

ABSTRACT
Physiologically and anatomically, humans and pigs share many similarities, which make porcine induced pluripotent stem cells (piPSCs) very attractive for modeling human cell therapy as well as for testing safety of iPSC based cell replacement therapies. To date, several integrative and non-integrative strategies have been reported to successfully generate piPSCs, but all resulting piPSCs had integration of transgenes. The use of integrative methods has the disadvantage of potential lack of silencing or inappropriate re-activation of these genes during differentiation, as well as uncertainty regarding disruption of important genomic regions caused by integration. In our study, we performed a non-integrative vector based reprogramming approach using porcine fetal fibroblasts. The resulting four piPSC lines were positive for pluripotency marker and when subjected to in vitro and in vivo differentiation assays, all four lines formed embryoid bodies, capable to differentiate into all three germ layers, and three out of the four cell lines formed teratomas. PCR analysis on genomic and plasmid DNA revealed that the episomal vectors were undetectable in six out of eight subclones derived from one of the piPSC lines (piPSC1) above passage 20. These piPSCs could potentially be ideal cell lines for the generation of porcine in vitro and in vivo models. Furthermore, subsequent analyses of our new transgene independent piPSCs could provide novel insights on the genetic and epigenetic necessities to achieve and maintain piPSCs.
KEYWORDS: Porcine induced pluripotent stem cells, non-integrative strategies, in vitro differentiation, transgene-free piPSCs
Introduction
Somatic cells can be reprogrammed to an embryonic stem cell (ESC) -like state by expression of defined transcription factors, which are associated with pluripotency. Such cells are termed induced pluripotent stem cells (iPSCs). Since the first murine iPSCs (miPSCs) were successfully established by expressing four transcription factors, Oct4, Sox2, c-Myc, and Klf4 in 2006 [1], numerous reports have been published describing the derivation of iPSCs from various species, such as mouse [2,3], human [4,5], rat [6,7], cattle [8–10], sheep [11–13], goat [14], monkey [15], and dog [16]. This technology is considered to be of high potential for regenerative medicine and in vitro modeling of diseases. In regards to porcine iPSCs (piPSCs), these cells can be used to model transplantation experiments in an animal model that is much closer to humans than rodents and provide evidence of safety and benefits of such endeavors. This is highly relevant since Wu and colleagues [17] showed that human iPSC could potentially be used to generate xeno-transplantable human tissues in ungulates.
To date, several integrative strategies, based on retroviral or lentiviral vectors, have been used to generate piPSCs [18–25]. These piPSCs display normal karyotypes, express telomerase activity as well as numerous cell surface markers and genes that are characteristic for human and murine ESC. They also are capable of differentiating into the three primary germ layers, both in vitro and in vivo. One of the most conclusive experiments to verify pluripotency is the generation of chimeric embryos. So far, it has only been possible to show true pluripotency via reproducible chimeric offspring generation, including germline transmission for mouse and rat iPSCs [26,27]. Until now only one group has reported success in generating porcine chimeras. In this study piPSCs derived from porcine mesenchymal stem cells were transduced with six human reprogramming factors (OCT4, SOX2, KLF4, LIN28, NANOG, and c-MYC). Subsequently, piPSC generated from this approach were used to produce chimeric offspring [28] and germline transmission of these piPSCs was confirmed [29] even though the resultant offspring were aberrant potentially due to epigenetic defects.
Until now it has not been possible to generate integration-free piPSCs using non-integrative methods, whilst in contrast several integration-free methods have been employed successfully in hiPSCs. These include: adenovirus [30], sendai virus [31,32], episomal vectors [33–38], direct protein delivery [39] and mRNA delivery [40,41]. These techniques allow generating clinical-grade hiPSCs without any manipulation of the genome in the future. Obviously, in order to use these hiPSCs in cell replacement therapies a full characterization and assessment of potential risks is needed. Until recently there has been focus on comparing the hiPSC to murine naïve iPSC. As mentioned earlier, only chimeric embryo experiments, including germline transmission can ultimately confirm the true naïve pluripotent nature of iPSC. This conclusion is supported by the fact that in early studies, miPSCs showed pluripotency characteristics but failed to produce chimeric offspring [1,26]. Due to ethical reasons such chimera experiments are not possible using hiPSCs. Therefore, it could potentially be informative to use piPSCs and generate chimeras with these. This could tell us if iPSC from a species, phylogenetically closer to humans, behave similar to rodents and has germline chimeric potential or at least potential to contribute to some tissues in chimeric embryos.
Safety in using iPSCs or their derivatives for potential cell replacement therapies is also an issue. It is anticipated that iPSCs could be used for personalized medicine. One potential scenario would be to perform biopsies of patient material, which would undergo gene correction via CRISPR technology, and be used for the generation of integration free iPSCs. These would subsequently be differentiated into desired progenitor/cell populations or organ and transplanted in order to replace damaged and malfunctioning cells. However, a major hurdle is the safety of the iPSC-derived cells following transplantation. Several groups have investigated the genomic stability of iPSCs and ESCs and have observed a significant amount of cell lines, which are chromosomally instable. This instability may lead to uncontrolled growth and potentially cancer [42]. Obviously, this is a concern if iPSCs are to be used for cell replacement therapy in humans. Pigs resemble human anatomy and physiology more closely than mice and rats and this makes them consequently more suitable for the assessment of risks and potentials of iPSC-based therapy [43].
With regard to piPSCs, both the development of integration-free reprogramming strategies as well as improved conditions for maintenance of the reprogrammed cells still need to be established. In order to be able to compare the potential and risks of hiPSCs and piPSCs, it is crucial to generate piPSCs with the same integration-free techniques as used in hiPSCs. So far only few groups have attempted to generate integration-free piPSCs by using episomal vectors [38,44], but in these cases, the derived iPSCs retained the episomal plasmids, which was most likely caused by genomic integration of the plasmid DNA, or due to retention of the episomal plasmids following extended culture. Whether these cells were dependent on exogenous pluripotency gene expression or whether the episomal plasmids were silenced was not clearly demonstrated, but it could be concluded that these cells were not integration-free.
Here, we report for the first time an integration-free reprogramming approach resulting in transgene-free porcine intermediate type piPSCs, devoid of the introduced episomal DNA. The resulting piPSCs were cultured in iPSC medium supplemented with bFGF and 2 inhibitors (2i) (PD0325901 and CHIR99021), which target mitogen-activated protein kinase kinase (MEK) and glycogen synthase kinase 3beta (GSK3β), respectively, and which have already been proven to be beneficial in the generation of hiPSCs [45] and have been used by other groups for piPSCs [18,44]. Our stable transgene-free piPSCs could serve as ideal cell lines for modeling iPSC-based cell replacement therapy in the pig.
Results
Reprogramming porcine fibroblasts to piPSCs
The electroporation procedure was optimized previously in-house for hiPSC reprogramming using an episomal GFP plasmid (Addgene plasmid #27082 [46];). A single pulse at 1300 V for 30 ms was found to have the highest transduction efficiency for porcine embryonic fibroblasts. GA1 fibroblasts were electroporated with three episomal plasmids (pCXLE-hOCT3/4-shp53; pCXLE-hSK; pCXLE-hUL) [34]. Seven days after electroporation, cells were split 1:2 onto mitomycin C-treated MEFs and cultured in in bFGF-dependent medium supplemented with MEK/ERK (PD0325901) and GSK-3β (CHIR99021) inhibitors [18,44] (Figure 1(a)).
Figure 1.
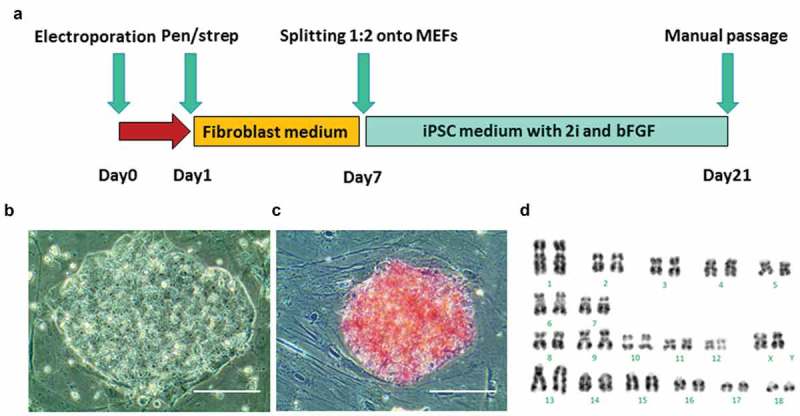
Generation of piPSCs from GA1 fibroblasts. (a), schematic of the piPSC induction protocol is shown. (b), morphology of a representative piPSC colony derived from GA1. Scale bar represents 100 µm. (c), alkaline phosphatase (AP) staining shows piPSC colony was positive for AP activity. Scale bar represents 100 µm. (d). G-band staining of piPSCs demonstrates a normal karyotype at passage 10 (38, XX).
Colonies with a human ESC-like morphology appeared on the feeders 13 days after electroporation. On day 21, twelve ESC-like colonies were manually transferred onto new MEFs and expanded. After four days culture, four piPSC lines, designated piPSC1, piPSC2, piPSC7, piPSC8, were selected for further culture and detailed analysis. Each of the four piPSC lines exhibited a human ESC-like morphology and grew as flat and compact colonies with clear boundaries, prominent nucleoli and a high nuclei-to-cytoplasm ratio (Figure 1(b)). All four piPSC lines could routinely be passaged using TrypLE Select without change in morphology or proliferation rate and were positive for AP (Figure 1(c)). Line piPSC1, which was karyotyped, exhibited a normal karyotype with 19 pairs of chromosomes (38, XX) (Figure 1(d)). All four piPSC lines could be continuously cultured beyond 20 passages and no difference in morphology could be observed in line piPSC1, piPSC2 and piPSC8 during this period. A certain degree of spontaneous differentiation was noted in line piPSC7 at passage 20. At this late passage, all piPSC lines had retained their AP positivity in the majority of cells within the piPSC colonies. Only line piPSC7 showed significantly larger AP negative areas (Supp. Figure 1). This indicates that piPSC7 is composed of a more heterogeneous cell population and could also have larger variation in regards to the presence of the episomal plasmids. These results in combination with the following observations promoted us to generate subclones of the most promising piPSC line as described later.
Expression of endogenous pluripotency genes in piPSC lines
Immunocytochemistry (ICC) was performed on all four piPSC lines for expression of pluripotency transcription factors and cell surface makers at passage 10. The four piPSC lines were positive for NANOG (Figure 2(a,b)), SOX2 (Figure 2(c,d)), and OCT4 (Figure 2(e,f)), but negative for cell surface makers SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81. In contrast the majority of cells in the piPSC within the colonies stained positive for SSEA-1 (Figure 2(g,h)). To investigate whether the four piPSC lines continue to express the pluripotency and cell surface makers following long-term culture, we additionally performed ICC on all four piPSC lines at passage 20. The ICC confirmed a small, but significant increase in expression levels of NANOG (Figure 2(i,j)), SOX2 (Figure 2(k,l)), and OCT4 (Figure 2(m,n)) in all four lines compared to colonies assessed at passage 10 (Supp. Table 1). Again, the lines remained negative for cell surface makers SSEA-3, SSEA-4, TRA-1-60, TRA-1-81, but large areas of the colonies stained positive for SSEA-1 (Figure 2(o), P; Supp.Table 1). This result is consistent with a previous report that piPSCs with human ESC-like morphology express SSEA-1 [19], which is interesting since SSEA1 expression cannot be found in human ESCs, but in mouse ESCs. This indicates that piPSCs share commonalities with both human and mouse ESCs. The staining for the other pluripotency factors, NANOG, SOX2 and OCT4, increased at passage 20, indicating a stabilization of the pluripotency phenotype of the transgene-free porcine intermediate type piPSC.
Figure 2.
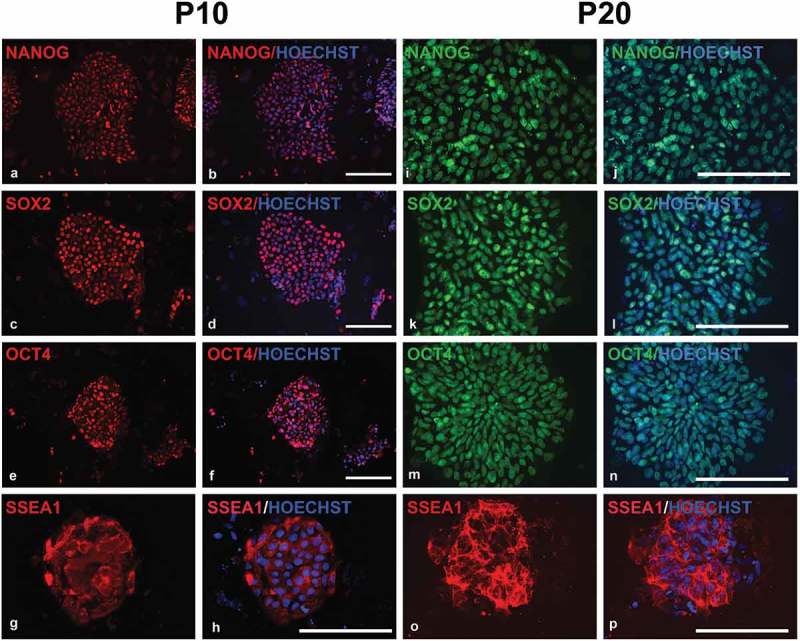
Fluorescent microscopy images of immunocytochemistry on piPSC1s at passage 10 and 20 for pluripotency marker expression. The piPSCs were positive for NANOG (a, b; i, j), SOX2 (c, d; k, l), OCT4 (e, f; m, n), and SSEA-1 (g, h; o, p). Scale bars represent 100 µm.
Table 1.
Porcine- and plasmid-specific oligonucleotides.
| Gene | Species | Sequence 5´ – 3’ | Product size (bp) | Reference No. (NCBI) |
|---|---|---|---|---|
| OCT4 | Porcine | 5´- AGGTGTTCAGCCAAACGACC −3’ 5´- TGATCGTTTGCCCTTCTGGC −3’ |
335 | NM_001113060.1 |
| SOX2 | Porcine | 5´- CCAGAAGAACAGCCCAGA −3’ 5´- GCAGCGTTCGCAGCCG −3’ |
203 | NM_001123197.1 |
| LIN28 | Porcine | 5´- CAGAGTAAGCTGCACATGGAGG −3’ 5´- GTAGGCTGGCTTTCCCTGTG −3’ |
325 | NM_001123133.1 |
| NANOG | Porcine | 5´- CGAATGAAATGTAAGAGGT −3’ 5´- CCAGCTCTGATTACCCCAC −3’ |
162 | NM_001129971.1 |
| OCT3/4 (pla) | Human | 5´- CATTCAAACTGAGGTAAGGG −3’ 5´- TAGCGTAAAAGGAGCAACATAG −3’ |
124 | Addgene plasmid #27077 |
| SOX2 (pla) | Human | 5´- TTCACATGTCCCAGCACTACCAGA −3’ 5´- TTTGTTTGACAGGAGCGACAAT −3’ |
114 | Addgene plasmid #27078 |
| KLF4 (pla) | Human | 5´- CCACCTCGCCTTACACATGAAGA −3’ 5´- TAGCGTAAAAGGAGCAACATAG −3’ |
156 | Addgene plasmid #27078 |
| LIN28 (pla) | Human | 5´- AGCCATATGGTAGCCTCATGTCCGC −3’ 5´- TAGCGTAAAAGGAGCAACATAG −3’ |
251 | Addgene plasmid #27080 |
| GAPDH | Porcine | 5´- TCGGAGTGAACGGATTTG −3’ 5´- CCTGGAAGATGGTGATGG −3’ |
219 | NM_001206359.1 |
| GAPDH* | Porcine | 5´- TCGGAGTGAACGGATTTG −3’ 5´- CCTGGAAGATGGTGATGG −3’ |
158 | NC_010447.4 |
*GAPDH primers were specific for porcine genomic DNA.
Quantitative RT–PCR analysis showed that the expression of endogenous genes such as pOCT4, pSOX2, pLIN28 and pNANOG had been reactivated and were significantly up-regulated in the four piPSC lines at passage 10 compared to the parental porcine fibroblasts (PEFs) (Figure 3(a)). As expected we observed clonal variation in regards to gene expression of the endogenous pluripotency markers. The expression of endogenous pOCT4 and pNANOG varied more than pSOX2 and pLIN28 expression between different piPSC lines. For example, the expression of endogenous pOCT4 in line piPSC7 was 7–8 folds higher than that in line piPSC2 (Figure 3(a)).
Figure 3.

Quantitative real-time PCR for the endogenous pluripotency markers pOCT4, pSOX2, pLIN28 and pNANOG was performed in the four piPSC lines at passage 10 and 20. Relative expression is shown as the fold change (calculated using 2 −ΔΔCT). The result showed the expression of endogenous genes pOCT4, pSOX2, pLIN28 and pNANOG were up-regulated in the four piPSC lines at passage 10 (a) and passage 20 (b) compared with the porcine embryonic fibroblasts (PEFs), which served as starting material (n = 3). Mean+ s.e.m. with asterisk* indicates statistically significant differences (p < 0.05).
The endogenous gene expression level in the four piPSC lines at passage 20 was also analyzed by quantitative RT–PCR. The results revealed that the endogenous genes pOCT4, pSOX2, pLIN28 and pNANOG remained highly expressed in all four piPSC lines compared to fibroblasts (Figure 3(b)). For each of these piPSC lines at passage 20, the expression of the endogenous pOCT4 and pNANOG remained at increased levels, but varied amongst clones and passage number. Line piPSC1 showed a slight reduction in the expression of pOCT4 and a slight increase in pNANOG at passage 20 compared to passage 10. Line piPSC2 displayed a different trend showing a decrease in pOCT4 expression at passage 20, but an even larger increase in pNANOG expression. Lines piPSC7 and piPSC8 displayed a similar decrease in pOCT4 and pNANOG expression in passage 20 compared to passage 10. Expression of pSOX2 and pLIN28 was significantly up regulated in passage 20 compared to passage 10 in all piPSC lines. These findings show a robust increase in pluripotency marker expression in all piPSC lines compared to the parental fibroblast lines confirming the successful generation of piPSCs. Interestingly, it also shows that expression not only varies amongst clones, but also within the same clones at different passages.
in vitro differentiation analysis
The in vitro differentiation potential of the four piPSC lines at passage 10 was investigated via EB formation. All four piPSC lines formed EBs in suspension culture for 7 days (Figure 4(a)). When placed on gelatin-coated culture plates and cultured in fibroblast medium, EBs attached to the substrate and differentiation could be observed two days after plating (Figure 4(b)). The EBs were allowed to differentiate for 14 days (Figure 4(c)), and the expression of lineage specific marker was confirmed by ICC showing staining for alpha-fetoprotein (AFP, endoderm) (Figure 4(d)), smooth muscle actin (SMA, mesoderm) (Figure 4(e)) and β-III tubulin (ectoderm) (Figure 4(f)), which demonstrated their capability of differentiating into all three germ layers. We next examined the in vitro differentiation capacity of all four piPSC lines at passage 20. As observed at passage 10 all lines formed large EBs in suspension culture (Figure 4(a)). Spontaneous differentiation was evident when these EBs were plated on gelatin-coated culture plates and cultured in fibroblast medium for 14 days (Figure 4(b,c)). ICC experiments confirmed the existence of different cell types positive for AFP (Figure 4(d)), SMA (Figure 4(e)), and β-III tubulin (Figure 4(f)). In all cases isotype controls were used as negative controls for the ICC experiments.
Figure 4.
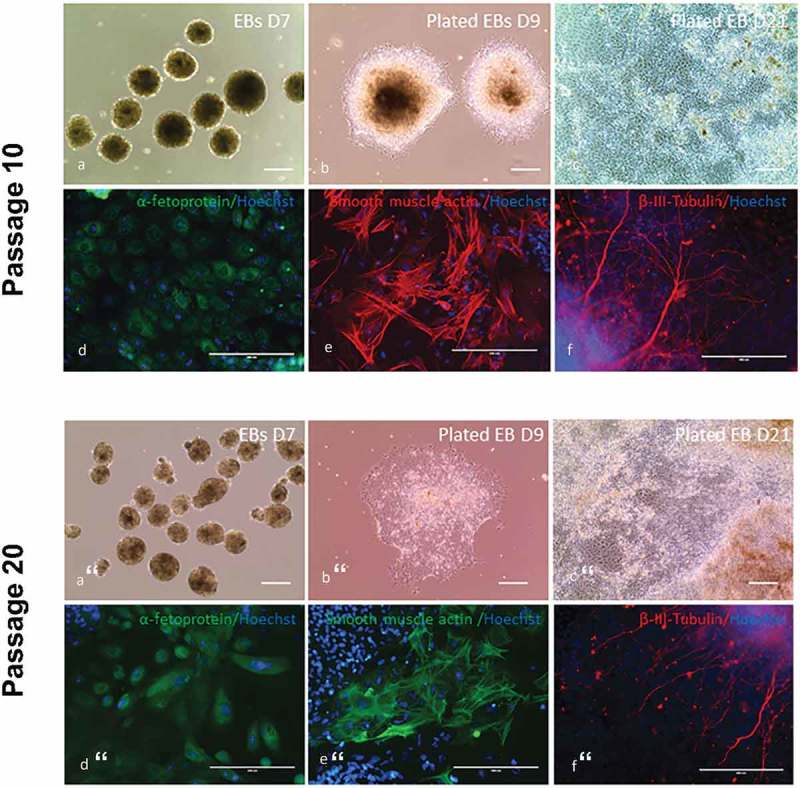
In vitro differentiation assay was performed in the four piPSC lines at passage 10 by using embryoid bodies (EBs) formation and fluorescence immunocytochemistry. (a), EBs formed in suspension culture for 7 days. (b), on day 7, EBs were plated on gelatin-coated plate in fibroblast culture medium for differentiation. D9 represents 2 days after plating. (c), D21 represents 14 days after plating. Scale bar represents 100 µm. (d-f), immunocytochemistry staining of plated EBs on day 21 with antibodies against alphafetoprotein (endoderm), smooth muscle actin (mesoderm) and β-III-tubulin (ectoderm) demonstrated their capability of differentiating into all three germlayers. Scale bar represents 200 µm.
In vitro differentiation assay was also performed in the four piPSC lines at passage 20 by using EBs formation and fluorescence immunocytochemistry. (a), EBs formed in suspension culture for 7 days. (b), D9 represents 2 days after plating EBs on gelatin-coated plate in fibroblast culture medium for differentiation. (c), D21 represents 14 days after plating. Scale bar represents 100 µm. (d-f), immunocytochemistry staining of plated EBs on day 21 with antibodies against alphafetoprotein (endoderm), smooth muscle actin (mesoderm) and β-III-tubulin (ectoderm). Scale bar represents 200 µm.
The in vitro differentiation experiments showed that episomal derived piPSCs were able to differentiate into cell types of all three germlayers at both early and late passages, confirming their pluripotent potential. Production of in vitro chimeras by injecting piPSC1 sublones into 5 days old porcine blastocysts, made by parthenogenetic activation, showed that it was possible to detect the cells by their red DIO fluorescence 48 hours after injection (39/68). The experiment showed that the piPSCs integrated into both the inner cell mass and trophectoderm of the blastocysts (Supp. Figure 2)
in vivo differentiation analysis
Teratoma assays were performed in order to assess the pluripotency potential of the original heterogeneous piPSC1 at passage 20 and the sub-clones 1,4,5 and 6 (piPSC1 SC1, SC4, SC5 and SC6; above passage 20) derived as single cell expansions from piPSC1. Six NOD/SCID mice were injected with either piPSC (piPSC1, piPSC1 SC1, piPSC1 SC4, piPSC1 SC5 and piPSC1 SC6) or PEFs from GA1. The original piPSC1 line from which the sub-clones were established developed 1 tumor with homogenously looking undifferentiated connective tissue and therefore failed to produce a conclusive teratoma. Five out of the six NOD/SCID mice injected with piPSC1 SC1 developed teratomas. One out of the six NOD/SCID mice injected with piPSC1 SC4 developed a teratoma. Three out of the six NOD/SCID mice injected with piPSC1 SC5 developed teratomas; and none of the six NOD/SCID mice injected with piPSC1 SC6 developed teratomas. No teratomas developed in the mice injected with GA1 PEFs. All teratomas were assessed via H&E staining and revealed tissue and structures representing the three germ layers. An example for the teratoma generated using piPSC1 SC5 is shown in Figure 5(a-b). The overall structure of the teratoma including different tissue types is shown in Figure 5(c). Higher magnification images reveal tubular structures lined with cuboidal epithelia representing potential endoderm (Figure 5(d)), lipocytes representing mesoderm (Figure 5(e)) and neuroepithelial-like epithelial rosettes representing potential ectoderm (Figure 5(f)), indicating the pluripotent nature of our piPSC. Moreover, when we analysed the teratomas in regards to their cell origin via CENP-A ICC, the majority of the tumor tissue was devoid of mouse cells, indicated by the absence of CENP-A expression, and therefore of porcine origin (Figure 5(g)). CENP-A positive cells were only present at the areas were the teratomas formed borders with the mouse tissue (Figure 5(h)).
Figure 5.
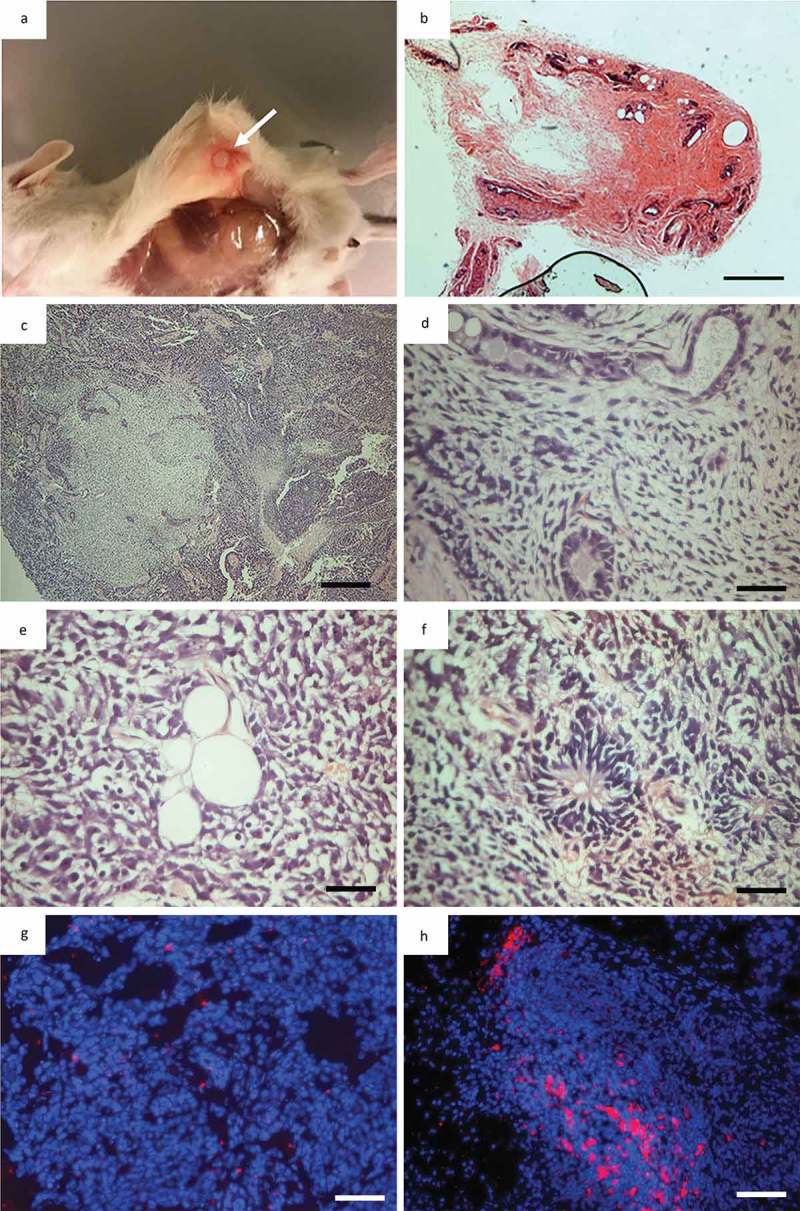
Teratoma formation in immunodeficient mice. (a), NOD/SCID mouse 8 weeks after subcutaneous injection of iPSC into left flank, arrow indicates tumor localtion; (b), Necropsy of NOD/SCID mouse with a tumor in its left flank (arrow indicate tumor) 8 weeks after iPSC injection; (c), Overview picture of haematoxylin and eosin (h&e) staining of tumor; (d), Endothelial-like epithelia representing endoderm; (e), Lipocytes representing mesoderm; (f), Neroepithelial-like epithelia representing ectoderm; (g), Overlay of a hoechst staining (Blue) and CENP-A staining red of a sample taken in the midle of a teratoma. Showing almost no murine tissiue (CENP-A positive); (h), Overlay of a hoechst staining (Blue) and CENP-A staining red of a sample taken on the the edge of a teratoma showing a significan amout of murine tissue (CENP-A) positive.
Taken together these teratoma experiments validate the pluripotent characteristics of the majority of our sub-clones derived from piPSC1 and moreover these are of true porcine origin verified by CENP-A ICC.
Analyses of potential integration or persistence of episomal plasmids
To examine whether episomal vectors persisted in our four piPSC lines, we performed PCR on total DNA (containing both genomic DNA and episomal DNA) using plasmid-specific primers to amplify plahOCT4, plahSOX2, and plahLIN28, which detect only the human transgenes [34]. Primers specific for porcine genomic GAPDH were employed as a positive control. The result showed that in all four piPSC lines exogenous plahKLF4, and plahLIN28 were detectable at passage 10, indicating that either genomic integration of the three episomal plasmids had occurred or they persisted in an episomal form (Figure 6(a)). The PCR analysis showed very weak signals for the plahOCT4 PCR product, indicating that less if any copy numbers of the episomal plasmid harboring hOCT4 have been retained. The expression ofplahKLF4, and plahLIN28 was also detected in the four lines at passage 10 and for plahOCT4 at a very low level by RT–PCR analysis with plasmid-specific primers (Figure 6(b)), indicating not only the presence of the transgenes, but also expression from these plasmids.
Figure 6.
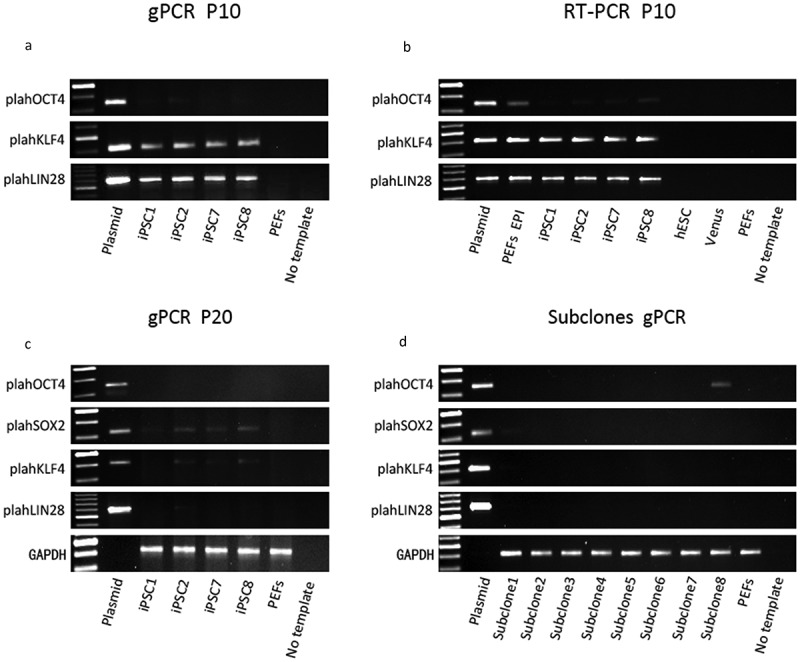
Integration and expression analysis of the three episomal plasmids in the four piPSC lines at passage 10 and passage 20. (a), genomic PCR (gPCR) with plasmid-specific primers for hOCT4, hKLF4 and hLIN28 confirmed the retention of the three episomal plasmids in the four piPSC lines at passage 10. (b), expression of the exogenous markers hOCT4, hKLF4 and hLIN28 was detected in the four piPSC lines at passage 10 by RT-PCR. (c), gPCR with plasmid-specific primers demonstrated the existence status of the three plasmids varied in the four piPSC lines at passage 20. Plasmid-specific primers for hOCT4, hSOX2, hKLF4 and hLIN28 were employed. (d), gPCR was performed to detect episomal vectors in the eight subclones (passage 26), which were derived from piPSC1. Episomal plasmids and cDNA obtained from the episomal transfected PEFs (one day after transfection with the three episomal plasmids) (PEFs EPI) served as positive controls. PEFs and a no template control served as negative controls. cDNA from human ESCs and lentiviral derived Venus piPSCs served as negative controls in RT-PCR.
It has previously been reported that oriP/EBNA1 (Epstein-Barr nuclear antigen-1)–based episomal vectors are spontaneously lost in the majority of human fibroblast derived hiPSCs at 11–20 passages [34]. Therefore, we analyzed further whether the episomal vectors were lost in our four piPSC lines over time in culture. We collected total DNA from the four piPSC lines at passage 20 and performed genomic PCR with plasmid-specific primers for amplifying plahOCT4, plahSOX2, plahKLF4 and plahLIN28 (Figure 6(c)). PCR analysis demonstrated the absence of plahOCT4 in all four piPSC lines, indicating that the episomal plasmid pCXLE-hOCT3/4-shp53 was efficiently lost during culture. However, plahSOX2 was still detected in lines piPSC2, piPSC7 and piPSC8 and at very low levels in piPSC1. The PCR for plahLIN28 showed that a product was still detectable in piPSC2, piPSC7 and piPSC8. This suggests that the two plasmids pCXLE-hSK and pCXLE-hUL are integrated or persisting in these lines. For piPSC1 we only observed a very faint PCR product for plahSOX2, pointing towards a nearly complete loss of this plasmid and the total loss of the other two plasmids. Overall, the PCR products of the transgenes examined were visible but much weaker compared to the PCR results at passage 10, indicating that the gradual decrease of the episomal plasmids had occurred during reprogramming. As mentioned earlier it appears as if the generated piPSC lines are not completely homogenous suggesting that some cells within the piPSC colonies have lost the episomal plasmids, whilst a few retained these. In order to establish clonal episomal-free piPSCs, eighteen subclones, derived from single cells from piPSC1, were isolated at passage 20. Eight subclones were further expanded for PCR analysis. PCR with plasmid-specific primers demonstrated loss of the plasmids in the majority of the subclones (subclone 2, 3, 4, 5, 6, 7), with the exception of weak plahSOX2 in subclone 1 and plahOCT4 in subclone 8 (Figure 6(d)). Hence, most subclones (2–7) were completely transgene-free and sustained pluripotency and self-renewal based on their endogenous pluripotency gene expression. This result not only proves successful generation of transgene-free piPSCs but also underlines the importance of subcloning for piPSCs in order to achieve integration-free piPSCs.
Discussion
In this study we established piPSCs by expression of human pluripotency factors via use of non-integrative episomal plasmids. To our knowledge these piPSCs are the first integration-free piPSCs generated using an episomal approach of reprogramming.
We reprogrammed porcine embryonic fibroblasts with episomal plasmids containing the following sequences: hOCT3/4-shp53, hSOX2, hKLF4, hLIN28 and hL-MYC [34]. L-MYC has been reported to be more potent and specific than c-MYC in generating hiPSCs [34], which might be an advantage over the classical use of c-MYC used as piPSC reprogramming factor. We performed a single transfection on porcine fibroblasts, which deviates from the approach of the other group who reported that their fibroblasts were electroporated twice within four days [44]. Although a second round of transfection may introduce higher levels of reprogramming factors into the cells and increase the efficiency of reprogramming, we decided to employ only one round of electroporation in order to reduce the potential risk of inducing chromosomal abnormality and integration of the episomal plasmids. Another problem associated with increased plasmid copy numbers is that potentially more cell cycles are needed before the plasmids are completely eliminated. The resulting four piPSC lines were found to be positive for AP activity. PiPSC1 were karyotyped at passage 10 and exhibited a normal karyotype. Genes encoding the endogenous pluripotency transcription factors NANOG, OCT4, SOX2 and LIN28 were expressed in all four lines, but the relative increase of endogenous OCT4 expression were low in the transgene-free iPSC in comparison to other porcine iPSCs [38]. This can indicate that the cells are more differentiated than naïve miPSC but Wu and colleagues [17] recently reported that a human intermediate type iPSC cultured with bFGF, Activin and CHIR99021 (FAC-hiPSCs), resulted in a more differentiated state than the naïve state and contributed to a higher degree to interspecies chimeras compared to naïve type human iPSC. Our piPSC line is also dependent on bFGF and CHIR9902, can survive single-cell dispersion and to a similar degree as the FAC-hiPSC contribute to teratomas and chimeric blastocyst indicating some similarities.
At the protein level, we were able to show the presence of NANOG, SOX2 and OCT4 in all piPSC colonies. Regarding SSEA-1 expression, large subpopulations of cells within the piPSC colonies were positive, but certain colony areas did not show staining. Unlike murine and human pluripotent stem cells, verified cell surface markers of pluripotency have not yet been well defined in piPSCs and variation of such marker have been noted. Previously, several authors have reported that SSEA-4 expression is characteristic of piPSCs, which would be in accordance with human ESCs and hiPSCs [18,21,24]. A single author also found SSEA-3 expressed in piPSCs [24]. However, another group reported their piPSCs, derived from porcine fetal fibroblasts, were positive for SSEA-1, but negative for SSEA-4 [19]. Our piPSCs only expressed SSEA1, which is consistent with Ezashi’s report that piPSCs displaying a human ES-like morphology express SSEA-1 [19]. Interestingly, the abundance of SSEA1 positive cells increased with prolonged passaging and we were able to observe a higher ratio of SSEA-1 positive cells at passage 20 than at passage 10. This could be indicative of stabilization of the state within these piPSC colonies over time, and potentially points to the fact that SSEA-1 could be an important cell surface marker for piPSC.
When subjected to in vitro differentiation, all four lines formed embryoid bodies, and immunostaining of the resultant cell outgrowths with antibodies against β-III tubulin (ectoderm), SMA (mesoderm), and AFP (endoderm) demonstrated their capability of differentiating into all three germ layers. No obvious differences were observable with respect to the differentiation potential of piPSCs at passage 10 vs. passage 20. This is an interesting observation and might indicate that piPSCs at later passages seem to be more stable.
In order to assess the transgene status with respect to genomic integration or plasmid persistence in our piPSCs, PCR analysis on total DNA extractions, which included genomic DNA and episomal plasmid DNA, were performed. These revealed that in all four lines at passage 10, that at least two of the three episomal plasmids were still present. Additionally, transgene expression was detected using the human specific primers for the pluripotency genes present on the episomal plasmids. Previous reports on piPSCs have shown that it is impossible to generate piPSCs without maintenance of the exogenous gene expression [18,19,21,24,44,47]. In our case however it is not clear if the transgene expression is required to maintain pluripotency at passage 10. Variation within the expression of pluripotency markers such as endogenous pOCT4, pNANOG, pSOX2 and pLIN28 in our piPSCs between passage 10 and 20 might indicate that a stable state is not yet fully established at passage 10. It could be possible that at passage 10, these piPSCs are still reliant on the transgene expression, which gradually decreases over time and passages. A previous study reported that the episomal vectors were spontaneously lost in the majority of hiPSC clones 10–20 passages after reprogramming. However, at passage 20 the episomal vectors were significantly diminished in all piPSC lines, especially hOCT3/4-shp53, which was completely undetectable in all piPSC lines. It needs to be noted that episomal vectors can be fragmented and that these fragments can integrate into the cells genome as reported by Du et al, 2015 [38]. However our results are consistent with the previous report in hiPSCs, confirming that oriP/EBNA1 (Epstein-Barr nuclear antigen-1) – based episomal vectors were prone to be eliminated over a few passages [34]. It is likely thus to assume that the episomal plasmids were not integrated but persisted in a plasmid form. Nevertheless, two plasmids pCXLE-hSK and pCXLE-hUL were still detected in passage 20 cells. This promoted us to select the piPSC1, which showed the weakest PCR products for hSOX2 (detecting presence of pCXLE-hSK) and hLIN28 (detecting presence of pCXLE-hUL). However, subcloning of piPSC1 colonies into single cells revealed that 6 out of 8 subclones were completely free of episomal vector DNA. We were hereby able to prove that our piPSCs are free of the reprogramming constructs. In our opinion one of the most striking findings was that subcloning appears to be crucial in order to obtain integration and episomal free piPSCs. To date, such a clonal expansion is not common practice but it might indeed prove to be extremely helpful to obtain transgene-free piPSCs. One drawback with this approach is that it takes considerable time to eliminate the reprogramming constructs from the cells. Therefore, improved methodologies that could help to eliminate the transgenes faster would be of benefit.
The importance of subcloning of piPSC was emphasized in the in vivo teratoma experiments. Our results showed that the parental pNF did not result in tumors, piPSC1 formed a tumor with undifferentiated connective tissue and not all sub-clones were able to generate teratomas. SC6, failed to produce teratomas, whilst the other sub-clones 1, 4 and 5 consistently produced teratomas containing porcine cells and tissues representing all three germ layers. It is interesting that SC1 was able to form teratomas even though it retained plahSOX2 (Figure 6(d)), but the amount of plahSOX2 was extremely low. Indicating that either very low amount of retained plasmid does not affect teratoma formation or the plasmid was transcriptionally inactivated. Overall we propose that isolation of single cells from heterogeneous piPSC colonies and expansion of those as sub-clones is crucial. The teratoma experiments underlines the importance to isolate single cells from heterogeneous piPSC colonies and to expand these as sub-clones. Because, only transgene free piPSC, which have activated their endogenous gene expression of pluripotency genes and the potential to form teratomas consisting of tissue derived from all three germ layers can be considered as transgene free pluripotent piPSC.
In regards to personalized medicine one of the biggest problems right now is the safety of cells transplanted into the patients. Pigs could be an ideal model to test these safety issues based on their remarkable anatomical and physiological similarity to humans [18,21,43,48]. We have now developed karyotypically normal piPSCs, devoid of transgenes, that may be optimal for testing safety and prospects for cell-based therapy in a pig model.
In conclusion, integration-free intermediate type piPSCs were generated for the first time and the cell lines appear to be completely independent of their reprogramming vectors existence and expression. A strategy for subcloning of the initial cell lines showed efficient for establishing integration-free lines, which remained competent with respect to self-renewal and expression of pluripotency transcription factors and other markers. Derivatives of these cells could be of use for testing in transplantation studies and for helping bring iPSCs closer for use in the clinic, however we still need to prove that the cells can contribute to chimeric offspring.
Materials and methods
Cell culture and piPSC reprogramming
Porcine embryonic fibroblasts were derived from 24-day-old Göttingen minipig embryos by using a standard protocol [49]. One embryonic fibroblast cell line, termed GA1 (Göttingen minipig fibroblast cell line A1), was used in this study. Cells were used from passage 1 onwards and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich, St. Louis, MO) containing 1% penicillin/streptomycin (pen/strep; Sigma-Aldrich) and 10% fetal bovine serum (FBS; Hyclone). Cells were maintained in 5% CO2 and cultured in a humidified environment at 38.5°C. Three episomal plasmids [34], pCXLE-hOCT3/4-shp53 (Addgene plasmid #27077), pCXLE-hSK (Addgene plasmid #27078) and pCXLE-hUL (Addgene plasmid #27080) were purchased from Addgene, Inc. (Cambridge, MA) and purified with the PurelinkTM HQ Mini Plasmid purification kit (K2100-01; Invitrogen, Carlsbad, CA). Passage 1 GA1 fibroblasts were trypsinized by Trypsin-EDTA (Sigma-Aldrich) and 1 × 105 cells were electroporated with 1 μg of the three episomal plasmids combination (1:1:1) using a NeonTM electroporation device (Life Technologies, Carlsbad, CA) with a single pulse at 1300 V for 30 ms. Cells were then plated in DMEM (Sigma-Aldrich) containing 10% fetal bovine serum (FBS; Hyclone) without antibiotics. The medium was replaced with the addition of pen/strep on Day 2, and on Day 7 cells were trypsinized by Trypsin-EDTA (Sigma-Aldrich) and passaged onto mitomycin C–treated (Sigma-Aldrich) CF1 mouse embryonic fibroblasts (MEFs) (Millipore, Billerica, MA) and cultured in DMEM/F12 medium (Sigma-Aldrich) supplemented with 20% KnockOut Serum Replacement (Invitrogen), 1 x pen/strep (Sigma-Aldrich), 1 x nonessential amino acids (Sigma-Aldrich), 100 µM β-mercaptoethanol (Life Technologies), 20 ng/mL human recombinant basic fibroblast growth factor (bFGF) (Prospec, East Brunswick, NJ), and two kinase inhibitors; 1 µM PD0325901 (Sigma-Aldrich), which inhibits MEK signal pathway, and 3 µM CHIR99021 (Sigma-Aldrich), a GSK3β inhibitor.
On day 21, twelve ESC-like colonies were manually cut into pieces using a 0.5 ml syringe (Terumo, Elkton, MD) and transferred onto new MEFs in single wells of 24-well plates. After four days culture, these colonies were dissociated with TrypLE Select (Gibco; Life Technologies) and transferred from 24-well plates into single wells of 6-well plates. Four piPSC lines, designated piPSC1, piPSC2, piPSC7, piPSC8, were selected for further culture and detailed analysis. The four piPSC lines were routinely passaged every 4–5 days with TrypLE Select (Gibco; Life Technologies) and the culture medium was changed daily. The four piPSC lines were cryopreserved from passage 3 and onward.
Subcloning was performed on piPSC1 at passage 10 aiming to derive piPSC clones devoid of episomal vectors. Eighteen individual colonies were manually picked from piPSC1, dispersed into single cells using acutase and individual piPSC plated onto fresh MEFs in single wells of 24-well plates. After one more passage, eight subclones were selected for further expansion and analysis, termed subclone 1–8.
Fluorescence immunocytochemistry
Cells from the four piPSC lines at passage 10 and passage 20 were cultured on Lab-Tek chamber slides (Thermo/Nunc) and fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature. They were permeabilized with 0.1% Triton-X 100 (Sigma-Aldrich) in PBS for 30 min at room temperature. Permeabilization was not performed when cells were immunostained for cell surface makers. After washing two times with PBS, cells were incubated in blocking buffer [5% normal donkey serum (Sigma-Aldrich) in 0.25% BSA/PBS] for 1 h at room temperature. Primary antibodies were diluted in 0.25% BSA/PBS and cells were cultured in diluted primary antibodies overnight at 4°C with gentle rotation. Primary antibodies used were as follows: Rabbit polyclonal anti-NANOG (1:400; Peprotech, #500-P236, lot 0405M322RB), goat polyclonal anti-OCT4 (1:250; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), mouse monoclonal anti-SOX2 (1:50; R&D Systems, Minneapolis, MN, USA), mouse monoclonal SSEA-1 (1:50; Biolegend, #125601, lot B134566), rat monoclonal SSEA-3 (1:100; Biolegend, #330302, lot B117272), mouse monoclonal SSEA-4 (1:200; Biolegend, #330402, lot B134263), mouse monoclonal TRA-1-60 (1:400; Biolegend, #330602, lot B133894), and mouse monoclonal TRA-1-81 (1:100; Biolegend, #330702, lot B1136798). Primary antibody isotypes were used as negative control. Isotype antibodies were diluted in 0.25% BSA/PBS at the following dilutions: Mouse IgG2a 1:10 (Biolegend, #ⅹ0943, lot 00017485)(SOX2), Mouse IgM, k, 1:100 (Biolegend, #401601, lot B144089)(SSEA-1), Rat IgM, k, 1:100 (Biolegend, #400801, lot B144892)(SSEA-3), Mouse IgG3, 1:200 (Biolegend, #401301, lot B147516)(SSEA-4), Mouse IgM, k 1:400 (Biolegend, #401601, lot B144089)(Tra-1-60), Mouse IgM, k, 1:200 (Biolegend, #401601, lot B144089)(Tra-1-81). Secondary antibody negative controls were performed for NANOG and OCT4. Cells were washed two times with PBS and incubated in either fluorescein isothiocyanate (FITC)-conjugated or Cy3-conjugated secondary antibodies (Jackson Immunoresearch, Suffolk, UK), which were diluted at 1:200 in 0.25% BSA/PBS. Secondary antibodies were as follows: FITC-conjugated/Cy3-conjugated donkey anti-rabbit IgG (NANOG), FITC-conjugated/Cy3-conjugated donkey anti-mouse IgG (SOX2, SSEA-4, Tra-1-60, Tra-1-81), FITC-conjugated/Cy3-conjugated donkey anti-mouse IgM (SSEA-1), FITC-conjugated/Cy3-conjugated donkey anti-rat IgG (SSEA-3) and FITC-conjugated/Cy3-conjugated donkey anti-goat (OCT4). DNA labeling was performed by incubating the cells in 5 µg/mL Hoechst 33342 (Sigma-Aldrich) for 10 min. Fluorescent mounting medium (DAKO, Glostrup, Denmark) was utilized to mount the glass slides. Images were captured using a DMRB fluorescent microscope (Leica Microsystems, Wetzlar, Germany). The human ESC line (H1/WA01) purchased from WiCell, USA was used as a positive control for SSEA-3, SSEA-4, TRA-1-61, and TRA-1-81. The mouse ESC line derived in Hungary by Biotalentum Ltd. was used as positive controls for NANOG, OCT4, and SSEA-1.
Cell counts and statistical analysis
Manual cell counts were performed on triplicates of images of piPSC1 colonies stained with antibodies against NANOG, SOX2, OCT4 and SSEA1 at p10 and p20. Numbers of fluorescent-labeled cells out of total cells labeled with HOECHST within the colonies were compared between P10 and P20 for each AB using SAS enterprise 9.1 and Proc freq.
Alkaline phosphatase (AP) assay
piPSCs at passage 5 were fixed in 4% PFA for 30 min at room temperature. FastRed (Sigma-Aldrich; F8764) was dissolved in MilliQ H2O (1 mg/ml). Napthol phosphate (Sigma-Aldrich; 855) (40 µl/ml) was added into FastRed solution and mixed well just prior to using. piPSCs were incubated in fresh FastRed/Napthol mixture for 15-30 min in dark. Stained cells were washed two times with MilliQ H2O and stored in MilliQ H2O at 4°C. Images were taken using EVOS™ XL Core digital inverted microscope (Life Technologies).
Embryoid body assay and in vitro chimeric contribution
Cells from the four piPSC lines at passage 10 and passage 20 were dissociated with Accutase (Thermo Electron) and transferred to low cell binding plates (Sigma-Aldrich, Z721050-7EA, lot#3110) for suspension culture in DMEM/F12 medium (Sigma-Aldrich) containing 20% KnockOut Serum Replacement (Invitrogen), 1x pen/strep (Sigma-Aldrich), 1x nonessential amino acids (Sigma-Aldrich), 100 µM β-mercaptoethanol (Life Technologies), in absence of bFGF or any inhibitors. Embryoid bodies (EBs) were formed after 7 days in suspension culture and then were transferred onto 0.1% gelatin-coated 6-well plates in DMEM (Sigma-Aldrich) supplemented with 1% pen/strep (Sigma-Aldrich) and 10% fetal bovine serum (FBS; Hyclone). Some EBs spontaneously differentiated in iPSC medium in comparison to fibroblast medium. EBs were allowed to differentiate for 2 weeks and differentiated cells were subsequently stained with antibodies against β-III tubulin (ectoderm) (Sigma-Aldrich, T8660; 1:200), smooth muscle actin (mesoderm) (SMA; DAKO, M0851; 1:200), and alpha-fetoprotein (endoderm) (AFP; DAKO, A0008; 1:200). Samples were analyzed using EVOS™ fl digital inverted fluorescent microscope (Life Technologies).
Additionally, subclones from piPSC1 were harvested for in vitro differention tests. These were dyed with the red fluorescent membrane dye DIO (Life technologies D282) according to the manufacturer’s manual. 10–15 cells were injected into 5 days old porcine blastocysts (n = 68). These blasotocysts had been generated via parthenogenetic activation and the injected cells within the blastocysts were followed for 48 hours in the NIKON Biostation IM as described previously [50].
Teratoma formation
Female NOD/SCID mice (Taconic: NOD/MrkBomTac-Prkdc< scid> Female, Lot: 20130624-EBU020601C-HC-M) were purchased from Taconic (Silkeborg, Denmark) and housed in groups of no more than 8 at the Department for Experimental Medicine (Panum Institute, Copenhagen, Denmark). General animal welfare guidance was followed and the teratoma experiments were approved under the protocol number P 13-210 by the veterinary authority at Department of Experimental Medicine. piPSC1 (passage 20) and sub-clones 1, 4, 5, 6 from piPSC1 (passage 20<) as well as the parental GA1 fibroblasts (control group) were harvested using 1x TrypLE (Gibco, Cat. # A12177-01), counted, spun down, re-suspended in PBS with 1% BSA (Sigma-Aldrich, Cat. # A7906) and aliquotted at 1.5 million cells/250 µl. The cells were injected subcutaneously into the left flank of the mice using a 1 ml insulin syringe and 29G needle from Terumo. The mice were monitored for up to 3 months for tumor formation. The origin of the teratomas were examined by CENP-A staining (Mouse specific). The teratomas were fixed in 4% PFA overnight and embedded in OCT compound (Sakura FineTek, Cat. # 4583). Subsequently, the tumors were sectioned on a cryostat at 5 µM and mounted on slides. The sections were subjected to hematoxylin and eosin staining or immunocytochemistry with rabbit poly antibodies for CENP-A (Cell Signaling #2048) 1:800 as primary antibody and donkey anti rabbit (#711-165-15292681) 1:200 as secondary antibody.
Karyotyping
Cells from piPSC1 at passage 10 were treated with KaryoMAX colcemid (Life Technologies, Carlsbad, CA, USA) for 45 min and afterwards disassociated with TrypLE Select (Gibco; Life Technologies). After centrifugation, the supernatant was removed and the cell pellets were suspended in fresh fixative solution composed of 25% acetic acid and 75% methanol. Karyotyping was performed using G-band standard staining at Cell Guidance Systems (Babraham Research Campus, Cambridge, UK).
Analyses of integration or prolonged persistence of plasmids
For assessment of transgene integration and prolonged plasmid retention, cells from the four piPSC lines were harvested at passage 10 and passage 20 respectively and total DNA (genomic DNA and episomal plasmids) was isolated from these cells for polymerase chain reaction (PCR) using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions. DNA from parental GA1 fibroblasts was isolated as negative control. PCR was also performed on genomic DNA from the eight subclones derived from piPSC1. Total DNA from the subclones at passage 26 was isolated using the same kit as described above. Episomal plasmid-specific primers as previously published by Okita et al., 2011 were used for PCR [34](Table 1). GAPDH primers specific for porcine genomic DNA was designed and used in PCR at passage 20 and subclones PCR (Table 1).
RNA extraction and reverse transcription PCR (RT-PCR)
For assessment of exogenous and endogenous gene expression, piPSCs from the four cell lines were harvested at both passage 10 and passage 20 and lysed in 350 µl RLT buffer (Qiagen) containing β-mercaptoethanol (Sigma-Aldrich). Total RNA was extracted using the RNAeasy Micro Kit (Qiagen) according to the manufacturer’s instructions. First-strand cDNA synthesis was performed using a RevertAidTM First Strand cDNA Synthesis Kit (Fermentas/Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. A total of 1 µg RNA was used as a template for cDNA synthesis. First-strand cDNA was used as template for subsequent PCR analysis in a 50 µl total volume composed of 5 µl 10x Taq buffer (Fermentas/Thermo Scientific), 2 µl 25 mM MgCl2 (Fermentas/Thermo Scientific), 2 µl 10mM deoxyribonucleotide triphosphates (dNTPs; Fermentas/Thermo Scientific), and 1 µl 5U/µL Taq polymerase (Fermentas/Thermo Scientific) in RNase-free water. Plasmid specific primers as described by Okita et al., 2011 [34] were used for RT-PCR (Table 1). RT-PCR was also performed to detect the genes expression of endogenous porcine pluripotency genes. Porcine specific primers were designed to bind within the coding exons and were intron-spanning if possible (Table 1).
PCR reactions were performed by initially denaturing cDNA at 95 °C for 2 min followed by 35 cycles of denaturing at 95 °C for 1 min, annealing at 58 °C for 45 sec, and elongation at 72 °C for 1 min. A further extension was performed at 72 °C for 5 min.
Quantitative real-time PCR
RNA extraction and cDNA synthesis were performed as described above. The obtained cDNA from the four piPSC lines at passage 10 and passage 20 was used for quantitative real-time PCR (qRT-PCR) reactions. qRT-PCR was carried out in triplicate in 96-well optical reaction plates (Scientific Specialties, Inc.) using the LightCycler 480 SYBR Green I Master Kit (F-Hoffman La Roche, Basal, Switzerland). Each reaction well contained 5 µl SYBR Green Master Mix, 1 µl forward (10 µM) and 1 µl reverse primers (10 µM) to the target genes, 1 µl dH2O and 2 µl of diluted cDNA in a final reaction volume of 10 µl. Primers used for qRT-PCR are listed in Table 1. Three biological replicates were prepared for each cell line and control cell lines. The housekeeping gene GAPDH was used as an internal control to normalize the Ct values of target genes. Overall assessment of gene expression was performed by normalizing sample gene expression to gene expression of parental embryonic porcine fibroblasts. The relative expression level of the target genes was calculated using 2−ΔΔCt method. 2−ΔΔCt values served for standard deviations calculation and statistical analyses were also performed on the difference in Ct values using an independent, unpaired, two-tailed students t-test and significance was determined when p ≤ 0.05.
Funding Statement
Funding for this research was provided by: The Danish Council for Independent Research, Natural Sciences (FNU), grant number: 11-106627; US Department of Agriculture (USDA), grant number: 2011-67015-30688; Personal PhD fellowship to Dong Li sponsored by China Scholarship Council (CSC); and European Union through the Coordination and Support Action of the Horizon 2020 programe, Grant number 692299.
List of abbreviations:
ESC: embryonic stem cell
iPSCs: induced pluripotent stem cells
piPSCs: porcine induced pluripotent stem cells
miPSCs: murine iPSCs
hiPSCs: human iPSCs
2i: 2 inhibitors
MEK: mitogen-activated protein kinase kinase
GSK3β: glycogen synthase kinase 3beta
GA1: Göttingen minipig fibroblast cell line A1
EBs: Embryoid bodies
ICC: Immunocytochemistry
qRT-PCR: quantitative real-time PCR
bFGF: basic fibroblast growth factor
Acknowledgments
We would like to thank Tina Christoffersen for cell culture support, Anita Pacht for assistance with Q-PCR.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Takahashi K, Yamanaka S.. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- [2].Maherali N, Sridharan R, Xie W, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. [DOI] [PubMed] [Google Scholar]
- [3].Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. [DOI] [PubMed] [Google Scholar]
- [4].Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- [5].Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- [6].Li W, Wei W, Zhu S, et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell. 2009;4:16–19. [DOI] [PubMed] [Google Scholar]
- [7].Liao J, Cui C, Chen S, et al. Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell. 2009;4:11–15. [DOI] [PubMed] [Google Scholar]
- [8].Han X, Han J, Ding F, et al. Generation of induced pluripotent stem cells from bovine embryonic fibroblast cells. Cell Res. 2011;21:1509–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang B, Li T, Alonso-Gonzalez L, et al. A virus-free poly-promoter vector induces pluripotency in quiescent bovine cells under chemically defined conditions of dual kinase inhibition. PLoS One. 2011;6:e24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sumer H, Liu J, Malaver-Ortega LF, et al. NANOG is a key factor for induction of pluripotency in bovine adult fibroblasts. J Anim Sci. 2011;89:2708–2716. [DOI] [PubMed] [Google Scholar]
- [11].Li Y, Cang M, Lee AS, et al. Reprogramming of sheep fibroblasts into pluripotency under a drug-inducible expression of mouse-derived defined factors. PLoS One. 2011;6:e15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu J, Balehosur D, Murray B, et al. Generation and characterization of reprogrammed sheep induced pluripotent stem cells. Theriogenology. 2012;77:338–46 e1. [DOI] [PubMed] [Google Scholar]
- [13].Sartori C, DiDomenico AI, Thomson AJ, et al. Ovine-induced pluripotent stem cells can contribute to chimeric lambs. Cell Reprogram. 2012;14:8–19. [DOI] [PubMed] [Google Scholar]
- [14].Bao L, He L, Chen J, et al. Reprogramming of ovine adult fibroblasts to pluripotency via drug-inducible expression of defined factors. Cell Res. 2011;21:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu H, Zhu F, Yong J, et al. Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell. 2008;3:587–590. [DOI] [PubMed] [Google Scholar]
- [16].Shimada H, Nakada A, Hashimoto Y, et al. Generation of canine induced pluripotent stem cells by retroviral transduction and chemical inhibitors. Mol Reprod Dev. 2010;77:2. [DOI] [PubMed] [Google Scholar]
- [17].Wu J, Platero-Luengo A, Sakurai M, et al. Interspecies chimerism with mammalian pluripotent stem cells. Cell. 2017;168:473–486 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Esteban MA, Xu J, Yang J, et al. Generation of induced pluripotent stem cell lines from Tibetan miniature pig. J Biol Chem. 2009;284:17634–17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ezashi T, Telugu BP, Alexenko AP, et al. Derivation of induced pluripotent stem cells from pig somatic cells. Proc Natl Acad Sci U S A. 2009;106:10993–10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ezashi T, Matsuyama H, Telugu BP, et al. Generation of colonies of induced trophoblast cells during standard reprogramming of porcine fibroblasts to induced pluripotent stem cells. Biol Reprod. 2011;85:779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Montserrat N, Bahima EG, Batlle L, et al. Generation of pig iPS cells: a model for cell therapy. J Cardiovasc Transl Res. 2011;4:121–130. [DOI] [PubMed] [Google Scholar]
- [22].Montserrat N, de Onate L, Garreta E, et al. Generation of feeder-free pig induced pluripotent stem cells without Pou5f1. Cell Transplant. 2012;21:815–825. [DOI] [PubMed] [Google Scholar]
- [23].Ruan W, Han J, Li P, et al. A novel strategy to derive iPS cells from porcine fibroblasts. Sci China Life Sci. 2011;54:553–559. [DOI] [PubMed] [Google Scholar]
- [24].Wu Z, Chen J, Ren J, et al. Generation of pig induced pluripotent stem cells with a drug-inducible system. J Mol Cell Biol. 2009;1:46–54. [DOI] [PubMed] [Google Scholar]
- [25].Telugu BP, Ezashi T, Sinha S, et al. Leukemia inhibitory factor (LIF)-dependent, pluripotent stem cells established from inner cell mass of porcine embryos. J Biol Chem. 2011;286:28948–28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. [DOI] [PubMed] [Google Scholar]
- [27].Hamanaka S, Yamaguchi T, Kobayashi T, et al. Generation of germline-competent rat induced pluripotent stem cells. PLoS One. 2011;6:e22008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].West FD, Terlouw SL, Kwon DJ, et al. Porcine induced pluripotent stem cells produce chimeric offspring. Stem Cells Dev. 2010;19:1211–1220. [DOI] [PubMed] [Google Scholar]
- [29].West FD, Uhl EW, Liu Y, et al. Brief report: chimeric pigs produced from induced pluripotent stem cells demonstrate germline transmission and no evidence of tumor formation in young pigs. Stem Cells. 2011;29:1640–1643. [DOI] [PubMed] [Google Scholar]
- [30].Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27:2667–2674. [DOI] [PubMed] [Google Scholar]
- [31].Fusaki N, Ban H, Nishiyama A, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lieu PT, Fontes A, Vemuri MC, et al. Generation of induced pluripotent stem cells with cytotune, a non-integrating Sendai virus. Methods Mol Biol. 2013;997:45–56. [DOI] [PubMed] [Google Scholar]
- [33].Okita K, Nakagawa M, Hyenjong H, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. [DOI] [PubMed] [Google Scholar]
- [34].Okita K, Matsumura Y, Sato Y, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. [DOI] [PubMed] [Google Scholar]
- [35].Gonzalez F, Monasterio MB, Tiscornia G, et al. Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc Natl Acad Sci U S A. 2009;106:8918–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kaji K, Norrby K, Paca A, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Du X, Feng T, Yu D, et al. Barriers for deriving transgene-free pig iPS cells with episomal vectors. Stem Cells. 2015;33:3228–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim JB, Zaehres H, Arauzo-Bravo MJ, et al. Generation of induced pluripotent stem cells from neural stem cells. Nat Protoc. 2009;4:1464–1470. [DOI] [PubMed] [Google Scholar]
- [40].Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Warren L, Ni Y, Wang J, et al. Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci Rep. 2012;2:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Na J, Baker D, Zhang J, et al. Aneuploidy in pluripotent stem cells and implications for cancerous transformation. Protein Cell. 2014;5:569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roberts RM, Telugu BP, Ezashi T. Induced pluripotent stem cells from swine (Sus scrofa): why they may prove to be important. Cell Cycle. 2009;8:3078–3081. [DOI] [PubMed] [Google Scholar]
- [44].Telugu BP, Ezashi T, Roberts RM. Porcine induced pluripotent stem cells analogous to naive and primed embryonic stem cells of the mouse. Int J Dev Biol. 2010;54:1703–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lin T, Ambasudhan R, Yuan X, et al. A chemical platform for improved induction of human iPSCs. Nat Methods. 2009;6:805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rasmussen MA, Holst B, Tumer Z, et al. Transient p53 suppression increases reprogramming of human fibroblasts without affecting apoptosis and DNA damage. Stem Cell Reports. 2014;3:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Okita K, Yamanaka S. Induction of pluripotency by defined factors. Exp Cell Res. 2010;316:2565–2570. [DOI] [PubMed] [Google Scholar]
- [48].Fairbairn L, Kapetanovic R, Sester DP, et al. The mononuclear phagocyte system of the pig as a model for understanding human innate immunity and disease. J Leukoc Biol. 2011;89:855–871. [DOI] [PubMed] [Google Scholar]
- [49].Hall VJ, Kristensen M, Rasmussen MA, et al. Temporal repression of endogenous pluripotency genes during reprogramming of porcine induced pluripotent stem cells. Cell Reprogram. 2012;14:204–216. [DOI] [PubMed] [Google Scholar]
- [50].Secher JO, Freude KK, Li R, et al. Optimization of three-dimensional imaging on in vitro produced porcine blastocysts and chimeras for stem cell testing: a technology report. Stem Cells Dev. 2015;24:1141–1145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.