitute a hybrid haired skin graft.
-
53.
After the 45 to 60 min attachment period, remove medium containing slowly attaching cells, such as keratinocytes, and rinse attached cells once gently with Fb medium, and discard both lots of medium.
-
54.
Feed attached cells with Fb medium and incubate overnight.
-
55.
Gentle trypsinization will release the fibroblasts, but leave any remaining keratinocytes attached; therefore, wash cells twice with PBS; expose cells to Trypsin/EDTA (5 ml/T150) for 1 min, then remove excess Trysin/EDTA.
-
56.
Incubate cells for an additional 1-2 min until 90% of the cells have rounded up.
-
57.
Add Fb medium and gently agitate flask to release the fibroblast like cells; transfer released fibroblasts to a 50 ml centrifuge tube and determine cell count.
-
58.
Under these conditions of trypsinization, very few if any keratinocytes are released. The extent of keratinocyte contamination at this stage can be qualitatively assessed by rinsing still attached cells with PBS, feeding them LoCa medium, and continuing incubation. After 3-4 days, keratinocyte colonies can easily be distinguished from any remaining fibroblast colonies by their characteristic culture morphologies.
Preparation and purification of dermal (intermediate size) hair follicles TIMING up to 3 hrs for 10 dermal equivalents
-
59.
Wash 20g pellet, saved from step 48, once by resuspension in HiCa medium and centrifugation at 20g for 3 min to reduce fibroblast contamination.
-
60.
Resuspend this 20g pellet in HiCa medium at 5 mouse equivalents/4 ml and mix with an equal volume of 9% Ficoll.
-
61.
Layer 8 ml of the resulting suspension of dermal follicles in 4.5% Ficoll over 5 ml 9% Ficoll in conical 15 ml tubes.
-
62.
Centrifuge the tubes at 40g for 5 min in a swinging bucket centrifuge.
-
63.
Remove and discard supernatants.
-
64.
Either take follicles in the pellet through another round of purification through the discontinuous Ficoll gradient centrifugation, or wash directly 3 times by suspension in HiCa medium and centrifugation at 20g for 3 min to remove Ficoll. The resulting dermal HF preparation is virtually free single cells, and contains developing hair follicles of various length and in addition structures that appear to be follicle fragments (Fig. 3e). When cultured for 3 days in LoCa medium, most of the cells have keratinocyte morphology; however, some of the cells are elongated and probably represent follicle associated cells, such as dermal papilla cells, melanocytes, and fibroblasts (Fig. 3f).
Preparation and purification of hair follicle buds from primary keratinocytes TIMING 5 min
-
65.
Centrifuge primary keratinocyte preparation (from step 31) at 20g for 3 min. to sediment hair follicle buds (HF buds). Resuspend pellet in HiCa medium and repeat the low speed centrifugation. The combined supernates represent primary keratinocytes from which HF buds have been removed. Figure 2 shows the morphology of this primary keratinocyte population, representing interfollicular keratinocytes, shortly after plating (Fig. 2c) and 3 days later (Fig. 2d).
-
66.
Resuspend the crude hair follicle bud pellet from step 65 in HiCa medium at no more than 20ME (mouse equivalents) of per 10ml.
-
67.
The procedure diverges at this point, depending on whether buds are to be used for grafting (see option A) or for culture (option B). Option B takes advantage of selective rapid attachment of hair follicle buds to fibronectin/collagen coated dishes, allowing separation from suprabasal cell clusters that would otherwise contaminate the HF bud preparation.
A) Buds suitable for grafting TIMING up to 2 hrs for 20 epidermis equivalents
Add an equal volume of 9% Ficoll and mix.
Layer 20 ml of this hair follicle bud suspension in 4.5% Ficoll over 12 ml of 9% Ficoll in a 50 ml conical tube
Centrifuge at 40g for 5 min in a swinging bucket rotor with low deceleration speed.
Remove all but 4 ml of supernatant containing single cells and small cell clumps, and dilute the remainder (in 9% Ficoll) with an equal volume of HiCa medium.
Mix and layer the 8 ml of resulting HF bud suspension in 4.5% Ficoll over 5 ml of 9% Ficoll in a 15 ml centrifuge tube.
Centrifuge 5 min at 40g and remove all of the supernatant.
Wash pellet(s) of HF buds 3 times with HiCa medium by resuspension and centrifugation at 20g for 3 min. The resulting HF bud preparation is virtually free of interfollicular basal epidermal cells, but contains suprabasal cell clumps and some granular cells (Fig. 3a). After 3 days of culture in LoCa medium, the cells are indistinguishable by morphology (Fig. 3b) from primary keratinocytes (Fig. 2b).
B) Buds suitable for collagen matrix coculture or for monolayer cultures TIMING up to 3 hrs for 20 epidermis equivalents
Coat the required number of 150 mm culture dishes with the Fibronectin/Collagen mixture by wetting the culture surface of each dish with about 10 ml of the solution and removing the excess before proceeding to coat the next dish. Incubate dishes 30 min in the incubator before applying the cells.
Plate a maximum of crude HF buds from 20 epidermises in 20-30 ml of HiCa medium per culture dish, taking care to distribute the follicles evenly (see !CAUTION under step 38), and incubate at 36°C.
After 1 hour of incubation, remove unattached cells and gently rinse attached follicle buds 2 times with HiCa medium to remove unattached cells.
Add 10 ml of HiCa medium to each dish and gently lift follicles with a Costar cell lifter. Transfer released follicle bud suspension to a centrifuge tube and combine with residual follicle buds recovered in two 10ml rinses of the dish.
Sediment follicle buds at 20g for 3 min. Remove all but 4 ml of supernate per 10 ME of HF buds in the pellet.
Add an equal volume of 9% Ficoll and mix gently.
Layer 8 ml of the resulting HF bud suspension in 4.5% Ficoll over 5 ml 9% Ficoll in a 15 ml centrifuge tubes and centrifuge at 40g for 5 min in a swinging bucket rotor.
Remove supernatant containing mostly single cells, and wash pellet of HF buds with HiCa medium 3 times by resuspension and centrifugation at 20g for 3 min. The resulting purified HF bud preparation is virtually free of interfollicular basal or suprabasal epidermal cells or cell clumps (Fig. 3c). After 3 days of culture in LoCa medium, the cells are indistinguishable by morphology (Fig. 3d) from that of primary keratinocytes (Fig. 2b).
Keep follicle preparation on ice until ready to use for intended application.
Box 3: Grafting of epidermis derived cells (freshly isolated, cultured, genetically modified, or cell lines) with mesenchymal cells (freshly isolated or cultured dermal cells, or cell lines) on the backs of athymic nude mice to determine the in vivo potential of the cell combination to reconstitute skin or epidermal tumors.
Grafting procedures are performed in laminar flow hoods in the animal facility clean room reserved for holding immunodeficient mice in microisolator cages and for performing experiments with them.
SUPPLIES to be taken to the animal facility for grafting:
Sterile instruments and supplies: forceps, curved scissors, wound clips with autoclip applicator, gauze, pipette tips for a P200 Pipetman, assembled grafting chambers (Domes) in sterilizing pouch. (Grafting chambers consists of a lower rim that looks like a hat with part of the top cut off, and a hat shaped upper part with a rounded dome. The vertical sides of the lower part fit inside the dome with the rim under the rim of the upper part. As manufactured, the upper part requires a 2.5 mm hole to be cut through the dome area with a disposable punch biopsy tool. Undamaged grafting chambers removed from mice can be recycled.)
Other supplies: bottles of 70% ethanol, Betadine, and sterile PBS; several 100 mm culture dishes: one to contain PBS, others to serve as sterile surface for resting surgery instruments; sterile beaker for holding instruments in 70% ethanol while changing from one group of mice to another; P200 Pipetman; Bandguard (Schering Plough bandage protectant, cream formulation); cotton tip applicators.
Anesthetic solution prepared in enough quantity to sedate all mice to be grafted: one ml Pentobarbital (50-60 mg/ml depending on lot) added to a mixture of 7.2 ml distilled water and 0.8 ml 95% ethanol. This is enough anesthetic to sedate 20 mice. Mice are given 0.01 ml/g body weight of the anesthetic solution i.p. with a 1 ml tuberculin syringe and a 25-gage 5/8” long needle. Mice lose consciousness within 3-5 minutes and remain sedated for at least 30 min.
Ice bucket with tubes containing the pellets of cell mixtures to be grafted, each mouse receiving the contents of one tube.
Procedures in the tissue culture room prior to grafting TIMING up to 2 hrs for 2 groups of 5 mice each
Have ready all cells to be used in the grafting experiment as cell suspensions at concentrations of the order of 2-5 million cells/ml; for example freshly prepared or cultured keratinocytes, hair follicles, fibroblasts, or cell lines.
Set up and label 15 ml conical tubes corresponding to the number of mice to be grafted in the experiment, with a minimum of 5 mice per experimental groups.
Add the appropriate amount of cell suspension for each type of cell to be combined and applied to the graft site. For example 5-8 million freshly prepared primary keratinocytes plus 5 million primary fibroblaststs will reconstitute normal haired skin in the graft site. Depending on the epidermal tumor cell line, between 1 and 5 million tumor cells combined with 5 million 1-week cultured fibroblasts will result in a tumor at the graft site.
Sediment the cell mixtures at 230g for 4 min.
Carefully aspirate the supernatant medium leaving about 0.5 to 1 mm of medium above the pellet.
Keep tubes on ice in an ice bucket to be taken to the animal room for grafting.
Grafting procedure TIMING 2-3 hrs for 2 groups of 5 mice each
-
7.
Inject mice i.p. with 0.01 ml per gram of body weight of the prepared pentobarbital anesthetic solution (one group of mice at a time).
-
8.
Using gauze, wipe backs of the sedated mice first with Betadine (Fig. 4a), then with 70% ethanol and place them side by side on a clean towel.
-
9.
For each mouse, pull up skin over the flattest part of the back with forceps and remove a portion of lifted skin with curved scissors to create a 1 cm diameter hole (Fig. 4b,c).
-
10.
Moisten forceps with PBS and free an approximately 5 mm rim of the surrounding skin from underlying attachments (Fig. 4d).
-
11.
With forceps in one hand, pull the skin near the hole to create a slit like opening and with forceps in the other hand pick up a grafting chamber, first touch the lower rim area to the surface of the PBS contained in a Petri dish, then ease one side of the double rim under the skin on one side of the slit (Fig. 4e,f). Steadying the dome in that position with forceps, gently pull skin with forceps in the other hand over the rim on the other side (Fig. 4g). With practice, dome insertion is accomplished in 10 – 20 seconds/mouse.
-
12.
If the hole is too large and the dome is not snugly held in place, use one or two autoclips to clip skin to the rim to prevent the dome from popping out of the hole (Fig. 4h).
-
13.
Using the pipetman with tip in place, gently stir the cell pellet with overlying medium and pull the cell suspension into pipette tip (50 -150 μl) without pulling in air, and eject the cell suspension through the hole of the dome onto the graft area (Fig. 4i). In order to avoid giving a double dose of cells to a single mouse, it is helpful to place the cap of the tube from which the cells were removed next to the mouse that has received the cells, until all the mice in the group have received cells.
-
14.
Using a cotton tipped applicator, apply Bandguard cream on the outside of the dome around the hole without getting any of it inside the dome
-
15.
Using both hands, pick up each mouse at the scruff of the neck and the base of the tail and transfer it to a clean cage so that the mice lie side by side close to each other for warmth by body contact.
-
16.
Observe mice until they recover from anesthesia.
-
17.
Check mice daily.
-
18.
Remove domes 5-7 days after grafting. Graft areas are usually healed completely by 3 weeks. If the graft yields haired skin, hair will be generally visible above the skin surface at that time. If tumor formation is expected, these may be visible as early as 2 weeks after grafting.
? TROUBLESHOOTING
Step 16 and Step 25
Problem: Difficulty in separating dermis from epidermis and consequent low keratinocyte yield will be encountered if the mice are older than 2 days after birth, or if the trypsin is not optimally active. If the age of the pups is not an issue, histological sections of skin from newborn to 1 day-old mice that was fixed after floating overnight on trypsin (without separating dermis from epidermis) can give clues of why difficulties were encountered. Fully active trypsin will digest the basement membrane, loosen the smallest HF buds from the dermis, and disrupt the adhesion between cells of the lower epidermis. Consequently histological sections will show loose single epidermal cells, cell aggregates and hair follicle buds sandwiched between the dermis and the upper layers of the epidermis (granular and stratum corneum)18. Skin even from newborn mice when floated on partially active trypsin will also seem more difficult to separate into epidermal and dermal layers. Histology of such skin after floating on trypsin over night revealed separation within the lower spinous cell layers. Thus, many basal cells and hair follicle buds remained attached to the dermis when it was lifted up from the epidermis.
Solutions: use only newborn to 2-day old mice and skin from adult mice that is in the resting phase of the hair cycle. Use only freshly thawed trypsin kept less than one week at 4°C. Pre-test new trypsin lots on newborn mouse skin and verify that cell yield is in the expected range and that overnight attaching cells look healthy and abundant after removing unattached suprabasal cells on the day after plating.
Steps #20-34, #41-51, #59-64, #65-67A&B
Problem: Low cell yields for the various cell preparations due to inadvertent loss during centrifugation steps for reasons such as choosing an inappropriate centrifugation speed or discarding wanted fractions.
Solution: When performing procedures for the first time, examine aliquots of all fractions under the microscope in order to become familiar with the expected cell morphologies. At various stages during the preparation, compare cell morphologies to those of the final preparations shown in the top row panels of Figures 2 and 3.
ANTICIPATED RESULTS
Cell yields of various cell preparations
| Source | Cell Type | Yield | Units |
|---|---|---|---|
| newborn mouse epidermis | keratinocytes | 8-10 | million particles/epidermis |
| newborn mouse dermis | fibroblasts | 30-50 | million cells/dermis |
| adult mouse dorsal epidermis | keratinocytes | 5-10 | million cells/epidermis |
| adult mouse tail epidermis | keratinocytes | 5-10 | million cells/epidermis |
Yields of dermal HF and of HF buds isolated from the epidermal preparation have not generally been quantified by particle count, although that should be possible, particularly with new generation particle counters (e.g. Nexcelcom Cellometer Auto T4, recommended for heterogeneous cell populations). For grafting experiments, amounts were standardized based on mouse equivalents (ME) per grafted mouse18 (0.5 ME of dermal HF or 1 ME of HF buds per mouse). Standardization of amounts of HF buds used in coculture experiments was based on DNA content20 (15 ug DNA/well of 12-well cluster dish).
Phase contrast morphologies of cell preparations, Fig. 2 & 3
Freshly prepared primary epidermal keratinocytes show multiple cell morphologies reflecting various stages of differentiation; they also contain immature hair follicle buds, recognizable as compact multicellular packets of cells surrounded by a basement membrane like structure. Only basal cells and hair follicle buds in this preparation have the ability to attach and to proliferate in culture. The monolayer culture morphology of purified hair follicle buds and of purified developing follicles obtained from the dermis are essentially indistinguishable from that of primary keratinocytes.
Useful culture life of primary keratinocytes
Primary keratinocytes from newborn mice are routinely plated at 0.25 to 0.5 mouse equivalents (depending on mouse strain) per 60 mm tissue culture dish. On the following day the attached number of cells comprises approximately 25% of total cells (attached plus floating) and appear to be 40 to 50% confluent. Under low calcium conditions, the cells will become increasingly more crowded (day 4 to 5) and rounded floating cells will begin to appear above the dense areas, representing presumably daughter cells that were unable to gain attachment to the plastic culture surface during mitosis. In LoCa medium, the number of attached cells will begin to decrease between day 6 and 8 after plating as the cells become more flat and appear to senesce. For this reason, most experiments to study the biochemical and molecular biological responses to treatments with growth factors, cytokines, and pharmacologic agents or to the introduction of genes (by transfection or by retroviral or adenoviral transmission) are performed between day 3 and 7 after plating primary keratinocytes.
The usefulness of the grafting procedure is by no means restricted to the cell preparations explicitly covered in this overall protocol. Likewise, in addition to the specific uses for the various cell preparations cited here, there are potentially uses in the development of other in vitro models designed to answer specific biological questions. An obvious one is the establishment of three-dimensional organotypic cultures using the various epithelial and dermal fractions to model epidermal proliferation and differentiation in association with a dermal equivalent in vitro. Also, the dermal single cell fraction, seemingly composed primarily of fibroblasts, could potentially be induced to show other characteristic differentiated phenotypes, such as of sebocytes, adipocytes, muscle cells, and nerve cells, if provided with the required permissive physical and inductive environment.
MATERIALS
REAGENTS
Mice, neonatal or adult in the resting phase (telogen) of hair growth; athymic nude mice! CAUTION: All procedures involving live rodents must conform to National and Institutional regulations.
Cells; freshly isolated primary keratinocytes and fibroblasts.
0.25% Trypsin without EDTA (Mediatech Cellgro cat. # MI 25-050-C1 ).
0.25% Trypsin-1mM EDTA (Gibco/Invitrogen cat.# 25 200).
S-MEM without calcium chloride (Gibco/Invitrogen, custom made Formula # 97-0267DJ) or Calcium-free MEM Eagle with Earle’s BSS, glutamine, and non-essential amino acids (Lonza cat # 06-174G).
Fetal Bovine Serum (FBS), Pretested lots, treated with chelating resin to remove calcium (see: serum chelexing procedure).
Penicillin-Streptomycin (Gibco/Invitrogen cat.# 15140-122).
Antibiotic-Antimycotic (Gibco/ Invitrogen cat.# 15240-062).
DMEM without L-Glutamine (Lonza cat.# 12-614F).
GlutaMAX-1 200mM (Gibco/Invitrogen cat.# 35050).
Newborn Calf Serum; (e.g. Gemini Bio-Products, Cat.# 100-504)
Sodium Pyruvate 100mM (Gibco/Invitrogen cat.# 11360).
PBS, 1×, Dulbecco’s, without calcium and magnesium (Gibco/Invitrogen cat.# 14190).
Betadine (Express Medical Supply, cat.# Pur181501).
70% (vol/vol) Ethanol, 95% Ethanol
Calcium Chloride stock solution (see REAGENT SETUP).
Collagenase (Worthington cat.# 4196; see REAGENT SETUP).
DNASE 1 (Worthington cat# LS002139; see REAGENT SETUP) .
Ficoll PM400 (GE Healthcare Life Sciences cat# 17-0300-10; see REAGENT SETUP).
Anesthetic (Nembutal, see REAGENT SETUP).
Chelex-100 (Biorad, 200-400 Mesh, sodium form, cat.# 142-2842; see REAGENT SETUP)
Bovine Collagen, Type I, 3 mg/ml in 0.01 N HCl, previously obtained from Vitrogen Collagen Corp. and from Chemicon, currently available as PureCol from Inamed, Freemont, CA.
Fibronectin, BD Biosciences, cat# 354008
BSA, Sigma, Cat# A4378, 1× crystallized
EQUIPMENT
Autoclaved sterile dissecting scissors and forceps
Sterile 50ml tubes
Cell Strainer 100μm (BD Falcon)
Sterile Stender dish, small beaker, or crystallizing dish
Sterile funnel
100μ and 20μ Nytex filter cloth (Swiss multifilament Polyester Fabric from MARTIN Screen Printing and Sign Supply Co. Baltimore, MD), cut into 10-11 cm2 squares, folded twice to be ready for placing into a small sterile funnel, and autoclaved inside a sterilizing pouch
Sterile 125 ml and 250 ml Erlenmeyer flasks
Sterile grafting domes (Cole Equipment Co., 301-946-4746, Rockville, MD) with 2.5 mm hole bored through the top with a punch biopsy tool
Table top centrifuge with swinging buckets (e.g., Beckman GS-6 with GH-3.8 rotor)
Inverted Phase Contrast Microscope (e.g., Nikon ECLIPSE TE300 with CoolSNAP digital camera and IPLab image capturing program)
REAGENT SETUP
Calcium Chloride: prepare 0.25 to 0.3 M stock solution, filter sterilize, determine actual calcium concentration by atomic absorption.
DNASE: prepare 20,000 units /ml solution in PBS, sterilize with low protein binding 0.2μm filter; store frozen in small aliquots.
Collagenase : prepare 0.35% (wt/vol) in Fibroblast Culture Medium, agitate on rocker for 10 min, centrifuge at 2800g rpm for 10 min filter sterilize through low protein binding filters, first through 0.45μm with prefilter, then through a 0.2μm; store at 4°C.
Low Calcium medium (LoCa, 0.05 mM Ca2+): S-MEM supplement with 8% (vol/vol) chelexed serum (see chelexing procedure) plus 5 ml of Penicillin-Streptomycin per 500ml bottle.
High Calcium medium (HiCa, 1.3 mM Ca2+): Low Calcium medium plus 2.6 ml of 0.25 M calcium stock solution per 500 ml bottle.
Fibroblast culture medium (Fb medium): DMEM supplement with 10% (vol/vol) Newborn Calf Serum plus 5 ml GlutaMAX, 5 ml Sodium Pyruvate and 5 ml Penicillin-Streptomycin per 500 ml bottle.
Fibronectin/Collagen coating solution for tissue culture plates to enhance keratinocyte attachment: to 200 ml stirred Leibovitz medium add 2 mg fibronectin, 1 ml 2% (wt/vol) BSA in PBS, 4 ml 1M HEPES, 2 ml bovine collagen type I, 3 mg/ml; sterilize by filtration through low protein-binding 0.2μm filter. Store at 4°C.
Ficoll Stock solution (25% (wt/vol) in water): Ficoll PM400 is a very light powder. Weigh out 75g in a tared graduated plastic beaker and add 250 ml water and a stirring magnet; cover beaker with parafilm and allow to gently stir overnight until all Ficoll has gone into solution. Bring volume to 300 ml with stirring. Transfer to a glass bottle and mark the level of the liquid in the bottle. Sterilize by autoclaving and after cooling, adjust volume with sterile water if necessary. Store at 4°C.
Ficoll working solution (9% (wt/vol) in HiCa medium): add 1 volume of 10× PBS and 15 volumes of HiCa medium to 9 volumes of Ficoll stock solution.
Serum Chelexing Resin: Suspend Chelex-100 resin in 4 vol (vol/wt) of water with magnetic stirring until suspension is uniform, add 0.4 vol (vol/wt) of 0.2M sodium phosphate buffer, pH 7 and continue stirring for 30 min. Remove liquid by filtration through Whatman #1 filter paper on Buchner funnel. Suspend resin in 4 volumes of 70% (vol/vol) ethanol and stir for 30 min. Filter to remove ethanol. The resin when suspended in 2 volumes (original wt/vol) of sterile normal saline is ready for column packing. Leftover resin can be kept at 4°C and taken through the washing procedure with the next batch of resin.
Serum chelexing to remove Calcium, general guidelines: A 500 ml radial flow SUPERFLOW LAB COLUMN from Sepragen, well packed with washed Chelex-100 resin, efficiently removes calcium from 15L of fetal bovine serum (containing about 3mM calcium) at a flow rate of 1500-2000 ml/hr (ratio of serum volume to packed resin volume is approximately 30). C-columns with adapters that eliminate/minimize mixing volume above the resin bed (formerly from Pharmacia, currently from GE Healthcare) are available in various sizes and provide a good alternative for small volume serum chelexing needs. The smaller columns are convenient for chelexing the 50 ml test sera, generally supplied free by serum supply companies. After packing the column, wash resin by pumping 2 or more column volumes of sterile normal saline through it before starting the flow of serum. Monitor the column effluent for the appearance of serum then transfer the outflow tube to the serum collection vessel. Efficient chelexing reduces the calcium concentration in serum to 0.01mM or less, but this needs to be confirmed by atomic absorption analysis. Mix 5 volumes of efficiently chelexed serum with 1 volume of unchelexed serum to adjust the calcium concentration of the serum. (The calcium concentration in LoCa medium made with 8% (vol/vol) of this serum mixture will be close to 0.05 mM, the target value for LoCa medium. Because Chelex-100 resin also removes other divalent cations, mixing with untreated serum partially restores their concentrations as well.) Finally, filter sterilize the serum mixture first through 0.45 micron and then through 0.2 micron low protein binding filters. For large volumes, do serum filtration under nitrogen pressure supplied to a pressure tank containing the serum mixture and connected to the filters arranged in tandem. Store serum at −20°C.
PROCEDURES
Preparing newborn mice for skin removal TIMING 1 h for 10 mice
-
1
Place mice into culture dishes without crowding and transfer to a CO2 euthanizing chamber for 20 min.
-
2
Place dishes on ice until the next step.
-
3
Immerse euthanized mice in Betadine for 2 min, twice.
-
4
Rinse mice with de-ionized water.
-
5
Immerse mice in 70% Ethanol for 2 min, twice, then place dishes on ice until the next step.
Removal of newborn mouse skin TIMING 30 min for 10 mice
-
6
Perform skin removal procedures with sterile dissecting tools on the inside surface of the lid of a sterile 150 mm culture dish. CRITICAL STEP Ease of separation of epidermis from dermis after the trypsin treatment is assured by removing the skin from the mouse as an intact sheet with smooth edges that will float flat on the trypsin solution.
-
7
Cut off limbs just above wrist and ankle joints, leaving visible stumps (Fig. 1a,b).
-
8
Cut off tail close to body leaving an easily visible hole (Fig. 1c).
-
9
Grasp the body firmly between a pair of forceps, insert small sharp scissors through the hole at the tail, and cut the skin in a smooth fashion along the dorsal midline of the body to the tip of the nose (Fig. 1d,e); be careful not to cut through underlying fascia into the body cavity.
-
10
With forceps in each hand, gently loosen the skin away from the midline (Fig. 1f).
-
11
Grasp exposed body at the hips with one pair of forceps and use the other to gently pull the back skin over the back leg stumps (Fig. 1g).
-
12
Grasp the back skin now hanging on the ventral side of the mouse with one pair of forceps and gently pull away the whole skin from the body with one continuous motion while holding the skinless rear of the body with another pair of forceps (Fig. 1h,i). The released skin does not include the whisker pads. The latter could be removed at this stage with a scalpel blade or scissors for isolating whisker follicles, if desired.
-
13
Place the released skin on the culture surface of a clean 150 mm culture dish, dermis side down (Fig. 1j)
-
14
Spread out skin completely flat; if edges are curled under, try uncurling by pulling skin back and forth on the dish first and then flattening isolated wrinkles (Fig. 1j); proper stretching of skin at this stage makes floating it on trypsin much easier.
-
15
Place eight to ten stretched skins in one dish; the closed dish can be kept in a refrigerator for at least 2 hours before floating the skin on trypsin.
Fig. 1.
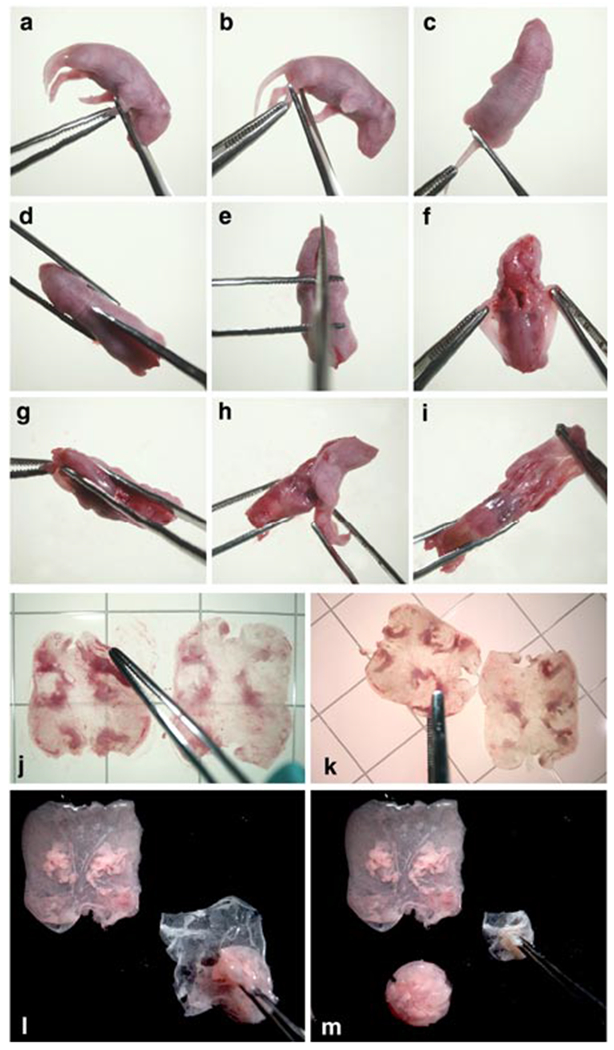
Steps in the procedure for obtaining epidermis and dermis from newborn mice: a. removal of lower forelimbs; b. removal of lower hind limbs; c. severing tail close to the body; d. cutting the skin along dorsal midline starting at the tail end of the body; e. ending the dorsal midline cut at the tip of the nose; f. loosening the skin on both sides of the dorsal midline cut; g. pulling skin over the hind leg stumps; h. gathering skin on the ventral side and pulling it toward the head; i. pulling the skin over the forelimb stumps and over the head; j. flattening skin, dermis side down, on the culture surface of culture dish; k. uncurling folded down edges of skin as necessary after transferring it, dermis side down, to the surface of the trypsin solution contained in culture dish; l. dermis being lifted above the adhering epidermis of skin, that had been floating overnight on trypsin and was transferred, epidermis side down, to the inner surface of a dry lid of a sterile culture dish and gently spread; m. gathering of epidermis to enclose loosely adhering cells prior to transfer to a container with HiCa medium; also shown is the dermis lifted in the previous panel and deposited on the lid surface.
Floating skin on cold trypsin TIMING 30 min for 10 skins
-
16
In a laminar flow hood set up the following: sterile forceps, freshly thawed cold Trypsin (0.25% without EDTA) and new culture dishes. Pour about 50 ml of Trypsin into a 150 mm dish.
!CAUTION The shelf life of thawed trypsin is limited. To assure easy separation of dermis from epidermis, always use freshly thawed trypsin. Though rare, it is possible to receive frozen trypsin lots of low activity. Pre-testing of new lots is advised.
? TROUBLESHOOTING
-
17
Grasp opposite ends of one side of the stretched skin with forceps, lift it, and transfer it so that the dermis side of the hanging skin touches the trypsin, then lower the rest of the skin and release the forceps; the skin will generally float perfectly flat (Fig. 1k)
-
18
If necessary, flatten curled-under edges using the rough side of one tip of forceps placed under the skin and gliding away from it (Fig. 1k) while steadying the skin with the other forceps placed gently on the center of the skin (not shown). Up to 10 skins can be floated in one dish. The tendency of skins is to float rather than to sink; therefore, carefully transporting dishes will generally not cause the skins to sink.
-
19
Store dishes with floating skins at 4°C overnight in a refrigerator or cold room.
Isolation of primary keratinocytes from newborn mouse epidermis TIMING 2 hrs for 10 skins
-
20
Place the following autoclaved or otherwise sterile or cleaned items into a laminar flow hood: dissecting scissors, forceps, 50ml conical tubes, Stender dish or similar container of required size, cell strainer and/or folded and sterilized square of coarse 100 micron Nytex, and funnel, cold HiCa medium with added antibiotic-antimycotic solution (2 ml/500ml), a dish with skins floating on trypsin, and several new 150 mm Falcon culture dishes. The bottoms of the new culture dishes can be used as sterile surfaces on which to rest dissecting instruments. The inner surface of the lids will be used for the separation of the dermis from the epidermis of the floating skins.
-
21
Add cold HiCa medium to the Stender or other suitable dish, 2-5 ml/epidermis, for the number of epidermises to be processed together.
-
22
Grasp each floating skin on opposite ends of one side with forceps, lift it up from the trypsin surface and, after very briefly allowing trypsin to drain from it, transfer it to a dry area on the dry inner surface of the lid of a culture dish, epidermis-side down. Eight to 10 skins can be accommodated in one lid. !CAUTION: in order for the epidermis to adhere to the inner surface of the lid, the lid must be dry and must not have been treated by the manufacturer for tissue culture use. Test with a drop of water: it will spread on a treated surface, but roll on an untreated surface.
-
23
Spread the skins gently to allow the epidermis to make contact with the plastic at various points around the edges (Fig. 1l).
-
24
For each skin, lift the dermis up straight above the epidermis (Fig. 1l), holding the epidermis down with forceps if necessary, and transfer dermis to a tube containing HiCa medium for later preparation of fibroblasts and of dermal hair follicles (steps 41-51 and 59-64) if planned, or to another receptacle for later disposal.
-
25
Fold the edges of each epidermis with forceps toward its center (Fig. 1m), and without letting the loosely attached keratinocytes escape onto the dish, transfer each packet of epidermis to the Stender dish containing HiCa medium.
? TROUBLESHOOTING
-
26
Mince the collected epidermises with scissors until pieces are small enough to enter the tip of a 10 ml pipette.
-
27
Triturate the resulting suspension by pipetting up and down 10 or more times, then transfer the suspension to a 50 ml conical tube, leaving most of the stratum corneum sheets behind.
-
28
Rinse the Stender dish with additional HiCa medium and transfer to the same tube.
-
29
Centrifuge the cell suspension at 150g for 5 min at 4°C.
! CAUTION First time users of all procedures involving cell preparations are advised to save all fractions and to examine aliquots under a phase contrast microscope before discarding any. This precaution serves to verify retention of the wanted cells, and gives assurance that fractions to be discarded are either virtually free of cells, or are not needed. It may also help in troubleshooting unexpected outcomes, such as low cell yield.
? TROUBLESHOOTING
-
30
Carefully aspirate the supernatant including any floating stratum corneum pieces that would reduce keratinocyte yield by entrapment during the filtration in the next step.
-
31
Resuspend the pellet in HiCa medium, and filter through a 100 μm cell strainer into a new 50 ml conical tube. Rinse the cell strainer with HiCa medium to release cells entrapped by remaining stratum corneum pieces. Limit the amount of epidermal material going through one BD cell strainer to no more than 30 mouse equivalents (keratinocytes from 30 skins).
-
32
Centrifuge cells again at 150g for 5 min at 4°C.
-
33
Aspirate the supernatant, resuspend pellet in HiCa medium to 1 mouse equivalent (ME)/ml of primary keratinocyte; ME = mouse equivalent = all the keratinocytes from one newborn mouse skin.
-
34
Count a suitable aliquot of cell suspension in 10 ml of Isoton with the Coulter Counter window open from 7μm to infinity to include large particles such as immature hair follicle buds. The particle count should be of the order of 8 to 10 million per ml. If desirable, a vital count of Trypan Blue-excluding cells could also be determined by using a hemocytometer.
PAUSE POINT The cell suspension can be kept at 4°C for several hours before plating.
Plating primary keratinocytes for culture TIMING 1 h for 10 mouse equivalents
-
35
Bring keratinocyte preparation in HiCa medium at 1 ME/ml from 4°C storage or ice bucket to laminar flow culture hood.
-
36
Initial plating of primary keratinocytes can be done either in 0.2 mM calcium medium or in LoCa medium (A or B)
-
(A)
Dilute suspension in HiCa with 7 volumes of LoCa ( 0.05 mM Ca) medium. The resulting calcium concentration will be approximately 0.2 mM and the concentration of cells will be 1/8 ME /ml.
-
(B)
pellet the desired number of cells by centrifugation at 150g for 5 minutes and suspend cell pellet in LoCa medium at 1/8 ME/ml
-
(A)
-
37
Plate desired amount of cell suspension into 60 mm or larger dishes as required for intended experiment: 4 ml/60 mm dish of this suspension will contain 0.5 ME or approximately 5 million particles per ml. For larger or smaller dishes calculate the volume needed so that the ME/unit area is approximately the same (e.g. 1 ME/100 mm dish; 3 ME per 150 mm dish); plating efficiency depends on the age and strain of the mice and needs to be determined by the experimenter. CAUTION: hair follicle buds which have a high plating efficiency are dense structures containing 50 to 150 cells per particle. They have a tendency to settle out rapidly under gravity. Therefore the cell suspension needs to be agitated (e.g. by pipetting the cell suspension up and down in the container) between each withdrawal of cell suspension for plating in order to maintain homogeneity within the population. Uniform distribution of cells over the tissue culture surface is enhanced by distributing the cell suspension during release of the cell suspension over the entire surface of the dish rather than by depositing it all in the center of the dish. Permitting the cells to settle in the culture dishes for a few minutes before carefully moving them to the incubator, also contributes to more uniform distribution of cells.
-
38
Incubate cells at 36°C in an atmosphere of 7% CO2 in air overnight.
-
39
On the next day, aspirate the medium containing unattached cells, rinse the attached cells with Ca and Mg free PBS, and add LoCa medium..
-
40
Feed cells every other day thereafter. Figure 2 shows the morphology of primary keratinocytes shortly after plating (Fig. 2a) and 3 days later (Fig. 2b). Cultures are generally confluent by day 3 or day 4.
Fig. 2.
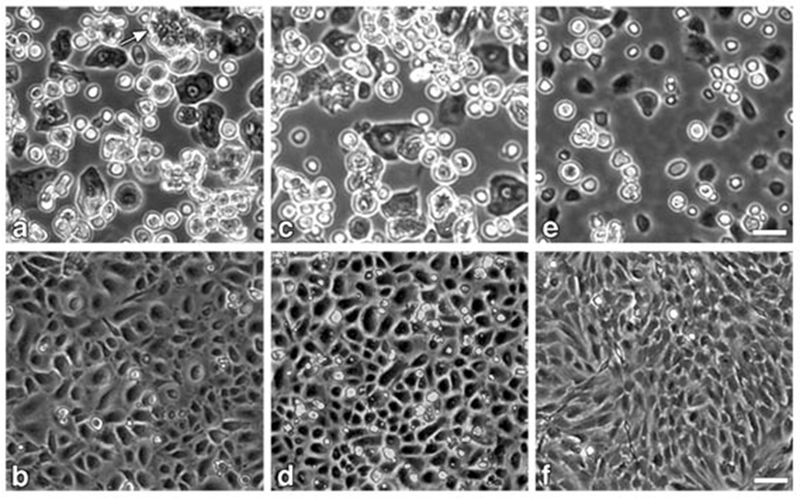
Phase contrast images of freshly prepared and cultured cell fractions from epidermis and dermis of newborn BALB/c mice: a, c, e: freshly prepared preparations captured through 10× objective; b, d, f: fractions cultured for 3 days, captured through 4× objective. a, b: total epidermal cell fraction plated at 0.5 mouse equivalent per 60 mm dish; white arrow near top edge of panel (a) points to an immature hair follicle bud; c, d: epidermal cell fraction after removing immature hair follicle buds by centrifugation at 20g, plated at 1 mouse equivalent per 60 mm dish; e, f: dermal cell fraction (fibroblasts) after removing dermal hair follicles by low speed centrifugation and final filtration through 20μ Nytex cloth, plated at 2.9 million cells per 60 mm dish. Epidermal fractions were plated in 0.2 mM Ca2+ medium overnight; attached cells were washed with PBS and switched LoCa medium; medium was changed on day 3 before image capture. Fibroblast fraction was plated in HiCa medium overnight followed by a medium change on day 1 and 3. Bars indicates 20 μm for top row panels and 50 μm for bottom row panels
Fig. 3.
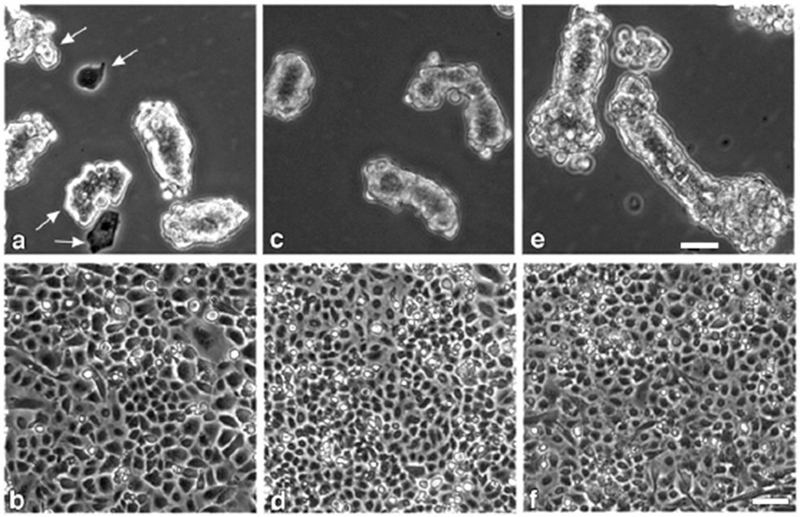
Phase contrast images of freshly prepared and cultured hair follicle fractions from epidermis and dermis of newborn BALB/c mice: a, c, e: freshly prepared hair follicle fractions captured through 10× objective; b, d, f: hair follicle fractions cultured for 3 days, captured through 4× objective. a, b: hair follicle bud preparation from epidermis suitable for grafting; white arrows in panel a point to contaminating suprabasal cell aggregates and to granular cells; plated at 0.75 mouse equivalents per 60 mm dish; c, d: more highly purified hair follicle bud preparations from epidermis devoid of extraneous cells and cell aggregates, suitable for collagen matrix co-culture experiments; plated at 5 mouse equivalents per 60 mm dish. Extremely high cell density on day 3 indicated underestimation of yield based on volume of final pellet. e, f: dermal hair follicles plated at 0.75 mouse equivalent per 60 mm dish. All hair follicle fractions were plated in 0.2 mM Ca2+ medium overnight, followed by washing attached cells with PBS and feeding with LoCa medium; medium was changed on day 3 before image capture. Bars indicates 20 μm for top row panels and 50 μm for bottom row panels.
Acknowledgements:
Many current and former members of the laboratory have been instrumental in refining the techniques described. Several of them are acknowledged by citing their publications in the text. Special thanks are owed to Christophe Cataisson, a current member of the laboratory, for contributing the images comprising Fig. 4.
Fig. 4.
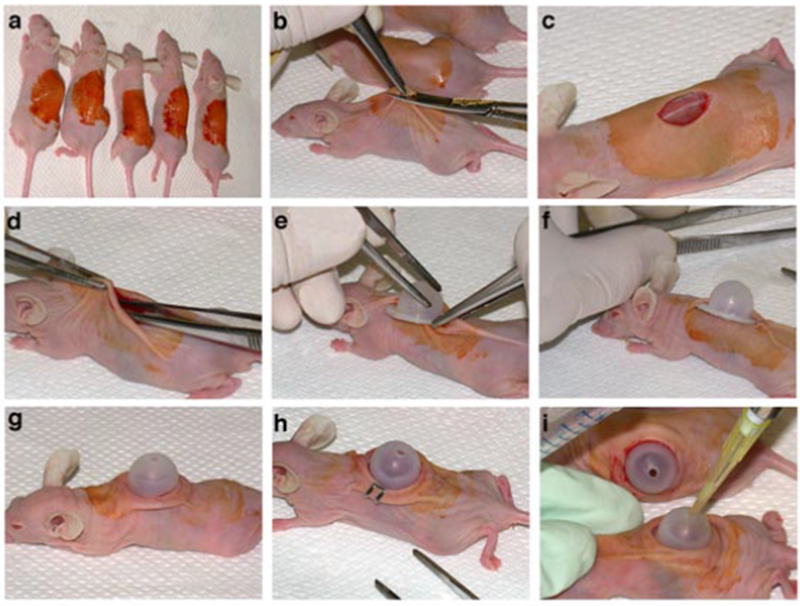
Steps in the procedure for grafting cells mixtures on the backs of athymic nude mice to determine their in vivo phenotype: a. decontamination of the graft area of sedated mice with Betadine (to be followed by wiping area with 70% ethanol); b. cutting an approximately 1 cm diameter piece of full thickness skin with curved scissors after lifting skin with forceps; c. appearance of graft area after removing the skin piece; d. lifting the skin and moistening the area under the skin around the graft area with PBS using forceps; e. sliding part of the PBS-moistened dome flange under the skin on one side of the graft area; f. appearance of partially inserted dome prior to pulling the skin on the other side over the remaining exposed flange; g. appearance of fully inserted and snugly seated dome; h. appearance of loosely fitting dome fastened to the skin with wound clips (clip on other side not visible); i. application of cell suspension to the graft area.
References
- 1.Hennings H et al. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell 19, 245–254 (1980). [DOI] [PubMed] [Google Scholar]
- 2.Hennings H Primary culture of keratinocytes from newborn mouse epidermis in medium with lowered levels of Ca2+ In Keratinocyte Methods. (eds.Leigh I & Watt FM) pp. 21–23. (Cambridge University Press, Cambridge, UK, 1994). [Google Scholar]
- 3.Yuspa SH, Kilkenny AE, Steinert PM, & Roop DR Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J.Cell Biol 109, 1207–1217 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dlugosz AA, Glick AB, Tennenbaum T, Weinberg WC, & Yuspa SH Isolation and utilization of epidermal keratinocytes for oncogene research In Methods in Enzymology, Vol 254, pp. 3–20, (Academic Press, New York, New York, USA, 1995). [DOI] [PubMed] [Google Scholar]
- 5.Kulesz-Martin MF, Koehler B, Hennings H, & Yuspa SH Quantitative assay for carcinogen altered differentiation in mouse epidermal cells. Carcinogenesis 1, 995–1006 (1980). [DOI] [PubMed] [Google Scholar]
- 6.Kilkenny AE, Morgan D, Spangler EF, & Yuspa SH Correlation of initiating potency of skin carcinogens with potency to induce resistance to terminal differentiation in cultured mouse keratinocytes. Cancer Res. 45, 2219–2225 (1985). [PubMed] [Google Scholar]
- 7.Kawamura H, Strickland JE, & Yuspa SH Association of resistance to terminal differentiation with initiation of carcinogenesis in adult mouse epidermal cells. Cancer Res. 45, 2748–2752 (1985). [PubMed] [Google Scholar]
- 8.Forslind B Particle probe analysis in the study of skin physiology. Scan.Electron.Microsc III, 1007–1014 (1986). [PubMed] [Google Scholar]
- 9.Elias PM et al. Formation of the epidermal calcium gradient coincides with key milestones of barrier ontogenesis in the rodent. J.Invest Dermatol. 110, 399–404 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Fusenig NE & Worst PK Mouse epidermal cell cultures. II. Isolation, characterization and cultivation of epidermal cells from perinatal mouse skin. Exp.Cell Res. 93, 443–457 (1975). [DOI] [PubMed] [Google Scholar]
- 11.Morris RJ, Haynes AC, Fischer SM, & Slaga TJ Concomitant proliferation and formation of a stratified epithelial sheet by explant outgrowth of epidermal keratinocytes from adult mice. In Vitro Cell Dev.Biol. 27A, 886–895 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Hager B, Bickenbach JR, & Fleckman P Long-term culture of murine epidermal keratinocytes. J.Invest Dermatol. 112, 971–976 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Yuspa SH, Koehler B, Kulesz-Martin M, & Hennings H Clonal growth of mouse epidermal cells in medium with reduced calcium concentration. J.Invest.Dermatol 76, 144–146 (1981). [DOI] [PubMed] [Google Scholar]
- 14.Kaighn ME, Camalier RF, Bertolero F, & Saffiotti U Spontaneous establishment and characterization of mouse keratinocyte cell lines in serum-free medium. In Vitro Cell Dev.Biol. 24, 845–854 (1988). [DOI] [PubMed] [Google Scholar]
- 15.Weissman BE & Aaronson SA BALB and Kirsten murine sarcoma viruses alter growth and differentiation of EGF-dependent balb/c mouse epidermal keratinocyte lines. Cell 32, 599–606 (1983). [DOI] [PubMed] [Google Scholar]
- 16.Backendorf C et al. Repair characteristics and differentiation propensity of long term cultures of epidermal keratinocytes derived from normal and NER-deficient mice. DNA Repair (Amst) 4, 1325–1336 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Castilho RM et al. Requirement of Rac1 distinguishes follicular from interfollicular epithelial stem cells. Oncogene 26, 5078–5085 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Weinberg WC et al. Reconstitution of hair follicle development in vivo: Determination of follicle formation, hair growth and hair quality by dermal cells. J.Invest.Dermatol 100, 229–236 (1993). [DOI] [PubMed] [Google Scholar]
- 19.Yuspa SH et al. Regulation of hair follicle development: an in vitro model for hair follicle invasion of dermis and associated connective tissue remodeling. J.Invest Dermatol 101, 27S–32S (1993). [DOI] [PubMed] [Google Scholar]
- 20.Scandurro AB et al. Immortalized rat whisker dermal papilla cells cooperate with mouse immature hair follicle buds to activate type IV procollagenases in collagen matrix coculture: correlation with ability to promote hair follicle development in nude mouse grafts. J.Invest.Dermatol 105, 177–183 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Roop DR et al. An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature 323, 822–824 (1986). [DOI] [PubMed] [Google Scholar]
- 22.Hansen LA et al. The epidermal growth factor receptor is required to maintain the proliferative population in the basal compartment of epidermal tumors. Cancer Res. 60, 3328–3332 (2000). [PubMed] [Google Scholar]