

Abstract
Rationale: Emerging evidence has suggested that sphingosine 1-phosphate (S1P), a bioactive metabolite of sphingolipids, may play an important role in the pathophysiological processes of cerebral hypoxia and ischemia. However, the influence of S1P on cerebral hemodynamics and metabolism remains unclear.
Material and Methods: Uniquely capable of high-resolution, label-free, and comprehensive imaging of hemodynamics and oxygen metabolism in the mouse brain without the influence of general anesthesia, our newly developed head-restrained multi-parametric photoacoustic microscopy (PAM) is well suited for this mechanistic study. Here, combining the cutting-edge PAM and a selective inhibitor of sphingosine kinase 2 (SphK2) that can increase the blood S1P level, we investigated the role of S1P in cerebral oxygen supply-demand and its neuroprotective effects on global brain hypoxia induced by nitrogen gas inhalation and focal brain ischemia induced by transient middle cerebral artery occlusion (tMCAO).
Results: Inhibition of SphK2, which increased the blood S1P, resulted in the elevation of both arterial and venous sO2 in the hypoxic mouse brain, while the cerebral blood flow remained unchanged. As a result, it gradually and significantly reduced the metabolic rate of oxygen. Furthermore, pre-treatment of the mice subject to tMCAO with the SphK2 inhibitor led to decreased infarct volume, improved motor function, and reduced neurological deficit, compared to the control treatment with a less potent R-enantiomer. In contrast, post-treatment with the inhibitor showed no improvement in the stroke outcomes. The failure for the post-treatment to induce neuroprotection was likely due to the relatively slow hemodynamic responses to the SphK2 inhibitor-evoked S1P intervention, which did not take effect before the brain injury was induced.
Conclusions: Our results reveal that elevated blood S1P significantly changes cerebral hemodynamics and oxygen metabolism under hypoxia but not normoxia. The improved blood oxygenation and reduced oxygen demand in the hypoxic brain may underlie the neuroprotective effect of S1P against ischemic stroke.
Keywords: Photoacoustic microscopy, Sphingosine 1-phosphate, Neuroprotection, Hypoxia, Ischemic stroke.
Introduction
Affecting about 800,000 people in the United States every year, stroke is a leading cause of death 1. Globally, stroke is the second leading cause of mortality, accounting for 11.8% of all deaths 2. With increased number of stroke incidents and the prevalence of stroke survivors, disability-adjusted life-years (DALYs) lost due to stroke is rapidly increasing, making it the third leading cause of DALYs lost worldwide 3. Ischemic stroke, caused by obstruction of vessel(s) that supplies blood to the brain, is the major type of stroke and accounts for 85% of all stroke incidents 4. Therefore, enormous efforts have been undertaken to explore preventions and treatments of this devastating disease.
Despite decades of intensive research, clinically applicable interventions for ischemic stroke remain very limited 5. Capable of re-establishing the cerebral blood flow by dissolving the blood clot, tissue plasminogen activator (tPA) is the only Food and Drug Administration-approved pharmacotherapy for acute ischemic stroke. However, owing to the increasing risk of hemorrhagic transformation over time, only a narrow therapeutic window (i.e., the first few hours after the stroke onset) is approved for the tPA treatment, which is not applicable to over 96% of stroke patients worldwide. Associated with reduced risks of hemorrhage, mechanical recanalization significantly extends the therapeutic window. However, this emerging approach has practical limitations, including limited accessibility 6 and the delay in treatment initiation. Thus, new pharmacotherapy strategies complementing currently available thrombolytic therapies are highly desired. Holding the potential to extend the viability of ischemic tissues for primary reperfusion interventions to initiate or be effective, neuroprotective agents are of particular interest and widely investigated 7.
Recently, sphingosine 1-phosphate (S1P), a bioactive metabolite of sphingolipids, has attracted increasing attention because of the newly observed neuroprotective effect against ischemic stroke 8. S1P is generated solely by sphingosine kinases (SphKs), which have two isoforms (SphK1 and SphK2). Thus, the S1P level can be regulated via the inhibition of SphK1 or SphK2 9. Recent studies have shown that elevated S1P is vasoprotective and neuroprotective 10 and that S1P promotes increased erythrocyte 2,3-bisphosphoglycerate and thus oxygen release as an adaptation to hypoxia 11, suggesting that the neuroprotective effect induced by the elevated S1P may be associated with cerebral hemodynamics and oxygen metabolism. However, the underlying mechanisms remain unclear.
Combining light and ultrasound for high-resolution imaging of optical absorption in vivo, photoacoustic imaging has emerged as an enabling technology in biomedicine 12, particularly in brain research 13. Taking advantage of the optical absorption of hemoglobin, the primary carrier of oxygen in the blood circulation, we have recently developed multi-parametric photoacoustic microscopy (PAM) for simultaneous imaging of blood perfusion, oxygenation and flow at the single-microvessel level 14 and in the awake mouse brain 15. Further, combining these hemodynamic parameters allows quantification of cerebral oxygen extraction and consumption 15. Uniquely capable of comprehensive assessment of cerebral hemodynamics and metabolism, multi-parametric PAM is well poised to investigate the hemodynamic and metabolic bases of S1P-mediated neuroprotection in the brain of awake behaving mice.
Herein, we describe the use of a selective SphK2 inhibitor to upregulate the blood S1P level. Then, the S1P-induced changes in cerebral hemodynamics and oxygen metabolism were investigated in the awake mouse brain under normoxia and hypoxia, using the multi-parametric PAM. Further, the neuroprotective effect of S1P against ischemic stroke was studied in an established mouse model of transient middle cerebral artery occlusion (tMCAO). The effects of both pre- and post-treatment were evaluated and compared.
Materials and Methods
Head-restrained Multi-parametric PAM
To measure cerebral hemodynamics and metabolism without the influence of general anesthesia, the head-restrained multi-parametric PAM 15 was used in this study. In this system (Figure S1a), two nanosecond-pulsed lasers at wavelengths of 532 nm and 558 nm (BX40-2-G and BX40-2-GR, Edgewave) were applied. These two beams were combined, collimated, filtered, and coupled into a single-mode optical fiber (P1-460B-FC-2, Thorlabs). Before the fiber-optic coupling, the laser energy was monitored by a photodiode (SM1D12D, Thorlabs) to compensate for pulse-to-pulse fluctuation. Then, the fiber output was launched into a home-made scanning head, in which the optical excitation and acoustic detection were confocally aligned through an achromatic doublet (AC127-025-A, Thorlabs) and a ring-shaped ultrasonic transducer (central frequency: 35 MHz; 6-dB bandwidth: 70%) to achieve maximum sensitivity. A field-programmable gate array was used to synchronize laser pulsing, mechanical scan, and acquisition of the signals from the photodiode and the ultrasonic transducer. A home-made apparatus consisting of an adjustable head plate and an air-floated spherical treadmill was used to restrain the head of the awake mouse, while allowing voluntary movement on the treadmill (Figure S1b and Figure S1c). The treadmill was floated by slightly compressed air (15 psi) to provide minimal resistance to mouse movement. More details of the head-restrained multi-parametric PAM can be found in our previous report 15.
Quantification of cerebral microvascular diameter, hemodynamics and oxygen metabolism
With the head-restrained multi-parametric PAM, cerebral hemodynamics and oxygen metabolism can be comprehensively quantified in awake mice. Based on the differential absorption spectra of oxy- and deoxy-hemoglobin, the oxygen saturation of hemoglobin (sO2) can be quantified through spectroscopic analysis of the dual-wavelength measurement at 532 nm and 558 nm 16.
Through statistical analysis of the fluctuation of PAM signal caused by the Brownian motion of red blood cells (RBCs), the average number of RBCs within the detection volume of PAM can be derived as
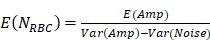 , ,
|
where 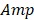 and
and 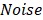 denote the amplitude of the signal and the noise of the PAM system, respectively.
denote the amplitude of the signal and the noise of the PAM system, respectively. 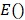 and
and 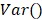 are the mean and variance operation, respectively. Further, given that on average each RBC contains ~15-pg hemoglobin 17, the total concentration of hemoglobin (CHb) can be estimated as
are the mean and variance operation, respectively. Further, given that on average each RBC contains ~15-pg hemoglobin 17, the total concentration of hemoglobin (CHb) can be estimated as
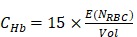 , ,
|
where 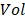 represents the detection volume of PAM and has been experimentally quantified to be 263 µm3 in our previous study 14.
represents the detection volume of PAM and has been experimentally quantified to be 263 µm3 in our previous study 14.
Furthermore, the speed of blood flow can be estimated by analyzing of the flow-induced decorrelation of adjacent photoacoustic A-line signals. Briefly, by fitting the experimentally measured decorrelation curve against the theoretical model of a second-order exponential decay, the decay constant can be calculated, from which the flow speed can be derived based on the linear relationship between these two parameters 18.
Moreover, we have developed a segmentation algorithm 15,19, which allows us to extract the structural and hemodynamic parameters at the single-vessel level. The segmentation procedure consists of four steps: (1) manually identify the boundaries of individual vessels; (2) refine the boundaries using Otsu's method 20; (3) divide the vessels into segments based on bifurcation points; (4) remove overlapping vessels, if any. With this segmentation algorithm, the blood flow speed and vessel diameter can be extracted at the single-vessel level, from which the volumetric blood flow of individual vessels can be calculated as
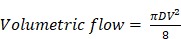 , ,
|
where D is the vessel diameter and V is the blood flow speed along the central axis of the vessel.
Further, the total cerebral blood flow (CBF) over the region of interest (ROI) can be estimated by summing up the volumetric flow of all feeding arteries. Since the oxygen extraction fraction (OEF) can be calculated using the measured sO2, the cerebral metabolic rate of oxygen (CMRO2) can be derived as
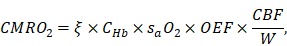 |
where 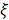 is the oxygen binding capacity of hemoglobin (1.36 mL of oxygen per gram hemoglobin),
is the oxygen binding capacity of hemoglobin (1.36 mL of oxygen per gram hemoglobin), 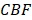 is the total volumetric flow through the region, and W is the tissue weight estimated by assuming an average cortical thickness of 1.2 mm 21 and an average tissue density of 1.05 g/ml 22. A more detailed description of the quantification of cerebral microvascular diameter, hemodynamics and oxygen metabolism can be found in our previous reports 14,15.
is the total volumetric flow through the region, and W is the tissue weight estimated by assuming an average cortical thickness of 1.2 mm 21 and an average tissue density of 1.05 g/ml 22. A more detailed description of the quantification of cerebral microvascular diameter, hemodynamics and oxygen metabolism can be found in our previous reports 14,15.
Animal Preparation for Awake-brain Imaging
The CD-1 mouse (male, 9-11 weeks old, Charles River Laboratory) was anesthetized by 1.5% isoflurane and kept at 37°C using a heating pad. After removing the hair in the scalp with a shaver and hair removal cream (Surgi Cream), a skin incision was made. Then, the periosteum was removed to expose the skull. Once the exposed skull was cleaned and dried, dental cement (C&B Metabond, Parkell Inc.) was applied to adhere a small nut (90730A005, McMaster-Carr) to the skull contralateral to the ROI. Ketoprofen (5 mg/kg) was subcutaneously injected to alleviate the pain caused by the surgical procedure. The mouse was returned to its home cage, when the cement was solidified and the nut was firmly adhered. In the following five consecutive days, the mouse was trained to acclimate the head restraint and attenuate possible stress.
One day before the PAM imaging, the skull above the ROI was carefully thinned following the established protocol 23. To avoid bleeding during the skull thinning, a mixed solution of lidocaine (1%) and epinephrine (1:100,000) was applied topically. According to the previous study 24, no severe inflammation or microglial activation was expected due to the relatively thick skull window (~100 μm). During the PAM imaging, the mouse brain was in contact against a temperature-controlled water tank, which was kept at 37 °C to maintain the brain temperature. The bottom of the water tank was sealed by an optically and acoustically transparent polyethylene membrane, and a thin layer of ultrasonic gel was applied between the thinned-skull window and the membrane for acoustic coupling.
Blood S1P Regulation and Measurement
The blood S1P levels were manipulated using our optimized selective SphK2 inhibitor (SLM6031434, [S-2-(3-[4-(octyloxy)-3-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide]), while the less potent R-enantiomer (SLM6081442, [R-2-(3-[4-(octyloxy)-3-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-yl) pyrrolidine-1-carboximidamide]) was used as a control. The synthesis and characteristics of SLM6031434 and SLM6081442 are detailed in our previous report 25. Groups of CD-1 mice (male, 9 weeks old, Charles River Laboratory) were injected intravenously with different doses of SLM6031434, while the vehicle group was injected with an equal amount of phosphate-buffered saline. After the injection, mice were bled via a tail nick at 1, 2 and 4 hours. The whole blood was acquired and processed immediately for liquid chromatography mass spectrometry (LC/MS) analyses to quantify the blood S1P level, following previously reported sample preparation protocols 26. The LC/MS analyses were performed using a Shimadzu Prominence LC and an AB Sciex 4000 QTRAP triple quadrupole mass spectrometer 25.
Transient Middle Cerebral Artery Occlusion
The mouse was anesthetized using isoflurane (1.5-2.0% for induction and 1.0-1.5% for maintenance). A feedback-controlled heating pad was used to maintain its body temperature during the surgery. A midline neck incision was made to expose the right common carotid artery and external carotid artery. The common carotid artery was temporarily clamped, and the external carotid artery was ligated with strings. Then, a small incision was made in the common carotid artery before the bifurcation to introduce a filament (Beijing CiNongtech Co.). The filament was carefully inserted into the internal carotid artery to occlude the takeoff point of the middle cerebral artery. After 90-minute occlusion, the filament was withdrawn for reperfusion. Successful occlusion (<20% of the baseline flow) and reperfusion (>70% of the baseline flow) were both confirmed using laser Doppler flowmetry.
Stroke Outcome Evaluation
The motor coordination was evaluated 24 hours before and 24 hours after the tMCAO. The mouse was placed on an accelerating rotarod. The speed of the rotarod was increased from 4 rpm to 40 rpm in 5 minutes. The latency and speed of the mouse falling off the rotarod were recorded. Each mouse was tested five times. The speed-latency index of each test was calculated as latency (in second) × speed (in rpm), and the mean index of the five trials was computed to reflect the motor coordination function before and after the MCAO. All mice were trained 5 times per day for 3 continuous days before formal tests. The 3-day training started 4 days before the tMCAO. The ratio of the speed-latency index measured 24 hours after the stroke onset to that measured before the onset was used to indicate the changes in the motor functions.
The neurological functions were evaluated 24 hours after the tMCAO, right before animal euthanasia. The tMCAO-induced neurological deficit in the left hemisphere was scored in a blind fashion according to an established 8-point scale: 0, no apparent deficits; 1, failure to fully extend the right forepaw; 2, decreased grip of the right forelimb; 3, spontaneous movement in all directions and contralateral circling only if pulled by the tail; 4, circling/walking to the right; 5, walking only if stimulated; 6, unresponsiveness to stimulation and with depressed level of consciousness; and 7, dead 27,28. Thus, higher neurological deficit scores indicate worse ischemic outcomes.
The mouse was euthanized for infarct volume analysis 24 hours after the tMCAO. Right after euthanasia, the brain was rapidly dissected and cut coronally into 1-mm-thick slices except for the first and last slices, which were 2-mm thick. Assessment of the infarct volume was performed with 2,3,5-triphenyltetrazolium chloride (TTC) staining. The infarct areas were quantified using the ImageJ (v1.60, National Institutes of Health), and the percentage of infarct volume in the ipsilateral hemisphere was calculated.
Statistical Analysis
The two-way analysis of variance (ANOVA) was used in Figure 1 to compare the readouts of different doses of SLM6031434 and the vehicle, at any of the given time points. The paired t-test was used in Figure 3 to examine the statistical significance of the hypoxia-induced changes and SLM6031434-induced changes. The One-way ANOVA and Tukey's honest significance test with correction for multiple comparisons was used in Figure 4 to analyze the time-dependent hemodynamic responses to SLM6031434. The unpaired t-test was used in Figs. 5b and 5c to compare the differences in infarct volume and behavior performance between the pre-treatment, post-treatment, and control groups. The one-way repeated measures analysis of variance with the Holm-Sidak test was used in Figure 5d to compare the differences in neurological deficient between the three groups. All data are shown in mean ± standard deviation. All p-values of <0.05 were considered statistically significant.
Figure 1.

Pharmacodynamics of the SphK2 inhibitor. (a) SLM6031434 level and (b) S1P level at 1, 2, and 4 hours in the whole blood after the injection of saline (Vehicle) or the indicated doses of SLM6031434. Sample size: n=4. *, p<0.05.
Figure 3.

Quantitative analysis of S1P-induced hemodynamic and oxygen-metabolic responses in the awake hypoxic mouse brain. (a) CHb, (b) arterial sO2, (c) venous sO2, (d) OEF, (e) average vessel diameter, (f) average blood flow speed, (g) regional CBF, and (h) regional CMRO2. Normoxia, before the injection of SLM6031434 under normoxia; Hypoxia, before the injection under hypoxia; Post-injection, 2 hours after the injection under hypoxia. Sample size: n=4. Data are shown as Mean ± SD. *, p<0.05.
Figure 4.

Dynamics of the S1P-induced responses in the awake hypoxic mouse brain. (a) Time-lapse PAM of the hemodynamic changes before (normoxia and hypoxia) and after the injection of SLM6031434 (0.5, 1, 1.5, and 2 hours). Time courses of (b) CHb, (c) arterial sO2, (d) venous sO2, (e) OEF, (f) average vessel diameter, (g) average blood flow speed, (h) regional CBF, and (i) regional CMRO2. Scale bar, 500 µm. Data are shown as Mean ± SD. *, p<0.05.
Figure 5.

S1P-induced neuroprotection against ischemic stroke. (a) Representative images of the TTC stain of brain coronal slices harvested 24 hours after the tMCAO. Statistical comparison of the (b) infarct volume, (c) motor functions, and (d) neurological deficit between the control group, pre-treatment group, and post-treatment group. Sample size: n=7. *, p<0.05.
Results
Pharmacodynamics of the SphK2 inhibitor
To examine the influence of SLM6031434 on the blood S1P level and to identify the optimal dosage, three different doses (0.05, 0.5, and 2 mg/kg) were evaluated. After the injection of SLM6031434, animals were bled at 1, 2, and 4 hours to measure the levels of blood S1P and the compound, using LC/MS analyses. As shown in Figure 1a, SLM6031434 increased the blood S1P level in a dose-dependent manner, and a dose of 2 mg/kg doubled the S1P level 2 hours after injection. Moreover, the S1P level remained elevated 4 hours after injection (Figure 1a) and the inhibitor was detectable in the blood even 6 hours later (data not shown), indicating that the S1P upregulation was continuously in effect (Figure 1b). Based on the LC/MS analyses, the dosage of 2 mg/kg was chosen for the studies on cerebral hemodynamics and neuroprotection.
Hemodynamic Responses to Blood S1P Increase in Hypoxic Brain
Given that acute tissue hypoxia is a major cause of injury in ischemic stroke, we studied the neuroprotective effect of S1P on cerebral hypoxia, which was induced by inhalation of mixed medical air and nitrogen with an oxygen concentration of ~12%. Specifically, cerebral hemodynamics, including CHb, sO2 and CBF, in the awake mouse was assessed before and 2 hours after the injection of 2-mg/kg SLM6031434, by the head-restrained multi-parametric PAM. The time point of 2 hours post-injection was chosen to be consistent with the pre-treatment study in ischemic stroke, in which the tMCAO was performed 2 hours after the injection of this compound.
As shown in Figure 2, following the switch from normoxia to hypoxia, decreases in both arterial and venous sO2 values were observed, accompanied by a noticeable increase in blood flow speed. These were in good agreement with our previous observations in the awake mouse brain 15. These mice were reimaged two hours after injection of either the active (SLM6031434) or inactive (SLM6081442) compound through a needle pre-implanted in the tail vein. In contrast to the lack of hemodynamic responses to the inactive compound (Figure 2a), the sO2 values in the mice treated with the active compound largely recovered while the blood flow remained unchanged (white arrows in Figure 2b). Interestingly, no hemodynamic response to the S1P upregulation was observed in the normoxic mouse brain (Figure S2).
Figure 2.

S1P-induced hemodynamic responses in the awake hypoxic mouse brain. Time-lapse PAM of CHb, sO2 and blood flow speed before the injection of (a) “inactive” compound SLM6081442 and (b) “active” compound SLM6031434. Normoxia, before the injection under normoxia; Hypoxia, before the injection under hypoxia; Post-injection, 2 hours after the injection under hypoxia. The white arrows in the 1st and 2nd rows of (b) highlight the changes in sO2 and blood flow speed, respectively. Scale bar, 500 µm.
We repeated this experiment in 4 mice for statistical analysis. As expected, CHb was not affected by either hypoxia or the active compound (Figure 3a). In contrast, the arterial and venous sO2 significantly dropped in response to hypoxia (from 93.7±0.9% to 77.8±3.3% for the arterial sO2 and from 70.3±0.7% to 45.4±3.2 % for the venous sO2). Increasing the blood S1P level with SLM6031434 was able to recover the sO2 values back to the corresponding baseline levels 2 hours after injection (90.8±4.4% for the arterial sO2 and 67±8.1% for the venous sO2, as respectively shown in Figure 3b and 3c). These sO2 changes resulted in increased OEF under hypoxia (from 0.25±0.01 to 0.42±0.04) and the subsequent decrease 2 hours after the injection of SLM6031434 (0.26±0.07). Interestingly, the increased S1P level in blood did not significantly change the average vessel diameter (Figure 3e) and blood flow speed (Figure 3f). As a result, the CBF was also unaffected by SLM6031434 (Figure 3g). Due to the decreased OEF and unchanged CBF, CMRO2 was found to decrease by 36.7 % in response to the injection of SLM6031434 (Figure 3h), suggesting that blood S1P lowered cerebral oxygen demand and thus mitigated the oxygen crisis under hypoxia.
Time Course of S1P-induced Hemodynamic Responses under Hypoxia
Given the observation that the blood S1P level increased gradually after the injection of SLM6031434 (Figure 1), we also investigated the time course of S1P-induced cerebral hemodynamic responses. To this end, the mice were first imaged under normoxia to obtain the baseline and then time-lapse monitored under hypoxia to reveal the dynamic responses. To allow for equilibrium under hypoxia, SLM6031434 was injected after imaging the hypoxic brain for 60 minutes. Then, the brain was monitored for 2 hours with an interval of 30 minutes (Figure 4a).
Again, this experiment was repeated in 4 mice for statistical analysis. The baseline values measured under normoxia were used to calculate the percentage changes of cerebral hemodynamics in response to hypoxia and SLM6031434 intervention. While CHb remained unchanged after the injection of the active compound (Figure 4b), both the arterial sO2 (Figure 4c) and venous sO2 (Figure 4d) gradually increased following the injection and eventually became significantly higher than the corresponding pre-injection values at 2 hours (from 83.0±4.0% pre-injection to 96.7±5.5% 2 hours post-injection for the arterial sO2 and from 64.5±4.7% pre-injection to 95.3±11.8% 2 hours post-injection for the venous sO2). Determined by the difference between the arterial and venous sO2, OEF did not experience significant change until 1.5 hours after the injection and eventually dropped from 167.5±19.0% pre-injection to 105.5±31.4% 2 hours post-injection (Figure 4e). In contrast to the dynamic response in blood oxygenation, the vessel diameter (Figure 4f), flow speed (Figure 4g), and CBF (Figure 4h) remained unchanged over the entire monitoring period. Given the lack of change in CBF, the time course of CMRO2 followed that of OEF, with significant changes observed after 1.5 hours post-injection (from 186.2±24.9% pre-injection to 151.7±30.3% at 1.5 hours and 118.3±11.5% at 2 hours; Figure 4i).
Neuroprotective Effect of S1P against Ischemic Stroke
Capable of improving blood oxygenation and reducing cerebral oxygen demand, increasing the blood S1P level was expected to protect the brain against ischemic stroke. To evaluate the neuroprotective effects of pre-treatment and post-treatment with SLM6031434, mice were randomly assigned into 3 groups (control, pre-treatment, and post-treatment). The mice in the pre-treatment group were administered with the active compound 2 hours prior to the tMCAO, while the post-treatment groups were injected immediately after the reperfusion. The control groups were subject to the same tMCAO procedure but without treatment. After finishing the injection and tMCAO, the mice were returned to their home cages and closely monitored until the completion of neurological evaluation and brain harvest.
As shown in the representative TTC pictures (Figure 5a), the infarct region is noticeably smaller in the pre-treatment mouse, compared to that in the control mouse. However, the post-treatment mouse did not show clear reduction in infarct volume. Indeed, statistical analysis (Figure 5b) showed significantly reduced infarct volume in the pre-treatment group (26.8±11.6%) compared to that in the control group (62.5±6.9%), while the improvement in the post-treatment group was not statistically significant (52.8±17.4%).
As shown in Figure 5c, although all mice demonstrated impaired motor coordination after brain ischemia with the speed-latency index ratio less than 0.5, the pre-treatment group showed much better motor coordination functions compared to the control and post-treatment groups. Furthermore, the neurological deficit score also suggested improvement of the stroke outcome in the pre-treatment group. As shown in Figure 5d, both the control group (average score: 4.4) and the post-treatment group (average score: 4.2) showed severely impaired neurological functions. Most of mice in these two groups only circled or walked to one side. In contrast, most mice in the pre-treatment group (average score: 2.9) still had spontaneous movement in all directions and did contralateral circling only when pulled by the tail.
Discussions
In previous studies 29-32, S1P has been reported to be neuroprotective against ischemic stroke. However, the underlying mechanisms remain incompletely understood. Recently, Sun et al. have found that elevated S1P levels in blood promote the oxygen release for adaptation to high-altitude hypoxia 11. Moreover, it has been demonstrated that the pathways involving S1P and its kinases are related to hypoxia signaling 33-35. Together, these observations suggest that the S1P-induced neuroprotection against brain hypoxia and ischemia may be mediated, in part, through the regulation in cerebral hemodynamics and oxygen metabolism. However, a direct experimental validation of this hypothesis is still missing, mainly due to technical limitations.
Capitalizing on the head-restrained multi-parametric PAM, we performed the first in vivo characterization of S1P-induced hemodynamic and oxygen-metabolic changes in the mouse brain without the confounding of general anesthesia. Using SLM6031434, a selective SphK2 inhibitor, we were able to gradually increase the blood S1P level and double it 2 hours after the injection. Our PAM studies showed that, in the hypoxic brain, the dynamic increase of blood S1P resulted in progressively increased arterial and venous sO2. In contrast, the vessel diameter and CBF remained unchanged. These findings suggest that the protective effect of S1P again cerebral hypoxia is likely mediated through changes in blood oxygenation rather than blood supply. Moreover, the decreased OEF (due to increased venous sO2) and unchanged CBF together resulted in reduced CMRO2, in response to the elevated blood S1P. Recent findings showed that increased blood S1P enhances the release of membrane-bound glycolytic enzymes to the cytosol, inducing glycolysis and the production of 2,3-bisphosphoglycerate 11. Also, other studies showed that S1P increases glucose uptake through trans-activation of insulin receptor 36. The S1P-induced shift in cerebral metabolism from oxidative phosphorylation to glycolysis may be responsible for the observed reduction in CMRO2 and help alleviate oxygen crisis in the hypoxic brain. Interestingly, under normoxia, the cerebral hemodynamics and oxygen metabolism were insensitive to changes in blood S1P. Although the underlying mechanism requires further investigation, it is reasonable to speculate that the S1P-induced metabolic re-programming may require activation of hypoxia-related signaling pathways.
We validated the neuroprotective effect of S1P against ischemic stroke in the established mouse model of tMCAO. Although significant improvement in the stroke outcome was observed in the pre-treatment group, no noticeable benefit was shown in animals treated with SLM6031434 immediately after the reperfusion. Given the relatively slow pharmacodynamics of this compound in eliciting the protective hemodynamic and oxygen-metabolic responses (e.g., it took 2 hours to reduce CMRO2 to near the baseline level, as shown in Figure 4), it may not be fast enough to protect the ischemic penumbra from permanent infarction when applied following reperfusion. To increase the translational potential of this neuroprotective agent, further studies are required to improve its pharmacodynamics.
Although S1P synthesis relies on two different forms of SphK, SphK1 and SphK2, our results showed that injection of the SphK2 inhibitor was sufficient to increase the blood S1P level within 4 hours. This is in contrast to S1P reduction induced by SphK1 inhibitors 37. Our results and previous studies, together, suggest that the blood S1P turnover is likely due to the role of SphK2 in the clearance of S1P from blood 25. However, the mechanism underlying the clearance remains obscure, and the role of SphK2 in this process remains unknown. Besides, the whole blood S1P was measured in this study, including S1P in different components such as erythrocyte, platelet and plasma. However, the roles of SphK1 and SphK2 in these components are different. For instance, SphK1 is responsible for erythrocyte S1P synthesis and most plasma S1P is derived from erythrocytes, while SphK2 is responsible for platelet S1P synthesis 38. Therefore, further studies are needed to better understand how SLM6031434 increases S1P in the blood and the molecular mechanisms of the S1P-induced hemodynamic responses.
In conclusion, we studied the hemodynamic mechanisms of the S1P-induced neuroprotection in the awake mouse brain using the head-restrained multi-parametric PAM. Our results showed the elevated blood S1P led to improved cerebral blood oxygenation and reduced oxygen demand under hypoxia, both of which alleviate the oxygen crisis during brain ischemia and hypoxia. Consistent with these findings, significantly improved neurological outcomes were observed in animals pre-treated with the SphK2 inhibitor but not in the post-treatment group. The failure to induce neuroprotective effects by post-treatment was likely due to the relatively slow pharmacodynamics of the compound to elicit S1P increase. Providing new insights into the S1P-mediated neuroprotection from the much understudied hemodynamic perspective, our work may facilitate the translation of this therapeutic strategy for brain hypoxia and ischemia.
Supplementary Material
Supplementary figures.
Acknowledgments
This work was supported in part by the National Institutes of Health (NS099261 to SH and GM121075 to KRL/WLS) and the American Heart Association (15SDG25960005 to SH). The authors thank Dr. Vlad Serbulea (UVa Pharmacology) for performing the LC/MS analyses.
Abbreviations
- S1P
sphingosine 1-phosphate
- PAM
photoacoustic microscopy
- DALYs
disability-adjusted life-years
- tPA
tissue plasminogen activator
- SphKs
sphingosine kinases
- SphK1/2
sphingosine kinase 1/2
- tMCAO
transient middle cerebral artery occlusion
- sO2
the oxygen saturation of hemoglobin
- RBCs
red blood cells
- CHb
the total concentration of hemoglobin
- CBF
cerebral blood flow
- OEF
oxygen extraction fraction
- CMRO2
the cerebral metabolic rate of oxygen
- LC/MS
liquid chromatography mass spectrometry: TTC: 2,3,5-triphenyltetrazolium chloride
- ANOVA
analysis of variance.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M. Heart Disease and Stroke Statistics—2017 Update. vol. 135; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GBD 2013 Mortality, Causes of Death Collaborators G 2013 M, C of D. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–71. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hankey GJ. Stroke. Lancet. 2017;389:641–54. doi: 10.1016/S0140-6736(16)30962-X. [DOI] [PubMed] [Google Scholar]
- 4.Musuka TD, Wilton SB, Traboulsi M, Hill MD. Diagnosis and management of acute ischemic stroke: Speed is critical. CMAJ. 2015;187:887–93. doi: 10.1503/cmaj.140355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans MRB, White P, Cowley P, Werring DJ. Revolution in acute ischaemic stroke care: a practical guide to mechanical thrombectomy. Pract Neurol. 2017;17:252–65. doi: 10.1136/practneurol-2017-001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molina CA. Reperfusion therapies for acute ischemic stroke: Current pharmacological and mechanical approaches. Stroke, vol. 42; 2011. [DOI] [PubMed] [Google Scholar]
- 7.Singhal AB, Lo EH, Dalkara T, Moskowitz MA. Ischemic stroke: Basic pathophysiology and neuroprotective strategies. Acute Ischemic Stroke. 2006:1–26. [Google Scholar]
- 8.Kim GS, Yang L, Zhang G, Zhao H, Selim M, McCullough LD. et al. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat Commun. 2015;6:7893. doi: 10.1038/ncomms8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spiegel S, Milstien S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 10.Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55:1596–608. doi: 10.1194/jlr.R046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun K, Zhang Y, D'Alessandro A, Nemkov T, Song A, Wu H. et al. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat Commun. 2016;7:12086. doi: 10.1038/ncomms12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L V, Hu S, Culver JP, Ntziachristos V, Holboke MJ, Yodh AG. et al. Photoacoustic Tomography: In Vivo Imaging from Organelles to Organs. Science (80- ) 2012;335:1458–62. doi: 10.1126/science.1216210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu S. Listening to the Brain With Photoacoustics. IEEE J Sel Top Quantum Electron. 2016;22:1–10. [Google Scholar]
- 14.Ning B, Sun N, Cao R, Chen R, Kirk Shung K, Hossack JA. et al. Ultrasound-aided Multi-parametric Photoacoustic Microscopy of the Mouse Brain. Sci Rep. 2015;5:18775. doi: 10.1038/srep18775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao R, Li J, Ning B, Sun N, Wang T, Zuo Z. et al. Functional and oxygen-metabolic photoacoustic microscopy of the awake mouse brain. Neuroimage. 2017;150:77–87. doi: 10.1016/j.neuroimage.2017.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang HF, Maslov K, Sivaramakrishnan M, Stoica G, Wang L V. Imaging of hemoglobin oxygen saturation variations in single vessels in vivo using photoacoustic microscopy. Appl Phys Lett. 2007;90:53901. [Google Scholar]
- 17.Everds N. The Laboratory Mouse. Elsevier; 2004. doi:10.1016/B978-012336425-8/50070-4. [Google Scholar]
- 18.Chen S-L, Xie Z, Carson PL, Wang X, Guo LJ. In vivo flow speed measurement of capillaries by photoacoustic correlation spectroscopy. Opt Lett. 2011;36:4017–9. doi: 10.1364/OL.36.004017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soetikno B, Hu S, Gonzales E, Zhong Q, Maslov K, Lee J-M. et al. Vessel segmentation analysis of ischemic stroke images acquired with photoacoustic microscopy. SPIE BiOS. 2012;8223:822345. [Google Scholar]
- 20.Sezgin Mehmet, Sankur B lent. Survey over image thresholding techniques and quantitative performance evaluation. J Electron Imaging. 2004;13:146–65. [Google Scholar]
- 21.DeFelipe J. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Front Neuroanat. 2011;5:29. doi: 10.3389/fnana.2011.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chong SP, Merkle CW, Leahy C, Srinivasan VJ. Cerebral metabolic rate of oxygen (CMRO_2) assessed by combined Doppler and spectroscopic OCT. Biomed Opt Express. 2015;6:3941. doi: 10.1364/BOE.6.003941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V. et al. Removable cranial windows for long-term imaging in awake mice. Nat Protoc. 2014;9:2515–38. doi: 10.1038/nprot.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan W-BB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc. 2010;5:201–8. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kharel Y, Morris EA, Congdon MD, Thorpe SB, Tomsig JL, Santos WL. et al. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J Pharmacol Exp Ther. 2015;355:23–31. doi: 10.1124/jpet.115.225862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaner RL, Allegood JC, Park H, Wang E, Kelly S, Haynes CA. et al. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009;50:1692–707. doi: 10.1194/jlr.D800051-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers DC, Campbell CA, Stretton JL, Mackay KB. Correlation Between Motor Impairment and Infarct Volume After Permanent and Transient Middle Cerebral Artery Occlusion in the Rat. Stroke. 1997;28:2060–6. doi: 10.1161/01.str.28.10.2060. [DOI] [PubMed] [Google Scholar]
- 28.Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–62. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brait VH, Tarrasón G, Gavaldà A, Godessart N, Planas AM. Selective Sphingosine 1-Phosphate Receptor 1 Agonist Is Protective Against Ischemia/Reperfusion in Mice. Stroke. 2016;47:3053–6. doi: 10.1161/STROKEAHA.116.015371. [DOI] [PubMed] [Google Scholar]
- 30.Kimura A, Ohmori T, Kashiwakura Y, Ohkawa R, Madoiwa S, Mimuro J. et al. Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain infarction. Stroke. 2008;39:3411–7. doi: 10.1161/STROKEAHA.108.514612. [DOI] [PubMed] [Google Scholar]
- 31.Iwasawa E, Ishibashi S, Suzuki M, Li F, Ichijo M, Miki K, Sphingosine-1-Phosphate Receptor 1 Activation Enhances Leptomeningeal Collateral Development and Improves Outcome after Stroke in Mice. J Stroke Cerebrovasc Dis; 2018. [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa Y, Suzuki H, Sozen T, Rolland W, Zhang JH. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke. 2010;41:368–74. doi: 10.1161/STROKEAHA.109.568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ader I, Malavaud B, Cuvillier O. When the sphingosine kinase 1/sphingosine 1-phosphate pathway meets hypoxia signaling: New targets for cancer therapy. Cancer Res. 2009;69:3723–6. doi: 10.1158/0008-5472.CAN-09-0389. [DOI] [PubMed] [Google Scholar]
- 34.Bouquerel P, Gstalder C, Müller D, Laurent J, Brizuela L, Sabbadini RA. et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis. 2016;5:e209. doi: 10.1038/oncsis.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnitzer SE, Weigert A, Zhou J, Brune B. Hypoxia Enhances Sphingosine Kinase 2 Activity and Provokes Sphingosine-1-Phosphate-Mediated Chemoresistance in A549 Lung Cancer Cells. Mol Cancer Res. 2009;7:393–401. doi: 10.1158/1541-7786.MCR-08-0156. [DOI] [PubMed] [Google Scholar]
- 36.Rapizzi E, Taddei ML, Fiaschi T, Donati C, Bruni P, Chiarugi P. Sphingosine 1-phosphate increases glucose uptake through trans-activation of insulin receptor. Cell Mol Life Sci. 2009;66:3207–18. doi: 10.1007/s00018-009-0106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kharel Y, Mathews TP, Gellett AM, Tomsig JL, Kennedy PC, Moyer ML. et al. Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem J. 2011;440:345–53. doi: 10.1042/BJ20110817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urtz N, Gaertner F, Von Bruehl ML, Chandraratne S, Rahimi F, Zhang L. et al. Sphingosine 1-phosphate produced by sphingosine kinase 2 intrinsically controls platelet aggregation in vitro and in vivo. Circ Res. 2015;117:376–87. doi: 10.1161/CIRCRESAHA.115.306901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures.