

Abstract
The vertebrate adaptive immune system has well defined functions in maintaining tolerance to self-tissues. Suppression of autoreactive T cells is dependent on the regulatory cytokine transforming growth factor-β (TGF-β) and regulatory T (Treg) cells, a distinct T cell lineage specified by the transcription factor Foxp3. Although TGF-β promotes thymic Treg cell development by repressing T cell clonal deletion and peripheral Treg cell differentiation by inducing Foxp3 expression, a recent study showed that TGF-β suppresses autoreactive T cells independent of Foxp3+ Treg cells. These findings imply that as an ancestral growth factor family member, TGF-β may have been co-opted as a T cell-intrinsic mechanism of self-tolerance control to assist the evolutionary transition of vertebrate adaptive immunity. Later, perhaps in placental mammals upon their acquisition of a TGF-β regulatory element in the Foxp3 locus, the TGF-β pathway was further engaged to induce peripheral Treg cell differentiation and expand the scope of T cell tolerance control to innocuous foreign antigens.
Keywords: TGF-β, Foxp3, Treg, adaptive immunity, autoimmunity, immune tolerance, evolution
1. Introduction
A salient feature of adaptive immunity is its recognition of pathogen-associated molecules (PAMs) rather than pathogen-associated molecular patterns (PAMPs) that are sensed by germline-encoded pattern recognition receptors of the innate immune system. To fulfill such a task, host moieties complementary to the varying PAMs need be acquired de novo, and the ensuing PAM recognition triggers a protective response that annihilates or limits the damaging effects of pathogens. As a testament of its importance, an array of adaptive immune system exists in major domains of life ranging from bacterial CRISPR/Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated genes) to B and T lymphocytes in jawed vertebrates.[1]
While the CRISP/Cas system acquires short nucleotide sequences (spacers) derived from bacteriophages resulting in a heritable PAM-specific response tailored to the pathogen,[2] adaptive lymphocytes with clonal expression of a diverse repertoire of antigen receptors develop prior to encountering cognate antigens.[3] Should an infection occur, pathogen-specific lymphocytes clonally expand and differentiate to effectors to curtail pathogen intrusion. Such a Darwinian strategy of adaptive immunity offers the host an advantage by producing an “army” of defenders preset to respond. However, the stochastic process of somatic gene recombination by which lymphocyte receptors are generated creates the inherent problem that some receptors are highly reactive to self-antigens. This problem is particularly pressing for the αβ-lineage of T lymphocytes as their development is dependent on T cell receptor (TCR) recognition of self-antigens presented by major histocompatibility complex (MHC) molecules.
Nonetheless, autoreactive T lymphocytes are tolerized in healthy individual. T cell tolerance control occurs in the thymus, a primary lymphoid organ that supports T cell development. As originally proposed in the clonal selection theory, deletion of auto-reactive T cells during thymic differentiation defines a mechanism of T cell central tolerance. The discovery of the autoimmune regulator (Aire) that promotes expression of “tissue-restricted” antigens in medullar thymic epithelial cells to induce T cell clonal deletion supports this hypothesis.[4] However, the purge of autoreactive T cells from the T cell repertoire is not an infallible process, and they are further kept in check via peripheral mechanisms of T cell tolerance.[5] Indeed, studies during the past three decades have revealed active modes of peripheral T cell tolerance control including those mediated by a subset of CD4+ regulatory T (Treg) cells and the regulatory cytokine Transforming Growth Factor-β (TGF-β).[6, 7]
2. Treg cells promote T cell tolerance
Thymic generation of Treg cells that suppress pathogenic autoreactive T cells was first implicated in neonatal thymectomy studies in the late 1960s.[8] Subsequent efforts at identifying such a cell-extrinsic mechanism of T cell tolerance revealed Treg cells as a distinct lineage with the forkhead box transcription factor Foxp3 as the lineage specification factor.[9] Indeed, Foxp3 deficiency results in autoimmune immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndromes in patients and lethal lymphoproliferative disorders in Scurfy mice, establishing a critical function for Treg cells in immune tolerance control.[10–15]
Many lines of investigation hve supported a role for high-affinity self-antigens in driving selection of thymic Treg (tTreg) cells.[7, 16] In fact, clonal deletion is associated with cognate antigen-triggered tTreg cell differentiation,[17, 18] implying an ingenious strategy of autoreactivity control by shunting pathogenic self-recognition to a dominant repressive pathway. Indeed, Aire-dependent expression of “tissue-restricted” antigens can not only trigger T cell clonal deletion, but also induce tTreg cell differentiation.[19]
In addition to thymic generation of Treg cells, naïve T cells can be converted to Treg cells in peripheral tissues (pTreg cells). Comparative genomics studies have revealed conserved noncoding DNA sequence (CNS) elements at murine and human Foxp3 gene loci.[20] Notably, the intronic CNS1 element is required for pTreg cell, but not tTreg cell differentiation, and mice devoid of CNS1 develop mucosal immunopathology,[21] In addition, CNS1-deficient female mice have higher embryo resorption rates when mated with allogenic males.[22] These findings support a critical function for pTreg cells to regulate immune responses to innocuous non-self antigens such as those associated with commensal microbes in mucosal tissues and allogenic fetuses.
3. TGF-β controls T cell tolerance
TGF-β belongs to a regulatory cytokine family with pleiotropic functions in many lineages of cell types, and controls numerous pathophysiological processes ranging from embryogenesis to carcinogenesis.[23] In mammals, three members of the TGF-β family (TGF-β1, TGF-β2, and TGF-β3) have been identified, which bind to a shared TGF-β receptor type I (TGF-βRI) and type II (TGF-βRII) complex with relatively high and low affinities for TGF-β1/TGF-β3 and TGF-β2, respectively.[23–26] TGF-β engagement of a tetrameric receptor complex made of two TGF-βRI and two TGF-βRII molecules stimulates their intrinsic kinase activities, resulting in phosphorylation and activation of Smad family of transcription factors. Active Smad2 and Smad3 proteins associate with a large number of transcription factor partners to mediate diverse biological effects of TGF-β.
A pivotal role of the TGF-β pathway in immune tolerance control was initially demonstrated in studies of TGF-β1-deficient mice.[27, 28] Later studies of mice with T cell-specific ablation of TGF-βRII or TGF-βRI, which manifest a neonatal lethal inflammatory disease similar to that of TGF-β1-deficient mice, reveal T cells as the critical target of TGF-β1-regulated immune tolerance.[29–31] Recent studies have suggested that TGF-β3 produced by CD4+CD25−Lag3+ T cells suppresses systemic autoimmune diseases in mice, although its definitive function remains to be determined with genetic loss-of-function experiments.[32, 33] Notably, Loeys-Dietz Syndrome patients with monogenic mutations of TGF-βRII or TGF-βRI suffer from inflammatory disorders including inflammatory bowel disease and autoimmune thyroiditis,[34] establishing a critical function for TGF-β in immune tolerance from mice to men.
4. TGF-β and Treg cell crosstalk in T cell tolerance control
The TGF-β signaling pathway plays an important role in the early development of tTreg cells with Foxp3+ T cells substantially reduced in the thymus of 3- to 5-day old mice lacking TGF-βRI.[31] A conserved Smad3-binding site is present in the CNS1 region of the Foxp3 gene.[35] However, mice devoid of the binding site or the entire CNS1 element exhibit normal tTreg cell differentiation,[20, 21] revealing that TGF-β does not promote tTreg cell development by regulating Foxp3 expression. Instead, TGF-β signaling promotes tTreg cell development by attenuating agonist antigen-triggered T cell clonal deletion, and thereby increasing the number of autoreactive precursor cells for differentiation to tTreg cells.[36]
Exposure of cognate antigen to mature naïve T cells under subimmunogenic conditions stimulates TGF-β production and converts CD4+ T cells to pTreg cells.[37] Inclusion of TGF-β in T cell culture induces Foxp3 expression and Treg cell differentiation,[38] which is dependent on Smad3 recruitment to the CNS1 element of the Foxp3 gene.[35] These findings demonstrate an important function for TGF-β signaling in inducing pTreg cell differentiation.
TGF-β regulation of tTreg cell development and pTreg cell differentiation represses pathogenic T cell responses. Furthermore, Treg cells could promote immune tolerance through TGF-β1 production and activation of the latent form of TGF-β1.[39, 40] The intertwined relationship between the TGF-β-dependent and Treg cell-mediated tolerance control raises the question of whether the two pathways fall into the same regulatory module, or have distinct mechanisms of T cell regulation.
5. TGF-β promotes T cell tolerance independent of Treg cells
The similarly severe autoimmune phenotype developed in T cell-specific TGF-βRII-deficient mice and Foxp3-deficient mice hinders the dissection of potentially independent mechanisms of T cell tolerance regulation by TGF-β and Treg cells. To this end, a transgenic mouse model of autoimmune diabetes has recently been used.[41] Membrane ovalbumin (mOva) expressed under the control of rat insulin promoter (RIP) was utilized as a model self-antigen, which can be recognized by transgenic OT-II T cells. In addition to pancreatic β cells, mOva is expressed in the thymus via Aire-dependent mechanisms, and triggers OT-II T cell clonal deletion as well as tTreg cell differentiation. Nonetheless, more than 95% peripheral OT-II T cells do not express Foxp3, and are potentially pathogenic autoreactive T cells.
Despite high pancreatic expression of mOva, OT-II RIP-mOva mice do not develop autoimmune diabetes.[36, 41] Induction of the T cell activation marker CD69 in OT-II T cells from pancreatic draining lymph nodes demonstrate that OT-II T cells are not ignorant of the mOva antigen.[41] These observations imply that active tolerance mechanisms are engaged to repress pathogenic autoreactive T cells. Indeed, in addition to the presence of Treg cells, elevated Smad2 and Smad3 phosphorylation was detected in OT-II T cells from pancreatic draining lymph nodes of OT-II RIP-mOva mice, implying enhanced TGF-β signaling.[41]
To dissect the individual contributions of Treg cells and TGF-β signaling in tolerance control, OT-II RIP-mOva mice were crossed to Foxp3-deficient or T cell-specific TGF-βRII-deficient background. Strikingly, loss of TGF-β signaling in T cells causes aggressive autoimmune diabetes with 100% penetrance at 2 month-of-age, while Foxp3 deficiency results in a much delayed disease with less than 50% penetrance at 5 month-of-age. These observations demonstrate that TGF-β promotes T cell tolerance primarily via Foxp3-indepednent mechanisms.[41]
The fulminant disease phenotype in TGF-βRII-deficient mice was associated with enhanced activation and differentiation of OT-II T cells from pancreatic draining lymph nodes. Notably, the proinflammatory cytokine GM-CSF was expressed at high level in TGF-βRII-deficient OT-II T cells that had infiltrated the pancreas. GM-CSF is a potent growth factor of inflammatory cells of the myeloid lineage including monocytes. Indeed, inactivation of GM-CSF receptor or depletion of inflammatory monocytes alleviates the disease. These findings reveal that TGF-β signaling supports immune tolerance by repressing the cross talk between autoreactive T cells and innate immune cells.[41]
6. An evolutionary perspective of immune tolerance control
According to the theory of major evolutionary transitions (METs), gaining complexity in the history of life depended on a small number of fundamental innovations to create new individuality for selection (e.g. the symbiogenetic origin of eukaryotes).[42] The emergence of a higher level of selection would then suppress evolution at the lower level (e.g. depriving “domesticated” mitochondria of autonomous replication). In this context, the vertebrate adaptive immune system can be considered as a radical evolutionary innovation, as it recreates Darwinian cell-level evolution in a multicellular organism.[1] Such an embedded lower level selection is inherently disruptive in terms of autoimmunity unless it is tightly controlled.
How was then adaptive immunity evolved in the first place in the lineage of vertebrates? The discovery of Rag1/2 recombinases in the invertebrate species sea urchins suggested that acquisition of the toolbox for somatic generation of antigen receptors preceded the origin of vertebrates.[43, 44] Intriguingly, a whole genome duplication event and a series of segmental genome duplications occurred close to the origin of vertebrates,[45] which might dramatically enhance regulatory complexity to ensure self-tolerance. Indeed, phylogenetic studies revealed that the Foxp family of transcription factors has a single orthologue in invertebrates, but four members in most vertebrates.[46] Similar to mammalian Foxp3, zebrafish Foxp3-like is selectively expressed in a Treg-like cell lineage, which appears first in the thymus and has an indispensable role in repressing immunopathology triggered by conventional T cells.[47, 48] These findings suggest that the autoreactive T cell-extrinsic mechanism of T cell tolerance control by Treg cells is well conserved in jawed vertebrates.
Could Treg cells then jumpstart the evolution of vertebrate adaptive immune system as initially suggested by Janeway,[49] and recently elaborated by Szathmary and colleagues?[1] Under the assumption that negative selection and Treg cell-mediated dominant immune suppression are two primary mechanisms of self-tolerance control,[1] it was reasoned that “To suffice alone, negative selection should be perfect; dominant tolerance just needs to be ‘good enough’ to have a statistically high chance of bringing self-reactive clones under control before they can do too much damage.” Nonetheless, the assumption that the emergence of Treg cells is the key to the evolution of adaptive immunity in vertebrates, raises the chicken and egg problem eloquently stated as “Clonal amplification of immune responses with stochastic (somatically diversified) targeting is unsafe without specific (dominant) tolerance; however, specific tolerance might not make much sense without stochastic immune targeting”.[1]
How might this dilemma be resolved? The demonstration of a Foxp3-independent autoreactive T cell-intrinsic mechanism of peripheral tolerance controlled by TGF-β might offer a solution.[41] The TGF-β superfamily evolved in early metazoans before the development of bilateral symmetry, while TGF-β itself evolved later at the emergence of deuterostomes (Fig 1A).[50] The single TGF-β present in lower deuterostomes such as sea urchin is an orthologue of mammalian TGF-β1,[50, 51] the very family member involved in T cell tolerance control.[6] In addition, phylogenetic analyses revealed that TGF-β3 evolved from TGF-β1 about 300 million years ago, and TGF-β2 diverged from TGF-β3 at a later time point.[25] It is thus conceivable that the evolutionarily ancient TGF-β signaling pathway, in particular TGF-β1, might be co-opted to suppress autoreactive lymphocyte responses prior to the advent of Foxp3+ Treg cells, and thus be a trigger of vertebrate adaptive immune system evolution (Fig 1B). In addition, TGF-β repression of thymic T cell clonal deletion could later assist the emergence of Treg cells to exert dominant immune tolerance (Fig 1B).[36] Notably, TGF-β signaling promotes T cell positive selection and homeostasis as well,[52] which might help establish a diverse repertoire of naïve T cells for immunosurveillance, a critical enabling property for the evolution of the adaptive immune system.
Figure 1. Stepwise acquisition of TGF-β and Foxp3+ Treg cell-dependent adaptive immune tolerance in evolution.
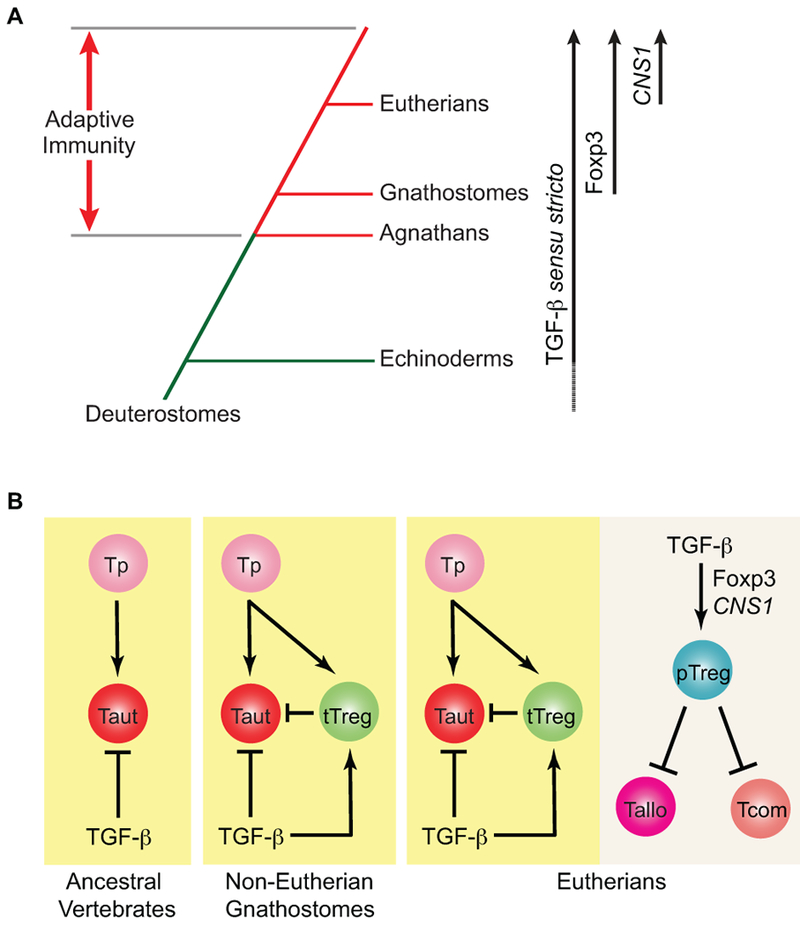
(A) Adaptive immunity was evolved in vertebrates about 500 million years ago with agnathans (jawless vertebrates) and gnathostomes (jawed vertebrates) utilizing distinct toolboxes to generate antigen-specific receptors. TGF-β sensu stricto was evolved in early Deuterostomes with definitive orthologues present in Echinoderms. Foxp3 emerged in jawed vertebrates with its acquisition of a TGF-β-regulated CNS1 element in Eutherians (placental mammals). (B) Due to the stochastic process of antigen receptor generation, autoreactive T cells (Taut) can be produced from T cell progenitors (Tp). In ancestral vertebrates, including jawless vertebrates, TGF-β signaling might represent the primordial mechanism to keep autoreactive T cells in check. In non-Eutherian jawed vertebrates, thymic Treg cells (tTreg), whose development is supported by TGF-β signaling via repression of T cell clonal deletion, emerged as a cell-extrinsic pathway working in concert with TGF-β signaling to suppress autoreactive T cells. In placental mammals, the CNS1 element was acquired in the Foxp3 gene locus, which mediated TGF-β-induced peripheral Treg cell (pTreg) differentiation to suppress allogenic T cells (Tallo) reactive to paternal antigens expressed in fetuses as well as commensal microbiota-reactive T cells (Tcom).
It is important to note that the TGF-β-responsive CNS1 element in the Foxp3 locus appears to be unique in placental mammals.[22] Indeed, pregnancy poses a special challenge to the maternal immune system, as the fetus and placenta could express foreign antigens from the paternal genome. Thus, the evolution of a TGF-β-induced CNS1-dependent pTreg cell differentiation pathway could endow a dominant immune tolerance pathway to allogeneic fetuses, and reinforce the reproductive fitness of placental mammals.[22] Such an immune tolerance mechanism may later be extended to commensals to foster symbiotic relationships with mammalian hosts, which enhances nutrient extraction and promotes host fitness. Therefore, TGF-β induction of pTreg cell differentiation is likely more recently evolved to extend the scope of immune tolerance control to innocuous foreign antigens (Fig 1B).
7. Conclusions
Aside from negative selection and Treg cell generation, autoreactive T cell-intrinsic mechanism of TGF-β regulation should now be considered as an essential, perhaps primordial, pathway of adaptive immune tolerance control, which might be a critical trigger of vertebrate adaptive immune system evolution prior to the acquisition of the Treg cell lineage. Notably, Foxp3 orthologue does not appear to be present in jawless vertebrates such as lamprey that utilizes a distinct gene conversion pathway to generate diverse variable lymphocyte receptors.[45, 53] It will be interesting to examine whether TGF-β signaling is primarily responsible for restraining self-reactive lymphocytes in these creatures.
Acknowledgements
We thank the M. Li lab for insightful discussions. This work was supported by NIAID (RO1 AI122264 to M.O.L), HHMI (Faculty Scholar Award to M.O.L.) and the Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748). The authors have no conflict of interest to declare.
Abbreviations:
- TGF-β
transforming growth factor-β
- Foxp3
Forkhead box protein p3
- CNS
conserved noncoding DNA sequence
- tTreg cell
thymic regulatory T cell
- pTreg cell
peripheral regulatory T cell
- PAM
pathogen-associated molecule
- RIP
rat insulin promoter
- mOVa
membrane ovalbumin
References
- [1].Muller V, de Boer RJ, Bonhoeffer S, Szathmary E, Biol Rev Camb Philos Soc 2017. [DOI] [PubMed] [Google Scholar]
- [2].Horvath P, Barrangou R, Science 2010, 327, 167. [DOI] [PubMed] [Google Scholar]
- [3].Burnet M, Nature 1964, 203, 451. [DOI] [PubMed] [Google Scholar]
- [4].Mathis D, Benoist C, Annu Rev Immunol 2009, 27, 287. [DOI] [PubMed] [Google Scholar]
- [5].Mueller DL, Nat Immunol 2010, 11, 21. [DOI] [PubMed] [Google Scholar]
- [6].Li MO, Flavell RA, Cell 2008, 134, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Josefowicz SZ, Lu LF, Rudensky AY, Annu Rev Immunol 2012, 30, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nishizuka Y, Sakakura T, Science 1969, 166, 753. [DOI] [PubMed] [Google Scholar]
- [9].Ziegler SF, Annu Rev Immunol 2006, 24, 209. [DOI] [PubMed] [Google Scholar]
- [10].Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, Nat Genet 2001, 27, 20. [DOI] [PubMed] [Google Scholar]
- [11].Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F, Nat Genet 2001, 27, 68. [DOI] [PubMed] [Google Scholar]
- [12].Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME, Nat Genet 2001, 27, 18. [DOI] [PubMed] [Google Scholar]
- [13].Khattri R, Cox T, Yasayko SA, Ramsdell F, Nat Immunol 2003, 4, 337. [PubMed] [Google Scholar]
- [14].Fontenot JD, Gavin MA, Rudensky AY, Nat Immunol 2003, 4, 330. [PubMed] [Google Scholar]
- [15].Hori S, Nomura T, Sakaguchi S, Science 2003, 299, 1057. [PubMed] [Google Scholar]
- [16].Li MO, Rudensky AY, Nat Rev Immunol 2016, 16, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ, Nat Immunol 2001, 2, 301. [DOI] [PubMed] [Google Scholar]
- [18].Apostolou I, Sarukhan A, Klein L, von Boehmer H, Nat Immunol 2002, 3, 756. [DOI] [PubMed] [Google Scholar]
- [19].Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA, Immunity 2016, 44, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY, Nature 2010, 463, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H, J Exp Med 2012, 209, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY, Cell 2012, 150, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blobe GC, Schiemann WP, Lodish HF, N Engl J Med 2000, 342, 1350. [DOI] [PubMed] [Google Scholar]
- [24].Massague J, Nat Rev Mol Cell Biol 2012, 13, 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Burt DW, Paton IR, DNA Cell Biol 1992, 11, 497. [DOI] [PubMed] [Google Scholar]
- [26].Lin HY, Moustakas A, Knaus P, Wells RG, Henis YI, Lodish HF, J Biol Chem 1995, 270, 2747. [DOI] [PubMed] [Google Scholar]
- [27].Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. , Nature 1992, 359, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S, Proc Natl Acad Sci U S A 1993, 90, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li MO, Sanjabi S, Flavell RA, Immunity 2006, 25, 455. [DOI] [PubMed] [Google Scholar]
- [30].Marie JC, Liggitt D, Rudensky AY, Immunity 2006, 25, 441. [DOI] [PubMed] [Google Scholar]
- [31].Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W, Nat Immunol 2008, 9, 632. [DOI] [PubMed] [Google Scholar]
- [32].Okamura T, Sumitomo S, Morita K, Iwasaki Y, Inoue M, Nakachi S, Komai T, Shoda H, Miyazaki J, Fujio K, Yamamoto K, Nat Commun 2015, 6, 6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morita K, Okamura T, Inoue M, Komai T, Teruya S, Iwasaki Y, Sumitomo S, Shoda H, Yamamoto K, Fujio K, Proc Natl Acad Sci U S A 2016, 113, E8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Naviglio S, Arrigo S, Martelossi S, Villanacci V, Tommasini A, Loganes C, Fabretto A, Vignola S, Lonardi S, Ventura A, J Crohns Colitis 2014, 8, 770. [DOI] [PubMed] [Google Scholar]
- [35].Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M, Nat Immunol 2008, 9, 194. [DOI] [PubMed] [Google Scholar]
- [36].Ouyang W, Beckett O, Ma Q, Li MO, Immunity 2010, 32, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H, Nat Immunol 2005, 6, 1219. [DOI] [PubMed] [Google Scholar]
- [38].Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM, J Exp Med 2003, 198, 1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li MO, Wan YY, Flavell RA, Immunity 2007, 26, 579. [DOI] [PubMed] [Google Scholar]
- [40].Worthington JJ, Kelly A, Smedley C, Bauche D, Campbell S, Marie JC, Travis MA, Immunity 2015, 42, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Oh SA, Liu M, Nixon BG, Kang D, Toure A, Bivona M, Li MO, Proc Natl Acad Sci U S A 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Szathmary E, Proc Natl Acad Sci U S A 2015, 112, 10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fugmann SD, Messier C, Novack LA, Cameron RA, Rast JP, Proc Natl Acad Sci U S A 2006, 103, 3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Litman GW, Rast JP, Fugmann SD, Nat Rev Immunol 2010, 10, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, Campbell MS, Yandell MD, Manousaki T, Meyer A, Bloom OE, Morgan JR, Buxbaum JD, Sachidanandam R, Sims C, Garruss AS, Cook M, Krumlauf R, Wiedemann LM, Sower SA, Decatur WA, Hall JA, Amemiya CT, Saha NR, Buckley KM, Rast JP, Das S, Hirano M, McCurley N, Guo P, Rohner N, Tabin CJ, Piccinelli P, Elgar G, Ruffier M, Aken BL, Searle SM, Muffato M, Pignatelli M, Herrero J, Jones M, Brown CT, Chung-Davidson YW, Nanlohy KG, Libants SV, Yeh CY, McCauley DW, Langeland JA, Pancer Z, Fritzsch B, de Jong PJ, Zhu B, Fulton LL, Theising B, Flicek P, Bronner ME, Warren WC, Clifton SW, Wilson RK, Li W, Nat Genet 2013, 45, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Andersen KG, Nissen JK, Betz AG, Front Immunol 2012, 3, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sugimoto K, Hui SP, Sheng DZ, Nakayama M, Kikuchi K, Dev Comp Immunol 2017, 73, 156. [DOI] [PubMed] [Google Scholar]
- [48].Kasheta M, Painter CA, Moore FE, Lobbardi R, Bryll A, Freiman E, Stachura D, Rogers AB, Houvras Y, Langenau DM, Ceol CJ, J Exp Med 2017, 214, 3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Janeway CA Jr., Proc Natl Acad Sci U S A 2001, 98, 7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hinck AP, Mueller TD, Springer TA, Cold Spring Harb Perspect Biol 2016, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lapraz F, Rottinger E, Duboc V, Range R, Duloquin L, Walton K, Wu SY, Bradham C, Loza MA, Hibino T, Wilson K, Poustka A, McClay D, Angerer L, Gache C, Lepage T, Dev Biol 2006, 300, 132. [DOI] [PubMed] [Google Scholar]
- [52].Ouyang WM, Oh SA, Ma Q, Bivona MR, Zhu JF, Li MO, Immunity 2013, 39, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Boehm T, Hirano M, Holland SJ, Das S, Schorpp M, Cooper MD, Annu Rev Immunol 2017. [DOI] [PubMed] [Google Scholar]