

Abstract
Inflammation has been implicated in the pathogenesis of hypertension and recent evidence suggests that isolevuglandin (IsoLG)-protein adducts play a role. Several hypertensive stimuli contribute to formation of IsoLG-protein adducts including excess dietary salt and catecholamines. The precise intracellular mechanisms by which these hypertensive stimuli lead to IsoLG-protein adduct formation are still not well understood; however, there is now evidence implicating NADPH-oxidase derived reactive oxygen species (ROS) in this process. ROS oxidize arachidonic acid leading to formation of IsoLGs, which non-covalently adduct to lysine residues and alter protein structure and function. Recent studies suggest that these altered proteins act as neo-antigens leading to an autoimmune state that results in hypertension. The goal of this mini-review is to highlight some of the hypertensive stimuli and the mechanisms contributing to IsoLG-protein adduct formation leading to inflammation and hypertension.
Keywords: Hypertension, inflammation, isolevuglandins
INTRODUCTION
Hypertension is a growing health care burden and is the leading cause of mortality due to myocardial infarction, stroke, heart failure, and chronic kidney disease. Recently, the American Heart Association (AHA) and the American College of Cardiology (ACC) developed new classification criteria that put nearly half of the American population in the hypertensive category (1). Unfortunately, the National Health and Nutritional Examination Study (NHANES) found that blood pressure is uncontrolled in approximately one-half of hypertensive patients and despite extensive research, the pathogenesis of essential hypertension remains elusive (2, 3).
In general, alterations in renal and systemic vascular function are required to sustain an elevation in blood pressure (4). In the past decade, it has become apparent that inflammation is important for the initiation and maintenance of these pathophysiologic changes and that both the innate and adaptive immune systems contribute to hypertension. Virtually all cells of the innate and adaptive immune system, including monocytes, dendritic cells (DCs), macrophages, T cells, and B cells have been implicated in the genesis of hypertension (5). Several studies have emphasized a role of CD8+ T cells, which are specifically activated by antigenic peptides presented in class 1 major histocompatibility complexes (MHC-I) (6). These studies combined suggest an autoimmune etiology of hypertension. However, the intra and intercellular processes required for the initiation of this autoimmune response are not characterized.
Activation of the immune system in hypertension occurs by multiple potential mechanisms (7). Sympathetic activation of immune cells occurs directly by the binding of catecholamines to adrenergic receptors expressed on immune cells (8). This has been shown to produce the elaboration of pro-inflammatory cytokines IL-1 β and IL-6 (9). Moreover, deletion of anti-oxidant enzymes in the brain enhances T-cell activation (10). Interestingly, elevated extracellular sodium concentrations have been shown to stimulate pro-inflammatory cytokine secretion by T-cells (11, 12). Sodium increases intracellular ROS by activation of NADPH oxidase (13). Finally, mechanical stretch induces the secretion of pro-inflammatory cytokines and the production of intracellular ROS from endothelial cells, leading to activation of inflammatory pathways (14). These studies suggest a central role of ROS in the initiation of inflammation and hypertension.
One consequence of ROS production is arachidonic acid peroxidation leading to formation of IsoLGs (5, 15-17). IsoLGs were first discovered in 1985 by Dr. Salomon who found that they form from non-enzymatic rearrangement of prostaglandin H2 as byproducts of the cyclooxygenase pathway (18). They are also formed as products of the isoprostane pathway by rearrangement of the prostaglandin H2-like endoperoxide intermediates (19). While the cyclooxygenase pathway yields only 2 gamma-ketoaldehydes, the isoprostane pathway yields 64 different IsoLG isomers. IsoLGs are highly labile and form covalent bonds to lysine residues leading to post-translational protein modifications (20) (Figure 1). The identification of the IsoLG-adducted peptides and the mechanism by which they are processed are unknown; however, recent studies have provided insight into their role in autoimmune mediated hypertension. In this review, we summarize findings providing insights into the mechanistic processes and the hypertensive stimuli contributing to IsoLG-protein adduct formation in hypertension.
Figure 1: IsoLG-protein adduct formation.
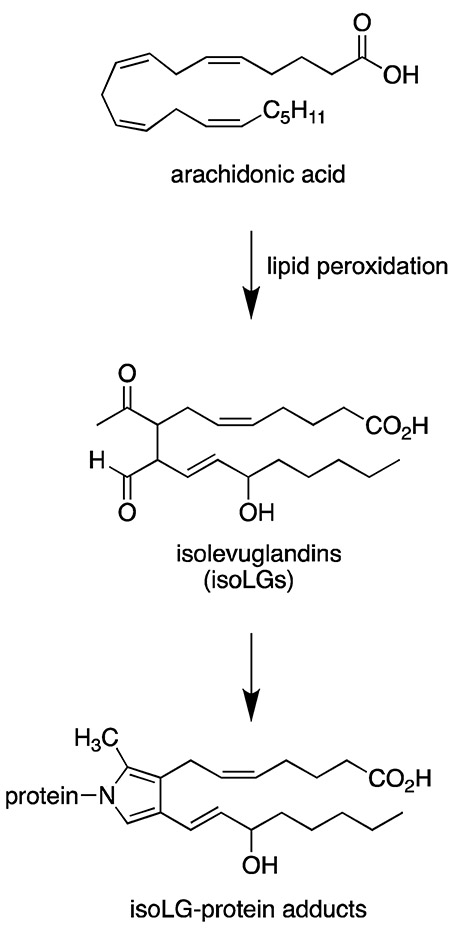
Isolevuglandins (IsoLGs) are formed from lipid peroxidation of arachidonic acid. They are highly labile and form covalent bonds with lysine residues of nearby proteins leading to formation of IsoLG-protein adducts.
ISOLEVUGLANDINS AND HYPERTENSION
IsoLG-protein adducts as biomarkers of hypertension:
IsoLG-protein adducts have been found to accumulate in inflammatory diseases including asthma and alcoholic liver disease (21, 22). In the past few years, we found that IsoLG-protein adducts are also increased in hypertension. Immunohistochemistry analysis using a single chain antibody that recognizes IsoLG-lysine adducts on any protein independent of the amino acid backbone (23) revealed a 2-fold increase of IsoLG-protein adducts in the heart and aorta of mice with angiotensin II-induced hypertension (15). Using flow cytometry, we found that angiotensin II infusion increases intracellular accumulation of IsoLG-protein-adducts in antigen presenting dendritic cells (DCs) and this was confirmed by mass spectrometry analysis (15). In addition to mice receiving angiotensin II infusion, accumulation of IsoLG-protein adducts has been found in other models of hypertension including deoxycorticosterone acetate (DOCA)–salt, L-Nitroarginine methyl ester (L-NAME)/high salt, as well as in transgenic mice overexpressing smooth muscle specific NADPH oxidase subunit p22phox (15, 24, 25).
Importantly, IsoLG-protein adducts increase in human hypertension. We found that patients with resistant hypertension had increased levels of plasma F2-isoprostanes compared to well-controlled and normotensive subjects (23.2 ± 11.5 vs. 18.3 ± 10.3 vs. 11.6 ± 7.0 pg/mL, p<0.05 respectively; adjusted for BMI, HbA1C, LDL, and statin use. IsoLGs and F2-isoprostanes are co-produced in the isoprostane pathway of lipid peroxidation (17, 19, 26). We also found that the IsoLG-protein adduct content in monocytes of humans with hypertension was 3-fold greater than those without hypertension (15). In addition, we observed a similar increase in IsoLG-protein adducts in a small population (< 0.5%) of circulating CD83+ which were CD19+ B cells (15). These studies suggest that IsoLGs are increased in hypertension and may serve as biomarkers of this disease.
IsoLG-protein adducts as mediators of hypertension:
There is compelling evidence that IsoLG-protein adducts mediate inflammation and contribute to hypertension. As mentioned above, IsoLGs once formed react with lysine residues on proteins forming lactam adducts and cross-linked proteins. Zagol-Ikapitte and Amarnath et al. have synthesized several compounds that scavenge IsoLGs in vitro at a rate more than 2 orders of magnitude greater than the potency of the reaction with the ε-amine of lysine (27, 28). One such compound is 2-hydroxybenzylamine (2-HOBA), and we have demonstrated that it prevents tissue accumulation of IsoLG-protein adducts in hypertension (15). We found that pre-treatment with 2-HOBA and several other IsoLG scavengers including 5-methyl-2-hydroxybenzylamine (5-Me-2-HOBA) and pentylpyridoxamine (PnPM) prevent hypertension with no apparent toxicity, while related compounds that exhibit low reactivity with IsoLGs such as N-methyl-2-HOBA (N-Me-2-HOBA), and 4-HOBA do not (15, 27). Importantly, the IsoLG scavengers demonstrated slow rate constants for reactions with malondialdehyde (MDA), another lipid modification, and these rates were unrelated to their ability to lower blood pressure (15). Of note, the effects of IsoLG scavengers on preventing hypertension cannot be attributed to inhibition of cyclooxygenase enzymes since Zagol-Ikapitte and Amarnath et al. demonstrated that none of these compounds inhibit cyclooxygenase enzymes (27). While these studies suggest that IsoLGs mediate hypertension and provide a potential therapeutic target, no studies demonstrating a lowering of already established hypertension have been done. This is especially important since it would mimic the clinical setting. In addition, studies describing the effects of infused IsoLGs on blood pressure have not been performed.
Mechanisms of IsoLG-mediated immune activation and hypertension:
Many potential mechanisms may underlie the role of IsoLG induced inflammation and hypertension. Specifically, IsoLGs might induce inflammation and hypertension, however, to date very few studies have been done. As mentioned above, IsoLGs are extremely reactive and covalently adduct to protein lysine residues which undergo further reactions to form extensive protein-protein and DNA cross-links (20, 29). The cellular fate of these cross-linked proteins is not clearly understood, however, Davies et al. have found that they are poorly degraded by the proteosome depending on the number of adducts that are present on the protein, and can inhibit the ability of the proteosome to degrade normal protein substrates (30). Proteosome inhibition induces NF-κB activation, which has been implicated in the pathogenesis of hypertension (31, 32). Thus, IsoLG-crosslinked proteins, could potentially promote an NF-κB-mediated inflammatory response leading to hypertension.
In 2014, we published a new mechanism underlying immune activation in hypertension mediated by IsoLGs. We found that hypertension causes a striking increase in formation of IsoLG-modified proteins in DCs which correlated with an increase in surface expression of B7 ligands CD80 and CD86 (15) (Figure 2). These B7 ligands interact with co-stimulatory molecules on T cells including CD28 and were previously shown to be critical for hypertension (33). This co-stimulatory signal, together with antigen presentation and cytokine production are required in the classical T cell activation pathway. We found that DCs that accumulate IsoLG-protein adducts produce pro-inflammatory cytokines IL-1β, IL-6 and IL-23 and induce T cell proliferation and production of TNF-α, IFN-γ and IL-17A which contribute to hypertension (5, 15, 16). Importantly, adoptive transfer of DCs from hypertensive mice prime hypertension to sub-pressor doses of angiotensin II, and this can be prevented if the donor mice are treated with the IsoLG scavenger 2-hydroxybenzylamine (2-HOBA) (15). Thus, IsoLG-adducted proteins may be presented by DCs as neoantigens that activate T cells. Activated T cells infiltrate the kidney and vasculature and release cytokines that promote vascular dysfunction and sodium/water retention, leading to hypertension (5).
Figure 2: Hypertensive stimuli leading to immune cell activation via IsoLG-protein adduct formation.
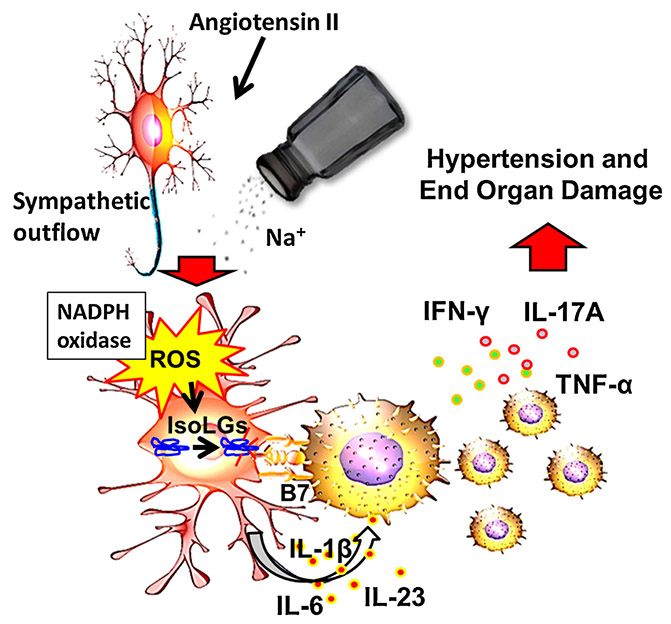
Hypertensive stimuli including angiotensin II, sympathetic outflow and excess dietary salt lead to increased NADPH oxidase-dependent production of reactive oxygen species (ROS) by antigen presenting myeloid derived cells. This leads to oxidation of arachidonic acid and formation of IsoLG-protein adducts. These are immunogenic and activate antigen presenting cells to produce inflammatory cytokines including IL 1b, IL6 and IL23. These myeloid cells activate T cell to proliferate and produce inflammatory cytokines including IL-17A, TNF-α and IFN-γ which lead to hypertension.
Numerous studies have found that oxidation leads to protein immunogenicity via mechanisms that are not well defined. While ROS production is increased in other cell types including vascular smooth muscle and endothelial cells, previous studies indicate that in the absence of T cells or the cytokine IL-17, hypertensive stimuli have minimal effects on vascular ROS production in vivo (5). This suggests that the adaptive immune system plays a very important role in signaling ROS production in these cells. Thus, it is conceivable that superoxide production in DCs might be causative in promoting neoantigen formation and ultimately T cell activation, which then leads to ROS production in vascular cells. Mice lacking either Nox2, or p22phox in DCs are protected against development of hypertension and IsoLG-protein adducts, suggesting that the NADPH oxidase is a major source of ROS that ultimately lead to the formation of IsoLGs (10, 15, 34). We found that ex vivo treatment of DCs with 1mM tert-butyl hydroperoxide (t-BHP) for 30 minutes led to IsoLG formation and surface expression of CD86 in DCs. T-BHP treated DCs promoted T cell proliferation and this was prevented by co-treatment with 2-HOBA. Adoptive transfer of t-BHP treated DCs augmented blood pressure elevation in response to low dose angiotensin II in recipient mice (15). These results confirm the concept that oxidative stress and IsoLG formation in DCs is sufficient to activate T cells and promote hypertension.
In addition to processing and presentation of intracellular IsoLG-protein adducts formed in response to increase oxidative stress to CD8+ T cells, we find that DCs likely process extracellular antigens and present them CD4+ T cells. Wu et al. found that DCs pulsed with aortic proteins from mice with vascular specific overexpression of the NADPH oxidase subunit p22phox had increased production of cytokines. Specifically, augmented levels of IL-1β, IL-6, TGF-β1, and GM-CSF were observed as were activated T cells of syngeneic mice (35). In addition, we found that exposing DCs to IsoLG-modified renal homogenates enhances their ability to drive CD8+ proliferation, likely through cross-presentation (15). Examination of T cell receptor usage revealed that an oligo-clonal population of CD8+ T cells in the kidney contributes to hypertension (36).
Another potential mechanism by which IsoLGs may contribute to inflammation in hypertension is through danger-associated molecular patterns (DAMPs) due to their injurious effect on forming protein-protein and DNA cross-links (20, 29). Antigen presenting cells recognize DAMPS via pattern recognition receptors and these have been implicated in hypertension and vascular dysfunction (37-41). However, the specific role of IsoLGs acting as DAMPS in hypertension remains to be elucidated.
In addition, IsoLG-adducted and crosslinked proteins could break immune tolerance and elicit an autoantibody response to normally non-immunogenic self-antigens. Previous studies have found that natural IgM antibodies are produced against lipid oxidation-specific epitopes which are thought to play an important protective role in preventing inflammatory reactions induced by the oxidatively modified lipids that they recognize (42, 43). These oxidation-specific epitopes are important targets of innate immunity in cardiovascular disease (42, 44). It is not known whether antibodies against IsoLGs contribute to hypertension.
Role of Angiotensin II in Promoting Isolevuglandin Formation:
The renin-angiotensin system plays a physiological role in controlling blood pressure and its dysregulation contributes to the development of hypertension (45). Angiotensin II, a key molecule in the renin-angiotensin system, is used as a hypertensive stimulus to induce experimental hypertension. It raises blood pressure through various mechanisms including vasoconstriction and secretion of aldosterone (45). Angiotensin II further acts on the kidneys to promote fluid retention/sodium reabsorption and on the brain to increase sympathetic out flow, fluid intake, and sodium appetite (46). In addition, recent studies have found that angiotensin II regulates the immune system (5). We recently found that angiotensin II-induced hypertension leads to production of NADPH oxidase-dependent superoxide in DCs resulting in increased lipid peroxidation IsoLG-protein adduct formation (15). Interestingly, we found that angiotensin II had a minimum effect on inducing IsoLG-protein adduct formation in DCs in vitro. These studies suggest that in vivo, angiotensin II may exert its effects on DCs indirectly, through other mechanism including augmentation of sympathetic outflow and increased sodium retention.
Renal Sympathetic Mechanisms and IsoLG-protein Adduct Formation in Hypertension:
In a recent study, we found that the kidney is a likely site for activation of DCs in hypertension. We discovered that hypertension induces formation of IsoLG-modified proteins in DCs, and that renal denervation prevents this (47). Moreover, DCs from mice that had undergone renal denervation could not confer hypertension to recipients (47). These data indicate that renal sympathetic nerves serve as a link between activation of the central nervous system and peripheral inflammation in hypertension.
There are two potential mechanisms that could lead to IsoLG-protein adduct accumulation in DCs. First, it is possible that DCs might themselves be activated by sympathetic nerves, which in turn could drive NADPH oxidase within DCs. We have tested the effect of several neurotransmitters on IsoLG-protein adduct formation in DCs and have found that norepinephrine, a major neural transmitter of efferent sympathetic nerves, dose dependently increases IsoLG-protein adduct formation in DCs in vitro. Further studies are needed to determine the specific adrenergic receptor involved in IsoLG-protein adduct formation in DCs. Other neurotransmitters released from sympathetic nerves, such as neuropeptide Y and substance P, have no effect on IsoLG-protein adduct formation in DCs (47).
The second mechanism by which the nervous system may induce IsoLG-protein adduct accumulation in DCs is by its effects on other cell types. The sympathetic nervous system may in fact activate NADPH oxidase in diverse cell types. Importantly, we have evidence that renal sympathetic nerves induce high levels of superoxide production in endothelial cells in the kidney (47). DCs intimately interact with endothelial cells, and in fact, the passage of monocytes through endothelial cells leads to their transformation to DCs (48). It is therefore possible that ROS generated by endothelial cells lead to extracellular accumulation of IsoLG-protein adducts that are then phagocytosed by DCs. Another scenario is that hydrogen peroxide produced by endothelial cells could activate the NADPH oxidase in DCs. This is known to occur in the case of vascular NADPH oxidase (49). Further research is needed to examine the specific mechanism by which sympathetic outflow induces IsoLG-protein adduct formation in DCs.
Excess Dietary Salt and its Role in Promoting IsoLG-protein Adduct Formation:
Extensive evidence demonstrates that excess dietary Na+ is epidemiologically linked to hypertension. Throughout most of human history, Na+ consumption was less than 0.6-0.8 g/day (50). The AHA recommends a maximum of 2.3 g/day of Na+ intake. However, the Center for Disease Control (CDC) recently reported that Americans consume more than double this amount, and less than 10% of the U.S. population observes the recommended guidelines. In the US, the average Na+ intake is 4 grams per day (50, 51). A meta-analysis by He et al estimated that modest reductions in Na+ intake would lower blood pressure and reduce the annual number of new cases of coronary heart disease and stroke in the U.S. by 20% (52). However, accomplishing these dietary goals is difficult, as more than 70% of dietary Na+ is added to food prepared outside of the home (53). Thus, the impact of excess dietary salt intake remains an area of intense interest, with unsolved questions and barriers hampering success of general recommendations on dietary salt intake.
Traditionally, interstitial Na+ has been considered to be in equilibrium with plasma. Moreover, a vast amount of literature suggests that increased Na+ intake results in volume retention and increased cardiac output leading to hypertension. However, there is growing evidence that Na+ can accumulate in the interstitium without commensurate increases in interstitial water, leading to concentrations exceeding those of plasma (54-56). Such high concentrations of Na+ polarize immune cells towards an inflammatory phenotype, enhancing interleukin (IL)-17 production (11, 57, 58). Likewise, high salt intake increases the percent of circulatory pro-inflammatory intermediate monocytes (59). These studies suggest that elevated Na+ increases inflammation and contributes to hypertension. Moreover, they suggest that therapeutic strategies to reduce tissue Na+ may reduce inflammation and hypertension.
Studies in our laboratory have found that elevated Na+ contributes to inflammation and hypertension by increasing IsoLG-protein adduct formation in antigen presenting cells. DOCA-salt hypertension is associated with activation of monocyte derived DCs leading to production of IsoLG-protein adducts (15). Recently, we found that elevated Na+ is a potent stimulus for IsoLG-protein adduct formation in DCs in mice. This occurs through amiloride sensitive Na+ transporters (13). The increased intracellular Na+ is exchanged for calcium (Ca2+) via the Na+/Ca2+ exchanger. Ca2+ activates protein kinase C (PKC) which in turn phosphorylates the NADPH oxidase subunit p47phox. This leads to activation of the NADPH oxidase, increased superoxide and derivative reactive oxygen species (ROS) production (13). Thus, diuretics such as amiloride may confer anti-inflammatory actions directly on immune cells.
Our studies indicate that the increased superoxide production in DCs is associated with accumulation of IsoLG-protein adducts (15). Exposure of DCs to elevated Na+ (190 mM NaCl) but not an equiosmolar concentration of mannitol increased formation of IsoLG-protein adducts when compared to the normal physiological concentrations (150 mM NaCl) (13). The high salt-induced formation of IsoLG-protein adducts is superoxide and NADPH oxidase-dependent since it was prevented in mice lacking the p22phox docking subunit of this enzyme complex in DCs. This increase in Na+ concentrations was associated with increased expression of CD86, a B7 ligand which indicates maturation and has been shown to be essential for hypertension (13, 33). High salt also increased DC production of proinflammatory cytokine IL-1β which contributes to priming T cells for production of IL-17A and mediates hypertension (13, 60, 61). We have previously shown that scavenging of IsoLGs prevents hypertension-induced production of IL-1β by DCs (15). Importantly, DCs exposed to high salt induced pro-hypertensive cytokine production in primed T cells and sensitized mice to a normally suppressor dose of angiotensin II leading to hypertension (13). These effects were prevented by scavenging of IsoLGs during exposure of DCs to high salt suggesting that high salt-induced activation of DCs is dependent on the formation of IsoLGs (13). While these studies are informative about the effect of elevated Na+ on immune activation and hypertension, they are only in vitro and the in vivo mechanisms by which tissue accumulation of salt activates immune cells are still not known.
Conclusions and future perspectives:
IsoLG-protein adducts play and important role in promoting inflammation and hypertension. Scavenging of IsoLGs blunts inflammation and the hypertensive response to various stimuli including angiotensin II, catecholamines, and excess salt (Figure 2). The specific peptides altered by IsoLGs in hypertension are not known and future research efforts should be directed toward this goal. Despite ongoing efforts by many laboratories, immunogenic peptides have not been identified for many diseases known to be caused by T cells. The involvement of the proteasome in the processing of IsoLG-adducted proteins also needs to be investigated. The proteasome plays a diverse role in the immune system. The ubiquitin-proteasome system assists with maintenance of cellular homeostasis by degrading molecules responsible for numerous processes. In antigen presenting cells, the proteasome facilitates the presentation of endogenously generated antigens into MHC-I. Interestingly, augmented auto-inflammation correlates with proteasome levels in autoimmune disease (62, 63). Importantly, Bortezomib, a proteasome inhibitor FDA approved for the treatment of multiple myeloma, reduced the expression of DC activation markers CD86, CD80, CD40, and CD83. Moreover, DCs treated with a proteasome inhibitor were unable to activate T-cells (64, 65). Given these findings, proteasome inhibition has been proposed as a therapy for autoimmunity and may be a novel therapeutic for hypertension and its cardiovascular complications. This is important because the current anti-hypertensive drugs are not sufficient to prevent end-organ damage. Even among patients with successful control of blood pressure, the cardiovascular risk remains greater than that of the normal population. The currently available drugs can be categorized into 5 classes including diuretics, adrenergic receptor antagonists, calcium channel blockers and drugs that block the renin/angiotensin system. Data from large clinical trials indicate that from 20 to 30% of patients have persistently elevated blood pressure despite taking 3 or more of these drugs (66). And yet no new drugs have been developed in the past 3 decades that fall outside of these categories.
Our research indicates that many forms of hypertension, including angiotensin II, DOCA-salt, and norepinephrine-induced hypertension are dependent on immune cell activation. This suggests that a common mediator, such as ROS may be responsible for linking the various hypertensive stimuli to inflammation. However, antioxidants have proven ineffective in reducing cardiovascular risk in large clinical trials. This might be in part because their rate constants (about 104 M/sec) is far below the rate constant of reactions of free radicals with endogenous antioxidants (109 to 1010 M/sec). For example, 1600 IU of vitamin E is required to suppress isoprostane levels (the precursor of IsoLGs) in humans (67). These very high doses have adverse effects in several clinical trials and therefore should not be used (68). Thus, scavenging IsoLGs, which are downstream of ROS may provide a more targeted approach for treatment of prevention of hypertension and cardiovascular disease.
ACKNOWLEDGEMENTS
Sources of Funding: This work was supported by the National Institutes of Health grant K01HL130497 and, American Heart Association Scientist Development Grant 17SDG33670829.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Whelton PK, Carey RM, Aronow WS, Casey DE Jr., Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, et al. 2017. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. [Google Scholar]
- 2.Joffres M, Falaschetti E, Gillespie C, Robitaille C, Loustalot F, Poulter N, McAlister FA, Johansen H, Baclic O, and Campbell N 2013. Hypertension prevalence, awareness, treatment and control in national surveys from England, the USA and Canada, and correlation with stroke and ischaemic heart disease mortality: a cross-sectional study. BMJ Open 3:e003423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf-Maier K, Cooper RS, Banegas JR, Giampaoli S, Hense HW, Joffres M, Kastarinen M, Poulter N, Primatesta P, Rodriguez-Artalejo F, et al. 2003. Hypertension prevalence and blood pressure levels in 6 European countries, Canada, and the United States. JAMA 289:2363–2369. [DOI] [PubMed] [Google Scholar]
- 4.1997. The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med 157:2413–2446. [DOI] [PubMed] [Google Scholar]
- 5.McMaster WG, Kirabo A, Madhur MS, and Harrison DG 2015. Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116:1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.High KP, Akbar AN, and Nikolich-Zugich J 2012. Translational research in immune senescence: assessing the relevance of current models. Semin Immunol 24:373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norlander AE, Madhur MS, and Harrison DG 2018. The immunology of hypertension. J Exp Med 215:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders VM 2012. The beta2-adrenergic receptor on T and B lymphocytes: do we understand it yet? Brain Behav Immun 26:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, and Harrison DG 2010. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res 107:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lob HE, Schultz D, Marvar PJ, Davisson RL, and Harrison DG 2013. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension 61:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, and Hafler DA 2013. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, and Kuchroo VK 2013. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496:513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, et al. 2017. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep 21:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jufri NF, Mohamedali A, Avolio A, and Baker MS 2015. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell 7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, et al. 2014. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 124:4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon KB, Davies SS, and Kirabo A 2017. Dendritic cells and isolevuglandins in immunity, inflammation, and hypertension. Am J Physiol Heart Circ Physiol 312:H368–H374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, and Roberts LJ 2nd. 1990. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A 87:9383–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salomon RG, and Miller DB 1985. Levuglandins: isolation, characterization, and total synthesis of new secoprostanoid products from prostaglandin endoperoxides. Adv Prostaglandin Thromboxane Leukot Res 15:323–326. [PubMed] [Google Scholar]
- 19.Brame CJ, Salomon RG, Morrow JD, and Roberts LJ 2nd. 1999. Identification of extremely reactive gamma-ketoaldehydes (isolevuglandins) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J Biol Chem 274:13139–13146. [DOI] [PubMed] [Google Scholar]
- 20.Iyer RS, Ghosh S, and Salomon RG 1989. Levuglandin E2 crosslinks proteins. Prostaglandins 37:471–480. [DOI] [PubMed] [Google Scholar]
- 21.Talati M, Meyrick B, Peebles RS Jr., Davies SS, Dworski R, Mernaugh R, Mitchell D, Boothby M, Roberts LJ 2nd, and Sheller JR 2006. Oxidant stress modulates murine allergic airway responses. Free Radic Biol Med 40:1210–1219. [DOI] [PubMed] [Google Scholar]
- 22.Mottaran E, Stewart SF, Rolla R, Vay D, Cipriani V, Moretti M, Vidali M, Sartori M, Rigamonti C, Day CP, et al. 2002. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic Biol Med 32:38–45. [DOI] [PubMed] [Google Scholar]
- 23.Davies SS, Talati M, Wang X, Mernaugh RL, Amarnath V, Fessel J, Meyrick BO, Sheller J, and Roberts LJ 2nd. 2004. Localization of isoketal adducts in vivo using a single-chain antibody. Free Radic Biol Med 36:1163–1174. [DOI] [PubMed] [Google Scholar]
- 24.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, et al. 2016. CD70 Exacerbates Blood Pressure Elevation and Renal Damage in Response to Repeated Hypertensive Stimuli. Circ Res 118:1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, and Harrison DG 2014. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 114:616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts LJ 2nd, Salomon RG, Morrow JD, and Brame CJ 1999. New developments in the isoprostane pathway: identification of novel highly reactive gamma-ketoaldehydes (isolevuglandins) and characterization of their protein adducts. FASEB J 13:1157–1168. [DOI] [PubMed] [Google Scholar]
- 27.Zagol-Ikapitte I, Amarnath V, Bala M, Roberts LJ 2nd, Oates JA, and Boutaud O 2010. Characterization of scavengers of gamma-ketoaldehydes that do not inhibit prostaglandin biosynthesis. Chem Res Toxicol 23:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies SS, Brantley EJ, Voziyan PA, Amarnath V, Zagol-Ikapitte I, Boutaud O, Hudson BG, Oates JA, and Roberts LJ 2nd. 2006. Pyridoxamine analogues scavenge lipid-derived gamma-ketoaldehydes and protect against H2O2-mediated cytotoxicity. Biochemistry 45:15756–15767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murthi KK, Friedman LR, Oleinick NL, and Salomon RG 1993. Formation of DNA-protein cross-links in mammalian cells by levuglandin E2. Biochemistry 32:4090–4097. [DOI] [PubMed] [Google Scholar]
- 30.Davies SS, Amarnath V, Montine KS, Bernoud-Hubac N, Boutaud O, Montine TJ, and Roberts LJ 2nd. 2002. Effects of reactive gamma-ketoaldehydes formed by the isoprostane pathway (isoketals) and cyclooxygenase pathway (levuglandins) on proteasome function. FASEB J 16:715–717. [DOI] [PubMed] [Google Scholar]
- 31.Cullen SJ, Ponnappan S, and Ponnappan U 2010. Proteasome inhibition up-regulates inflammatory gene transcription induced by an atypical pathway of NF-kappaB activation. Biochem Pharmacol 79:706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luft FC 2001. Angiotensin, inflammation, hypertension, and cardiovascular disease. Curr Hypertens Rep 3:61–67. [DOI] [PubMed] [Google Scholar]
- 33.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, and Guzik TJ 2010. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122:2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montezano AC, and Touyz RM 2014. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal 20:164–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, et al. 2016. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest 126:50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, et al. 2014. Oligoclonal CD8+ T Cells Play a Critical Role in the Development of Hypertension. Hypertension 64:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson JA, and Webb RC 2013. Potential role of Toll-like receptors in programming of vascular dysfunction. Clin Sci (Lond) 125:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, and Webb RC 2014. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 306:H184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, and Carvalho MH 2012. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 122:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarthy CG, and Webb RC 2016. The toll of the gridiron: damage-associated molecular patterns and hypertension in American football. FASEB J 30:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goulopoulou S, McCarthy CG, and Webb RC 2016. Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol Rev 68:142–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chou MY, Hartvigsen K, Hansen LF, Fogelstrand L, Shaw PX, Boullier A, Binder CJ, and Witztum JL 2008. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med 263:479–488. [DOI] [PubMed] [Google Scholar]
- 43.Binder CJ 2010. Natural IgM antibodies against oxidation-specific epitopes. J Clin Immunol 30 Suppl 1:S56–60. [DOI] [PubMed] [Google Scholar]
- 44.Bartolini Gritti B, and Binder CJ 2016. Oxidation-specific epitopes are major targets of innate immunity in atherothrombosis. Hamostaseologie 36:89–96. [DOI] [PubMed] [Google Scholar]
- 45.Atlas SA 2007. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm 13:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johns EJ 2005. Angiotensin II in the brain and the autonomic control of the kidney. Exp Physiol 90:163–168. [DOI] [PubMed] [Google Scholar]
- 47.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, et al. 2015. Renal Denervation Prevents Immune Cell Activation and Renal Inflammation in Angiotensin II-Induced Hypertension. Circ Res 117:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, and Muller WA 1998. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science 282:480–483. [DOI] [PubMed] [Google Scholar]
- 49.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, and Griendling KK 2002. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91:406–413. [DOI] [PubMed] [Google Scholar]
- 50.Lev-Ran A, and Porta M 2005. Salt and hypertension: a phylogenetic perspective. Diabetes Metab Res Rev 21:118–131. [DOI] [PubMed] [Google Scholar]
- 51.Frisoli TM, Schmieder RE, Grodzicki T, and Messerli FH 2012. Salt and hypertension: is salt dietary reduction worth the effort? Am J Med 125:433–439. [DOI] [PubMed] [Google Scholar]
- 52.He FJ, Li J, and Macgregor GA 2013. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst Rev 4:CD004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harnack LJ, Cogswell ME, Shikany JM, Gardner CD, Gillespie C, Loria CM, Zhou X, Yuan K, and Steffen LM 2017. Sources of Sodium in US Adults From 3 Geographic Regions. Circulation 135:1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Muller DN, Derer W, et al. 2009. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med 15:545–552. [DOI] [PubMed] [Google Scholar]
- 55.Kopp C, Linz P, Wachsmuth L, Dahlmann A, Horbach T, Schofl C, Renz W, Santoro D, Niendorf T, Muller DN, et al. 2012. (23)Na magnetic resonance imaging of tissue sodium. Hypertension 59:167–172. [DOI] [PubMed] [Google Scholar]
- 56.Kirabo A 2017. A New Paradigm of Sodium Regulation in Inflammation and Hypertension. Am J Physiol Regul Integr Comp Physiol:ajpregu 00250 02017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, Sun S, Zhang Z, Chen XQ, Qin M, et al. 2015. High salt primes a specific activation state of macrophages, M(Na). Cell Res 25:893–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jorg S, Kissel J, Manzel A, Kleinewietfeld M, Haghikia A, Gold R, Muller DN, and Linker RA 2016. High salt drives Th17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp Neurol 279:212–222. [DOI] [PubMed] [Google Scholar]
- 59.Zhou X, Zhang L, Ji WJ, Yuan F, Guo ZZ, Pang B, Luo T, Liu X, Zhang WC, Jiang TM, et al. 2013. Variation in dietary salt intake induces coordinated dynamics of monocyte subsets and monocyte-platelet aggregates in humans: implications in end organ inflammation. PLoS One 8:e60332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, and Mitchell BM 2013. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 97:696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, and Harrison DG 2010. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD, and Jansen G 2015. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res Ther 17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Egerer T, Martinez-Gamboa L, Dankof A, Stuhlmuller B, Dorner T, Krenn V, Egerer K, Rudolph PE, Burmester GR, and Feist E 2006. Tissue-specific up-regulation of the proteasome subunit beta5i (LMP7) in Sjogren’s syndrome. Arthritis Rheum 54:1501–1508. [DOI] [PubMed] [Google Scholar]
- 64.Zinser E, Rossner S, Littmann L, Luftenegger D, Schubert U, and Steinkasserer A 2009. Inhibition of the proteasome influences murine and human dendritic cell development in vitro and in vivo. Immunobiology 214:843–851. [DOI] [PubMed] [Google Scholar]
- 65.Moran E, Carbone F, Augusti V, Patrone F, Ballestrero A, and Nencioni A 2012. Proteasome inhibitors as immunosuppressants: biological rationale and clinical experience. Semin Hematol 49:270–276. [DOI] [PubMed] [Google Scholar]
- 66.Acelajado MC, and Calhoun DA 2010. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin 28:639–654. [DOI] [PubMed] [Google Scholar]
- 67.Roberts LJ 2nd, Oates JA, Linton MF, Fazio S, Meador BP, Gross MD, Shyr Y, and Morrow JD 2007. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radic Biol Med 43:1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Widder JD, and Harrison DG 2005. Can vitamin E prevent cardiovascular events and cancer? Nat Clin Pract Cardiovasc Med 2:510–511. [DOI] [PubMed] [Google Scholar]