

ABSTRACT
Sterile alpha motif and HD domain-containing protein 1 (SAMHD1) is a mammalian dNTP hydrolase (dNTPase) and functions as a negative regulator in the efficacy of cytarabine treatment of acute myeloid leukemia (AML). We have reported that SAMHD1 knockout (KO) increased the activity of phosphoinositide 3-kinase (PI3K) in AML-derived THP-1 cells and attenuated their ability to form subcutaneous tumors in xenografted immunodeficient mice. However, the functional significance of SAMHD1 in controlling AML leukemogenesis remains unclear. Previous studies show that in vitro transformation of Madin-Darby canine kidney (MDCK) epithelial cells by the Jaagsiekte sheep retrovirus (JSRV) envelope protein requires activation of the PI3K/Akt oncogenic signaling pathway. Using this cell transformation model, we demonstrated that ectopic expression of wild-type human SAMHD1 or a dNTPase-defective SAMHD1 mutant (HD/AA) significantly inhibited MDCK cell transformation, but did not affect cell proliferation. To visualize and quantify THP-1 cell growth and metastasis in xenografted immunodeficient mice, we generated luciferase-expressing stable SAMHD1 KO THP-1 cells and control THP-1 cells, which were injected intravenously into immunodeficient mice. Bioluminescence imaging and quantification analysis of xenografted mice revealed that SAMHD1 KO cell-derived tumors had similar growth and metastatic potential compared with control cells at 35 days post-injection. However, mice xenografted with SAMHD1 KO cells showed greater survival compared with mice injected with control cells. Our data suggest that exogenous SAMHD1 expression suppresses in vitro cell transformation independently of its dNTPase activity, and that endogenous SAMHD1 affects AML tumorigenicity and disease progression in vivo.
Keywords: SAMHD1, retrovirus oncogene, cell transformation, acute myeloid leukemia (AML), immunodeficient mice xenograft, PI3K-Akt signaling
Introduction
SAMHD1 is a deoxynucleoside triphosphohydrolase (dNTPase) involved in the regulation of dNTP homeostasis in mammalian cells [1–5]. Through its ability to degrade dNTPs, SAMHD1 restricts the replication of retroviruses and DNA viruses, thus blocking the virus life cycle [2,4]. Along with its role in viral infection, SAMHD1 exerts additional functions, including control of cell proliferation, apoptosis and tumor development [6]. SAMHD1 somatic mutations have been identified in solid and hematological malignancies, including glioblastoma, lung, colon, pancreatic and breast cancers, as well as medulloblastoma, myeloma and chronic lymphocytic leukemia (CLL) [7–16]. These mutations result in alterations of the DNA damage response, which leads to higher frequency of mutations, and increased resistance to chemotherapy [8,16–18]. Moreover, downregulation of SAMHD1 expression has been reported in T-cell leukemia, lymphoma, CLL, lung adenocarcinoma, and breast cancer [8,19–22]. Altogether, these observations suggest that SAMHD1 may have a tumor suppressor role in different cell types. However, the molecular mechanisms associated with these effects are not completely understood.
We have recently reported that, in cutaneous T-cell lymphoma derived-cells, exogenous SAMHD1 expression inhibits cell growth and induces apoptosis via the extrinsic apoptotic pathway and downregulation of the expression of the anti-apoptotic molecule cFLIPS (short form of cellular FLICE-like inhibitory protein) [23]. Conversely, SAMHD1 KO in acute myeloid leukemia (AML)-derived THP-1 cells enhances cell proliferation and migration via increased activation of the PI3K-Akt pathway, leading to increased phosphorylation and inhibition of nuclear localization of the cyclin-dependent kinase inhibitor p27 [24]. By using a subcutaneous xenograft mouse model, we have shown in vivo that subcutaneous tumors from SAMHD1 KO THP-1 cells have lower growth rate compared to cells expressing the endogenous protein, and this phenotype correlated with increased inflammation status in SAMHD1 KO versus control cells, as demonstrated by higher expression of the pro-inflammatory cytokine tumor necrosis factor α (TNF-α) [24].
The PI3K-Akt signaling pathway plays a key role in the regulation of cell cycle, apoptosis, cellular quiescence and senescence [25]. Activation of PI3K by growth factors is followed by induction of the serine-threonine kinase Akt, which in turn modulates the activity of a plethora of downstream targets, such as p27 (also known as Kip1), mTOR (mammalian target of rapamycin), FOXO (Forkhead family of transcription factor), thus positively modulating cell growth and survival [7,26]. This network is often overactive in cancers, including AML [26–30], and therefore significant effort has been devoted to the design of specific inhibitors which are currently tested in pre-clinical and clinical studies [30–32]. A few reports have shown that inhibition of the PI3K-Akt signaling potentiates the anticancer activity of the deoxycytidine analog cytarabine in AML and other cancers [28,33–35], suggesting that synergistic combination of PI3K-Akt inhibitors and other anticancer drugs can be a potential therapeutic option for AML. The PI3K-Akt signaling pathway can also be activated by viral and cellular oncogenes [25,36]. For instance, the envelope glycoprotein (Env) of the Jaagsiekte sheep retrovirus (JSRV), a retrovirus causing ovine pulmonary adenocarcinoma in sheep, can transform fibroblasts from mice, rats, chickens [37–41] and MDCK epithelial cells through activation of the PI3K-Akt pathway [42].
In this study, we show that exogenous SAMHD1 expression significantly inhibits in vitro transformation of MDCK cells induced by the Env of JSRV in a dNTPase-independent manner, but does not affect cell proliferation. Moreover, considering the important role of SAMHD1 in AML pathogenesis and treatment, we generated a physiologically relevant AML mouse model that allowed us to further investigate the role of SAMHD1 in AML development in vivo. Our findings suggest that endogenous SAMHD1 protein affects AML tumorigenicity and disease progression in vivo.
Materials and methods
Cell culture and treatments
MDCK epithelial cells (obtained from the American Type Culture Collection (ATCC), ATCC CCL-34) and human embryonic kidney 293T (HEK293T) cells were maintained in complete DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Transformed MDCK cells were grown under the same conditions using 5% FBS. All the cell lines utilized in this study were tested negative for mycoplasma contamination using universal mycoplasma detection kit (ATCC, #30–101-2K). AML-derived THP-1 cell lines with SAMHD1 KO and control cells were generated and cultured as described [43].
Generation of MDCK cell lines with stable SAMHD1 expression
To generate MDCK cell lines stably expressing SAMHD1 wild-type (WT) or HD/AA mutant, we first produced lentiviral vector particles by transiently transfecting HEK293T cells with plasmid DNA encoding pLenti-puro empty vector, hSAMHD1 WT or hSAMHD1 HD/AA, as well as pCMV delta R8.2 packaging plasmid and vesicular stomatitis virus G protein (VSV-G). Supernatants containing VSV-G-pseudotyped retroviral vectors were then harvested and used to transduce MDCK cells as described [23]. At 24 h post-infection, MDCK cells were selected in DMEM medium containing 4 μg/ml puromycin. Expression of SAMHD1 in MDCK cells was confirmed by immunoblotting as described [23].
Protein extraction and immunoblotting
Cells were pelleted, washed with ice-cold PBS and lysed using 1 × cell lysis buffer (Cell signaling, #9803). Protein samples were normalized by the bicinchoninic acid (BCA) assay (Pierce) and analyzed by SDS-PAGE followed by immunoblotting as described [24]. Immunoblotting was performed using the following antibodies: SAMHD1 (1:1000, ProSci, #1224), HA-11 (1:1000, Covance #901,501) and GAPDH (1:3000, Bio-Rad #AHP1628). Quantification of immunoblotting was performed using Quantity One software (Bio-Rad).
Cellular transformation assay
MDCK cells were seeded at 5 × 105 cells per well in 6-well plates and transfected with 2.5 μg of JSRV Env expression plasmid along with 2.5 μg pLenti puro empty vector or 2.5 μg plasmids encoding human SAMHD1 (hSAMHD1) or mouse SAMHD1 (mSAMHD1, isoform 1) by Lipofectamine 2000 (ThermoFisher Scientific) as per manufacturer’s protocol. At 24 h post-transfection, the cells were trypsinized and split into 6-cm dishes in the presence of 4 μg/ml puromycin and 1 mg/ml G418 (ThermoFisher Scientific). The medium with antibiotics was replaced every 4 days. Transformed foci were counted and cell image were obtained 4 weeks after transfection as described [42].
Cell proliferation assay
MDCK control or WT-SAMHD1 or HD/AA-SAMHD1 expressing cells were seeded in a 96-well plate at a density of 2 × 103 cells per well in 100 µl of culture media. Control or SAMHD1 KO- cells stably expressing firefly luciferase (fLuc) reporter were plated into 96-well plates at a density of 2.5 × 104 cells per well in 100 µl of culture media (4 replicates per condition). On the indicated time points, MTT assay was performed using the CellTiter cell proliferation assay kits (Promega) as described [23].
Cell cycle analysis
Parental, control and SAMHD1-expressing MDCK cells were plated at a density of 1 × 106 in 6-cm culture dishes in 5 ml media. At 24 h post-seeding, cells were collected and cell cycle analysis was performed using the Guava Cell Cycle Reagent (EMD Millipore, #4500–0220) following the manufacturer’s instructions. Briefly, the collected cells were plated in a 96-well plate (in triplicate), washed with ice-cold PBS, fixed with 70% ethanol, and stained with 200 µl of Guava Cell Cycle Reagent. Flow cytometry was performed using the Millipore Guava Easycyte Mini Flow Cytometer and Guava Cytosoft 4.2 software to determine the distribution of cells in G0/G1, S, and G2/M phases of cell cycle as described [24].
Caspase-3/7 activity assay
The activity of caspase 3/7 in control or SAMHD1 KO-fLuc cells was analyzed at 24 h after seeding of cells by the Caspase-Glo 3/7 assay (Promega) as described [24].
Generation of THP-1 control and SAMHD1 KO cell lines stably expressing firefly luciferase
To establish cell lines stably expressing fLuc reporter, lentiviral vectors were generated by transfection of HEK293T cells with pCDH-LTR-1-luc-EF1α-copGFP vectors along with the lentiviral packaging vectors as described [23]. The pCDH-LTR-1-luc-EF1α-copGFP reporter vector was created using restriction digestion and PCR cloning. Briefly, pCDH-CMV-MCS-EF1α-copGFP (SBI) was digested with SpeI and NheI to remove the cytomegalovirus (CMV) promoter. The fragment of the long terminal repeat of human T-cell leukemia virus type 1 (LTR-1) and luciferase-coding sequence were PCR amplified from an LTR-1-luciferase reporter plasmid [44] with SpeI and NheI restriction sites added via PCR primers. The resulting reporter vector was sequenced and verified for functionality using cotransfection with a Tax-1 expression vector. Successful transduction in control (Ctrl) and SAMHD1 KO cells was assessed via quantifying the number of GFP expressing cells (~95% positive for GFP, data not shown) via flow cytometry. Stable expression of fLuc was validated via luciferase assay (Promega).
Mouse injection, in vivo imaging, necropsy, and survival studies
All mouse experiments were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee at The Ohio State University (OSU). Female, 4–6 weeks old NSG (non-obese diabetic/severe combined immune deficient-gamma) mice were purchased from the Target Validation Shared Resource of the Comprehensive Cancer Center at OSU. The mice (n = 8 per group) were injected intravenously with Ctrl-fLuc or KO-fLuc cells (3 × 106 per mouse) and monitored tumorigenesis via whole-body bioluminescent imaging using the IVIS Spectrum In Vivo Imaging System (PerkinElmer). On the indicated days post-injection (dpi) of cells, each mouse was injected intraperitoneally with D-luciferin (150 mg/kg body weight; VivoGlo, P1041, Promega), and bioluminescent images were taken with a 10-min delay and 5-min exposure. Average radiance (p/s/cm2/sr) was quantified per mouse to determine the relative tumor growth and metastasis. Mouse necropsy and pathological evaluation were performed at 35 days post-injection by a board-certified veterinary pathologist at the Comparative Pathology & Mouse Phenotyping Shared Resource at OSU. Mouse survival analysis was performed using Graphpad Prism 5 software and presented as Kaplan-Meier curve.
Statistical analysis
Error bars displayed on the bar graphs represent standard deviations. P values were calculated based on the non-parametric Student’s t-test using Graphpad Prism 5 software. P < 0.05 is considered as statistically significant.
Results
SAMHD1 inhibits JSRV Env-induced in vitro transformation in MDCK cells
JSRV Env protein can transform MDCK epithelial cells via a mechanism that involves enhanced activation of the PI3K/Akt pathway [42]. However, the molecular mechanisms by which the Env protein activates PI3K/AKT signaling in these transformed cells remain unknown. More recently, we have shown that SAMHD1 can negatively regulate the PI3K-Akt-p27 signaling axis in AML-derived THP-1 cells, thus reducing cell proliferation and migration [24]. Therefore, we hypothesized that SAMHD1 expression can potentially inhibit JSRV Env-induced MDCK cell transformation in vitro. We generated MDCK cells that stably express human or mouse SAMHD1 (isoform 1) via lentiviral vector-mediated transduction. It has been shown that human and mouse SAMHD1 share structural and functional similarity, with 72–74% identity of amino-acid sequences [45–49]. Mouse SAMHD1 has two functionally similar isoforms that differ at their C-termini due to alternative splicing [49]. We tested mouse SAMHD1 isoform 1 because its mRNA expression levels in mouse tissues and organs are 7-fold higher than isoform 2 [49].
After transfection of these cells with a construct expressing JSRV Env protein, we validated SAMHD1 expression in MDCK cells (Figure 1(a)), and performed in vitro transformation assays to determine transformation efficiency. We observed that exogenous expression of human SAMHD1 in MDCK cells resulted in a 2.2-fold decrease in the JSRV Env-induced transformation relative to vector control cells (Figure 1(b,c)). Notably, transformation efficiency was also reduced of 1.7-fold in MDCK cells expressing isoform 1 of mouse SAMHD1 (Figure 1(b,c)). These results suggest that ectopic SAMHD1 expression inhibits JSRV Env-induced transformation of MDCK cells in vitro, and that this ability is conserved in human and mouse SAMHD1. Moreover, the inhibitory effect of cell transformation did not correlate with SAMHD1 expression levels (Figure 1(a,c)), consistent with the conclusion that SAMHD1-mediated inhibition of cellular transformation is likely regulated by a more complex mechanism.
Figure 1.
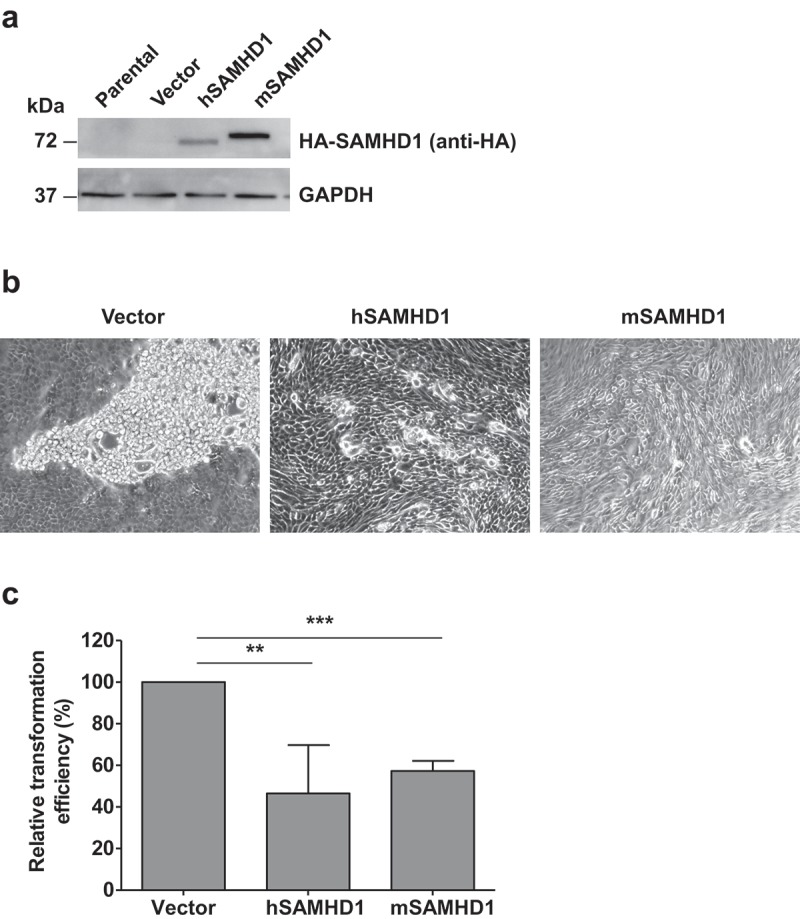
SAMHD1 inhibits JSRV Env-induced in vitro transformation of MDCK epithelial cells. MDCK cells were transfected with JSRV Env and empty vector or JSRV Env and plasmids expressing N-terminal hemagglutinin (HA)-tagged human SAMHD1 (hSAMHD1) or mouse SAMHD1 (mSAMHD1, isoform 1). (a) Exogenous SAMHD1 protein expression was assessed by immunoblotting using anti-HA antibodies. GAPDH was used as a loading control. (b) Cell images show the representative transformed foci 4 weeks post-transfection. Magnification, 10 × . (c) Relative transformation efficiency in MDCK cells from 3 independent experiments was quantified as presented. MDCK cells transfected with JSRV Env expression plasmid along with pLenti puro empty vector or plasmids encoding hSAMHD1 or mSAMHD1 (isoform 1). At 24 h post-transfection, the cells were split and cultured in the presence of puromycin (4 μg/ml) and G418 (1 mg/ml). The medium with antibiotics was replaced every 4 days. Transformed foci were counted and cell image were obtained 4 weeks after transfection (representative images are shown in b). Values of vector control were set to 100%. **, p = 0.0038; ***, p < 0.0001.
Inhibition of JSRV Env-induced transformation by SAMHD1 is independent of its dNTPase activity
We next focused on functional and mechanistic studies of human SAMHD1 considering that our goal is to better understand the role of human SAMHD1 in cell transformation and proliferation. To identify whether the dNTPase activity of SAMHD1 is required for its transformation inhibitory function, we stably expressed full-length human wild-type (WT)-SAMHD1 or the well characterized dNTPase-inactive mutant HD/AA-SAMHD1 (residues 206HD207 were mutated to 206AA207) [50] in MDCK cells (Figure 2(a)), and assessed the transformation induced by JSRV Env. Interestingly, loss of dNTPase activity of SAMHD1 did not alter its ability to inhibit transformation in these cells, as both WT- and HD/AA-SAMHD1 similarly reduced the JSRV-Env induced transformation efficiency relative to parental or vector control MDCK cells (Figure 2(b)). These results indicate that the transformation inhibitory function of SAMHD1 is independent of its dNTPase activity, suggesting that SAMHD1 may regulate transformation through alternative mechanisms.
Figure 2.
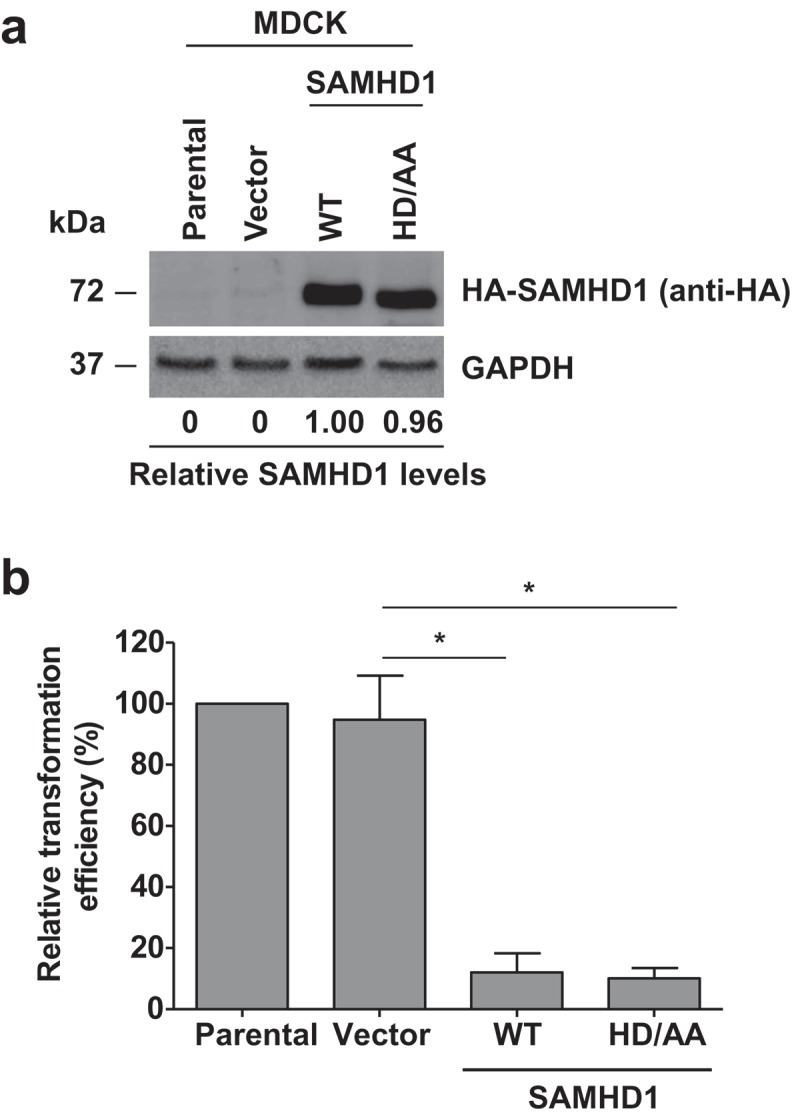
Inhibition of JSRV-Env-induced transformation by SAMHD1 is independent of its dNTPase activity. (a) Stable expression of human HA-tagged wild-type (WT) SAMHD1 and the dNTPase-defective HD/AA-SAMHD1 in the MDCK stable cell lines was detected by immunoblotting. GAPDH was a loading control. Relative levels of SAMHD1 were quantified by densitometry analysis and normalized by GAPDH levels. (b) MDCK cell lines stably expressing WT-SAMHD1 or HD/AA-SAMHD1 along with parental and vector control cells were transfected with a plasmid encoding JSRV Env. Four weeks post-transfection, transformed foci were counted and plotted. Presented data were obtained from 3 independent experiments. *, p ≤ 0.017.
SAMHD1 expression in MDCK cells significantly affects cell cycle distribution, but not cell proliferation
In our previous studies, we have reported that SAMHD1 inhibits growth and proliferation of cutaneous T-cell lymphoma-derived HuT78 cells and AML-derived THP-1 cells in vitro [23,43]. To test whether increased SAMHD1 expression can also modulate MDCK cell proliferation in addition to transformation, we performed MTT-based cell proliferation assays using parental, vector control, WT- and HD/AA-SAMHD1-expressing MDCK cells. As shown in Figure 3(a), expression of either SAMHD1 WT- or the dNTPase defective mutant did not significantly affect MDCK cell proliferation compared to parental or vector control cells, indicating that SAMHD1 does not regulate cell proliferation in these cells. Because SAMHD1 has been previously shown to regulate the cell cycle in HuT78 and THP-1 cell lines [23,43], we sought to determine whether SAMHD1 could modulate cell cycle in MDCK cells. Interestingly, stable expression of WT-SAMHD1, but not HD/AA-SAMHD1, resulted in a significant increase of the G1/G0 cell population and a significant decrease of S- and G2/M cell populations (Figure 3(b)). These results suggest that SAMHD1 may regulate cell cycle in MDCK cells by a dNTPase-dependent mechanism.
Figure 3.
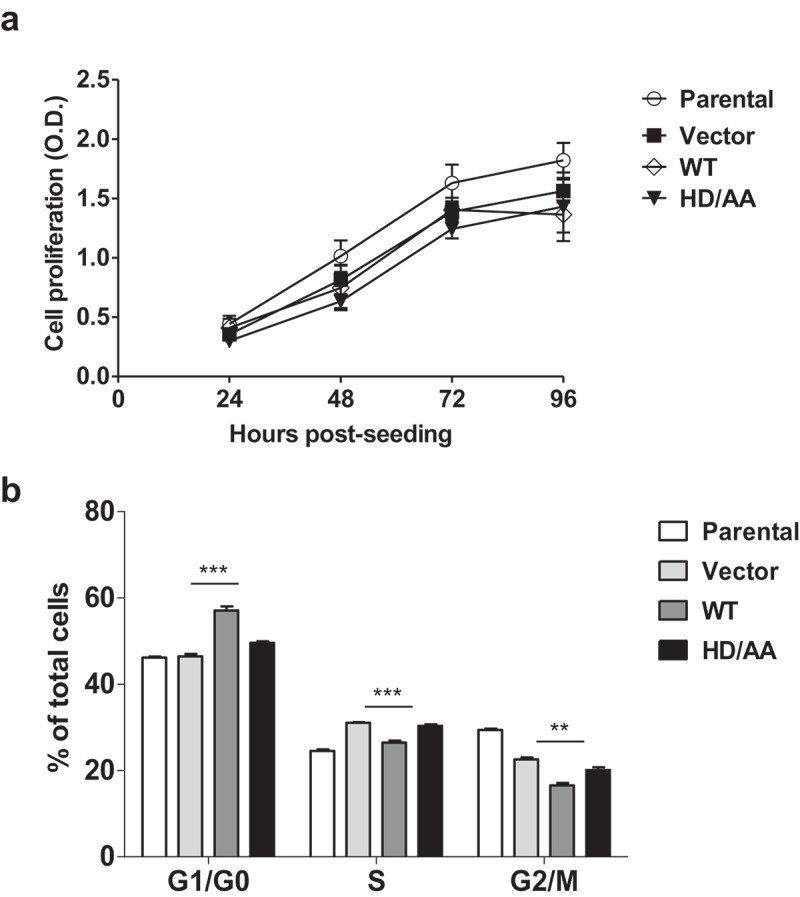
Stable SAMHD1 expression in MDCK cells significantly affects cell cycle distribution, but not cell proliferation. (a) MDCK cell lines stably expressing WT-SAMHD1 or HD/AA-SAMHD1 along with parental and vector control cells were analyzed by an MTT-based cell proliferation assay at the indicated time points. One representative experiment performed in three replicates is shown. OD, optical density (490 nm). (b) At 24 h post-seeding, MDCK cell lines were stained with propidium iodide and cell cycle analysis was performed via flow cytometry. Percentages of cells in G1/G0, S, and G2/M phases of cell cycle are presented. Each experiment was performed in three biological replicates. **, p = 0.0011; ***, p = 0.0006.
Establishment of intravenous AML mouse xenograft in vivo imaging model
We have recently described a xenograft mouse model where we subcutaneously injected THP-1 control or SAMHD1 KO cells in immunodeficient NSG mice [24]. In that mouse model, xenografted SAMHD1 KO cells did not lead to higher rate of subcutaneous tumor formation relative to control cells, most likely due to increased TNF-α-mediated inflammation in SAMHD1 KO cells that could influence tumor microenvironment and contribute to the phenotype observed in vivo [24]. Further review of the literature suggested that the subcutaneous xenograft system would not be an ideal model to analyze tumorigenicity of AML, which is characterized by malignancy of myeloid cells that build up primarily in the bone marrow and blood [51]. Therefore, we aimed to test the effect of SAMHD1 on AML-derived THP-1 tumor growth in vivo using an alternative mouse model via intravenous xenograft.
Using lentiviral vector-mediated transduction, we stably expressed firefly luciferase reporter (fLuc) in THP-1 control (Ctrl) and SAMHD1 KO cells [43]. We validated SAMHD1 levels and similar expression of fLuc in these cells by immunoblotting (Figure 4(a)) and a luciferase-based assay (Figure 4(b)), respectively. As expected based on our previously published data [43], SAMHD1 KO-fLuc cells demonstrated significantly higher cell proliferation (Figure 4(c)) and reduced caspase-3/7 activity (Figure 4(d)) relative to the Control-fLuc cells, confirming that the expression of the luciferase reporter gene did not affect the cellular growth phenotype. We then established and optimized intravenous mouse xenograft in vivo imaging model using NSG mice (female, 4–6 weeks old, n = 2 per group) and performed a necropsy study to evaluate tumorigenicity and metastasis. We observed the development of tumors within 3 weeks after intravenous cell injection, with metastasis clearly detectable at later time points (Figure 4(e)). Additionally, necropsy analysis on a mouse at 35 days post-injection of KO-fLuc cells demonstrated several large masses (size up to 2.2 × 1.0 × 1.0 cm) throughout the liver, caudal abdomen, and at the base of the tail (Figure 4(f)). Together, these results demonstrated that the intravenous mouse xenograft model via in vivo imaging system is a viable way to determine the effects of SAMHD1 KO on in vivo tumorigenicity of AML-derived THP-1 cell lines.
Figure 4.
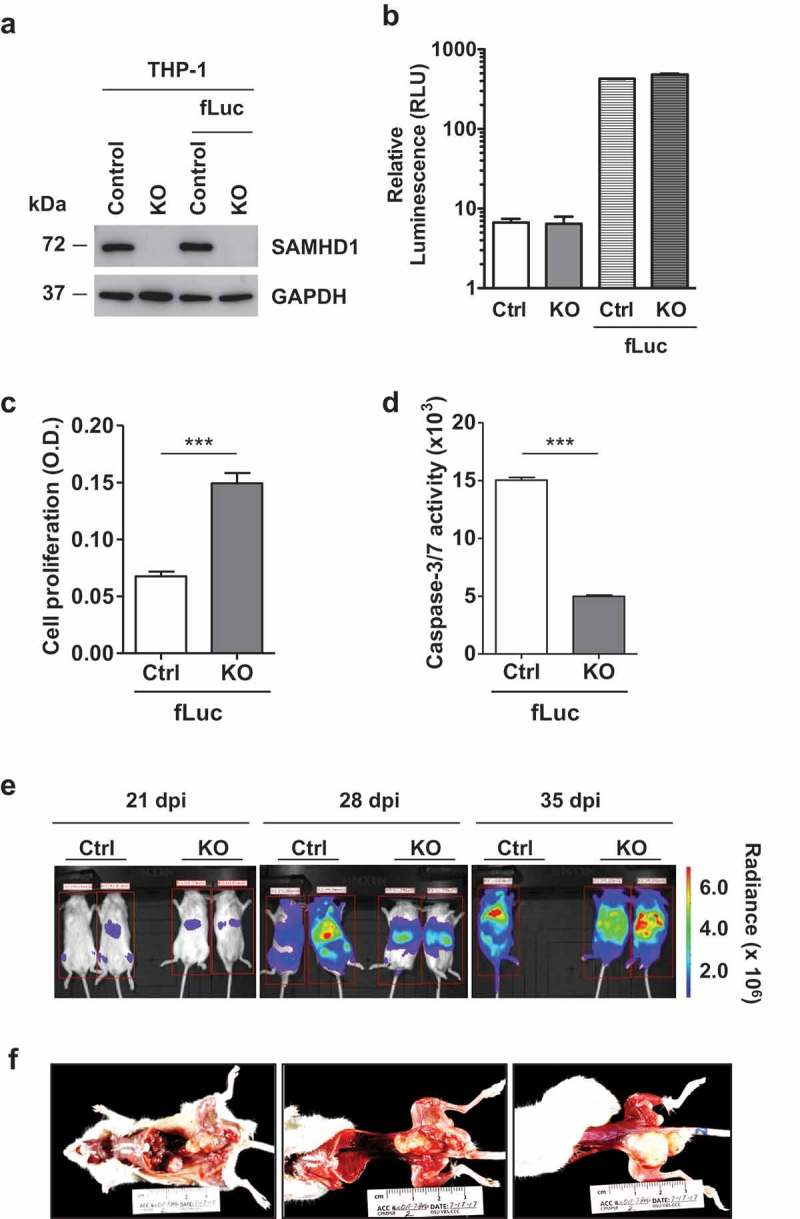
Establishment of intravenous AML mouse xenograft in vivo imaging model and pathological analysis. THP-1 control (Ctrl-fLuc) and SAMHD1 KO (KO-fLuc) cells stably expressing fLuc were analyzed (a) via immunoblotting to determine SAMHD1 expression and (b) via in vitro luciferase assays to detect levels of fLuc expression in comparison to parental cells (background is shown). At 24 h post-seeding, cells were analyzed for (c) proliferation using an MTT-based assay, and for (d) caspase-3/7 activity. ***, p ≤ 0.0002. (e) NSG mice were injected intravenously with THP-1 control (Ctrl)-fLuc or SAMHD1 KO-fLuc cells as described. On the indicated days post-injection (dpi), each mouse was injected intraperitoneally with D-luciferin, and bioluminescent images were taken at 10 min delay and 5 min exposure. Representative images are presented. One mouse injected with Ctrl-fLuc cells died due to tumor metastasis at 35 dpi. (f) Gross necropsy was performed on a mouse injected with SAMHD1 KO cells at 35 dpi.
SAMHD1 affects AML-derived leukemogenesis in vivo
To test the effect of SAMHD1 expression on leukemogenesis in vivo, we intravenously injected NSG mice with Ctrl-fLuc or KO-fLuc cells (3 × 106 cells per mouse; n = 8 mice/group), and monitored the rate of tumor growth, metastasis and survival via an in vivo bioluminescent imaging system (Figure 5(a)). Figure 5(b) shows the tumor growth rate in all the mice as measured by average radiance per animal at 21, 28, and 35 days post-injection (p/s/cm2/sr). As shown in Figure 5(c), at 28 days post-injection, no statistically significant difference was detected in tumor growth and metastasis rate between the two mouse groups, indicating that SAMHD1 KO in THP-1 cells did not significantly affect leukemogenesis in vivo. In contrast, mice injected with SAMHD1 KO cells survived longer than those injected with control cells (Figure 5(d)), indicating that SAMHD1 expression potentially affect survival in vivo.
Figure 5.
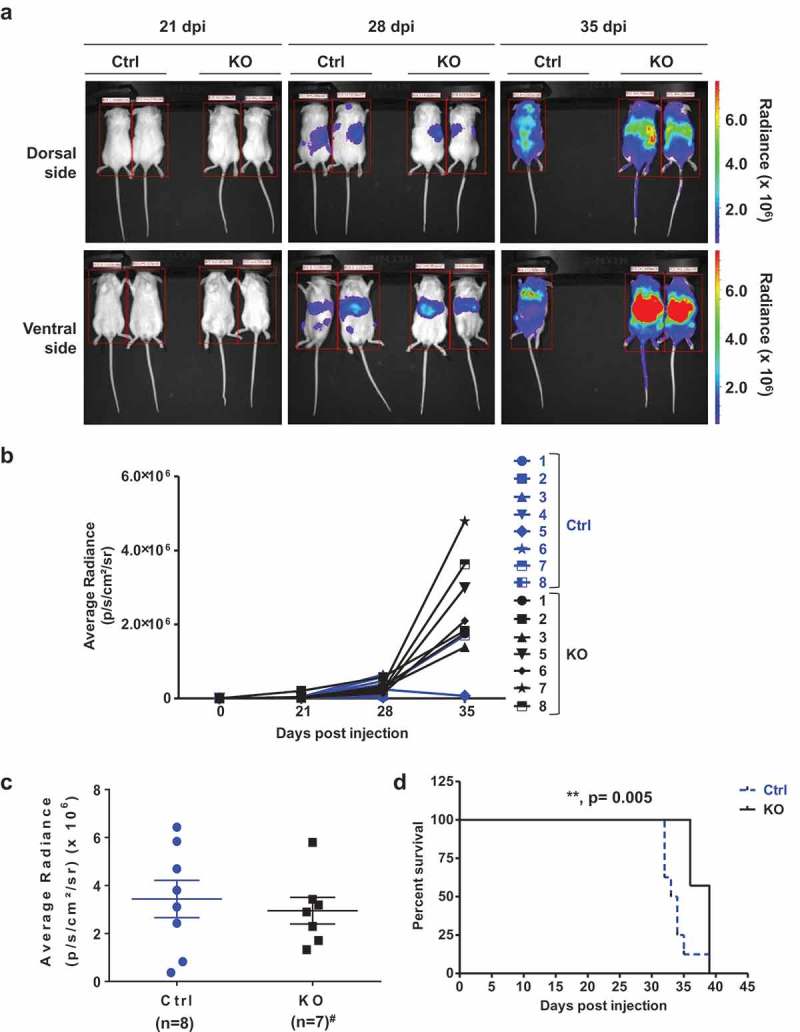
SAMHD1 affects AML-derived leukemogenesis in vivo. (a) NSG mice were injected intravenously with THP-1 Ctrl-fLuc (n = 8; 3 × 106 per mouse) or KO-fLuc cells (n = 7; 3 × 106 per mouse). On the indicated days post-injection (dpi) of cells, each mouse was injected intraperitoneally with D-luciferin, and bioluminescent images were taken at 10 min delay and 5 min exposure. Representative images are presented. (b) THP-1 cell-derived tumor growth was monitored by measuring the average radiance (p/s/cm2/sr) per mouse to determine the relative tumor growth and metastasis at 21, 28, and 35 dpi. (c) Average radiance from the Ctrl cell-injected mice (n = 8) and KO cell-injected mice (n = 7, #one mouse died due to non-experimental reasons) is presented at 28 dpi. (d) Percentage survival of mice (n = 8 for Ctrl and n = 7 for KO) was quantified using Kaplan-Meier curve survival analysis. **, p = 0.005.
Discussion
The potential inhibitory role of SAMHD1 in tumor development has emerged from recent studies showing that SAMHD1 is downregulated or mutated in cancers of solid, hematopoietic or lymphoid origin [8,13,15,16,20,37]. These lines of evidence support the hypothesis that SAMHD1 can act as a tumor suppressor. In line with this hypothesis, we have reported that SAMHD1 negatively affects proliferation of AML- and T-cell lymphoma-derived cells, and have identified the PI3K-Akt-p27 signaling pathway as the cellular network affected by SAMHD1, which leads to reduced AML cell growth and migration [23,24,43].
JSRV is an acutely transforming retrovirus known to induce tumors in sheep [42]. JSRV-induced transformation is mediated by the cytoplasmic tail of the viral Env protein, and in certain cell types, including MDCK, mouse, rat and chicken fibroblasts, transformation occurs through activation of the PI3K-Akt pathway [42]. To the best of our knowledge, the involvement of SAMHD1 in cellular transformation by oncogenes has never been investigated. We therefore examined whether SAMHD1 could affect the transformation potential of JSRV Env in MDCK epithelial cells.
In this study, we found that SAMHD1 overexpression in MDCK cells inhibits transformation by JSRV Env. Our data indicate that the SAMHD1 inhibitory effect does not rely on its dNTPase activity, suggesting that SAMHD1 inhibits Env-mediated transformation through a mechanism independent of the regulation of intracellular dNTP levels. Interestingly, neither SAMHD1 wild-type nor the dNTPase deficient mutant affect proliferation of non-transformed MDCK, implicating that SAMHD1 may not be directly involved in the regulation of cell proliferation in this cell type. Indeed, non-transformed cells such as MDCK have a lower proliferation rate compared to cancer cells, and would most likely present a different expression profile of key genes and proteins regulating cell growth. As SAMHD1-mediated control of proliferation can occur through direct or indirect effects on these modulators, non-transformed cells may respond differently than cancer cells to the effects of SAMHD1 on cell growth.
AML is an aggressive tumor of immature blood cells with a five-year survival rate of only 27.4% in the United States based on National Cancer Institute statistics (https://seer.cancer.gov/statfacts/html/amyl.html). Despite intensive research, current treatments based on combination of chemotherapy and nucleoside analog drugs are still ineffective due to high incidence of tumor relapse. SAMHD1 has been described as a negative biomarker in AML treatment because of its ability to interfere with the therapeutic activity of several drugs [18,52,53]. On the other hand, a positive correlation between SAMHD1 mRNA expression and long-term AML prognosis has been reported [54]. Furthermore, our recent study demonstrated that endogenous SAMHD1 protein levels are highly variable in peripheral blood mononuclear cells from 22 different AML patients, likely due to genetic heterogeneity of AML patients. Therefore, a clear understanding of SAMHD1 functions in AML is still lacking, and warrants further investigation.
Our recent studies performed in a subcutaneous xenograft mouse model of AML suggested that knockout of SAMHD1 reduces tumor growth in vivo [24], arguing against in vitro data showing that, in the absence of SAMHD1, AML-derived cell lines show higher proliferation rate compared to cells expressing the endogenous protein [43]. We observed an increased inflammation status in tumors from SAMHD1 KO cells, and hypothesize that this may contribute to reduced tumor growth in vivo [24]. However, given that AML is a hematological malignancy, a subcutaneous model may not fully reflect AML physiology. Therefore, in this study, we generated a xenograft mouse model by intravenous injection of THP-1 control and SAMHD1 KO cells expressing a firefly luciferase reporter into immunodeficient mice. THP-1 SAMHD1 KO cells showed similar tumor growth and metastasis compared to control cells. Interestingly, mice injected with KO cells showed greater survival rate than mice injected with control cells. Additional studies are required to confirm this phenotype and to further understand whether this change in survival is directly dependent of the absence or presence of SAMHD1 and the mechanisms involved.
Together, these results show that the effects of SAMHD1 in vitro and in vivo could be different. It is possible that in vivo the tumor microenvironment substantially affects the phenotype of the injected cancer cells, therefore resulting in different effects compared to in vitro observations. Additional studies are needed to shed light on these differences and clarify why the phenotype observed in vitro in SAMHD1 KO cancer cell lines cannot be recapitulated in vivo in SAMHD1 KO AML mouse models.
Funding Statement
This work was supported by National Cancer Institute under administrative supplement for non-AIDS defining cancers (Cancer Center Support Grant P30 CA016058) to LW. LW was partially supported by National Institutes of Health (NIH) grants AI120209 and GM128212. PLG was supported by NIH grant CA100730. SLL was supported by NIH grants AI112381 and GM132069.
Acknowledgments
We thank Dr. Baek Kim for sharing the THP-1 control and SAMHD1 KO cell lines, and the Wu lab members for valuable discussions. The pLenti-puro empty vector and the hSAMHD1 WT-expressing plasmid were a kind gift from Dr. Nathaniel Landau. We thank Dr. Corine St. Gelais for critically reading the manuscript. We also thank the Small Animal Imaging Core, Facility Target Validation Shared Resources, and Comparative Pathology & Mouse Phenotyping Shared Resource supported by the Ohio State University Comprehensive Cancer Center.
Authors’ contributions
LW conceived the study and designed experiments with SLL and KMK. KMK and ML performed experiments and analyzed data with SB, SLL and LW. ARP generated the pCDH-LTR-1-luc-EF1α-copGFP reporter vector, and PLG provided key reagents for in vivo experiments and assisted with editing the manuscript. KMK, SB and LW wrote the manuscript. All the authors read, edited and approved the manuscript.
Disclosure statement
All the authors declare that there are no conflicts of interest.
References
- [1].Goldstone DC, Ennis-Adeniran V, Hedden JJ, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480(7377):379–382. PubMed PMID: 22056990. [DOI] [PubMed] [Google Scholar]
- [2].Hrecka K, Hao C, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474(7353):658–661. PubMed PMID: 21720370; PubMed Central PMCID:PMC3179858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Franzolin E, Pontarin G, Rampazzo C, et al. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci U S A. 2013;110(35):14272–14277. PubMed PMID: 23858451; PubMed Central PMCID:PMC3761606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474(7353):654–657. PubMed PMID: 21613998; PubMed Central PMCID:PMC3595993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Powell RD, Holland PJ, Hollis T, et al. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286(51):43596–43600. PubMed PMID: 22069334; PubMed Central PMCID:PMC3243528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kohnken R, Kodigepalli KM, Wu L.. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer. 2015;14:176 PubMed PMID: 26416562; PubMed Central PMCID:PMC4587406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. PubMed PMID: 22810696; PubMed Central PMCID:PMC3401966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Clifford R, Louis T, Robbe P, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123(7):1021–1031. PubMed PMID: 24335234; PubMed Central PMCID:PMC3924925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schuh A, Becq J, Humphray S, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120(20):4191–4196. PubMed PMID: 22915640. [DOI] [PubMed] [Google Scholar]
- [10].Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. PubMed PMID: 21941284; PubMed Central PMCID:PMC3307543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. PubMed PMID: 18772397; PubMed Central PMCID:PMC2848990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. PubMed PMID: 18772396; PubMed Central PMCID:PMC2820389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435–439. PubMed PMID: 21163964; PubMed Central PMCID:PMC3110744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Lee W, Jiang Z, et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012;22(12):2315–2327. PubMed PMID: 23033341; PubMed Central PMCID:PMC3514662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. PubMed PMID: 23415222; PubMed Central PMCID:PMC3575604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rentoft M, Lindell K, Tran P, et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci U S A. 2016;113(17):4723–4728. PubMed PMID: 27071091; PubMed Central PMCID:PMC4855590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Daddacha W, Koyen AE, Bastien AJ, et al. SAMHD1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep. 2017;20(8):1921–1935. PubMed PMID: 28834754; PubMed Central PMCID:PMC5576576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Herold N, Rudd SG, Sanjiv K, et al. SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle. 2017;16(11):1029–1038. PubMed PMID: 28436707; PubMed Central PMCID:PMC5499833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang JL, Lu FZ, Shen XY, et al. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem Biophys Res Commun. 2014;455(3–4):229–233. PubMed PMID: 25449277. [DOI] [PubMed] [Google Scholar]
- [20].de Silva S, Hoy H, Hake TS, et al. Promoter methylation regulates SAMHD1 gene expression in human CD4+ T cells. J Biol Chem. 2013;288(13):9284–9292. PubMed PMID: 23426363; PubMed Central PMCID:PMC3610999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kohnken R, Kodigepalli KM, Mishra A, et al. MicroRNA-181 contributes to downregulation of SAMHD1 expression in CD4+ T-cells derived from Sezary syndrome patients. Leuk Res. 2017;52:58–66. PubMed PMID: 27889686; PubMed Central PMCID:PMC5195900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].de Silva S, Wang F, Hake TS, et al. Downregulation of SAMHD1 expression correlates with promoter DNA methylation in Sezary syndrome patients. J Invest Dermatol. 2014;134(2):562–565. PubMed PMID: 23884314; PubMed Central PMCID:PMC3844041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kodigepalli KM, Li M, Liu SL, et al. Exogenous expression of SAMHD1 inhibits proliferation and induces apoptosis in cutaneous T-cell lymphoma-derived HuT78 cells. Cell Cycle. 2017;16(2):179–188. PubMed PMID: 27929746; PubMed Central PMCID:PMC5283819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kodigepalli KM, Bonifati S, Tirumuru N, et al. SAMHD1 modulates in vitro proliferation of acute myeloid leukemia-derived THP-1 cells through the PI3K-Akt-p27 axis. Cell Cycle. 2018;17(9):1124–1137. PubMed PMID: 29911928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu P, Cheng H, Roberts TM, et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. PubMed PMID: 19644473; PubMed Central PMCID:PMC3142564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. PubMed PMID: 22358332. [DOI] [PubMed] [Google Scholar]
- [27].Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. PubMed PMID: 19629070. [DOI] [PubMed] [Google Scholar]
- [28].Park S, Chapuis N, Tamburini J, et al. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica. 2010;95(5):819–828. PubMed PMID: 19951971; PubMed Central PMCID:PMC2864389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015;3:2 PubMed PMID: 26056603; PubMed Central PMCID:PMC4452048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lindblad O, Cordero E, Puissant A, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016;35(39):5119–5131. PubMed PMID: 26999641; PubMed Central PMCID:PMC5399143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Martelli AM, Evangelisti C, Chiarini F, et al. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1(2):89–103. PubMed PMID: 20671809; PubMed Central PMCID:PMC2911128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Polivka J Jr., Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142(2):164–175. PubMed PMID: 24333502. [DOI] [PubMed] [Google Scholar]
- [33].Bortul R, Tazzari PL, Billi AM, et al. Deguelin, A PI3K/AKT inhibitor, enhances chemosensitivity of leukaemia cells with an active PI3K/AKT pathway. Br J Haematol. 2005;129(5):677–686. PubMed PMID: 15916691. [DOI] [PubMed] [Google Scholar]
- [34].Sampath D, Cortes J, Estrov Z, et al. Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood. 2006;107(6):2517–2524. PubMed PMID: 16293603; PubMed Central PMCID:PMC1895741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sato S, Fujita N, Tsuruo T. Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene. 2002;21(11):1727–1738. PubMed PMID: 11896604. [DOI] [PubMed] [Google Scholar]
- [36].Diehl N, Schaal H. Make yourself at home: viral hijacking of the PI3K/Akt signaling pathway. Viruses. 2013;5(12):3192–3212. PubMed PMID: 24351799; PubMed Central PMCID:PMC3967167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Allen TE, Sherrill KJ, Crispell SM, et al. The jaagsiekte sheep retrovirus envelope gene induces transformation of the avian fibroblast cell line DF-1 but does not require a conserved SH2 binding domain. J Gen Virol. 2002;83(Pt 11):2733–2742. PubMed PMID: 12388809. [DOI] [PubMed] [Google Scholar]
- [38].Maeda N, Palmarini M, Murgia C, et al. Direct transformation of rodent fibroblasts by jaagsiekte sheep retrovirus DNA. Proc Natl Acad Sci U S A. 2001;98(8):4449–4454. PubMed PMID: 11296288; PubMed Central PMCID:PMC31855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rai SK, Duh FM, Vigdorovich V, et al. Candidate tumor suppressor HYAL2 is a glycosylphosphatidylinositol (GPI)-anchored cell-surface receptor for jaagsiekte sheep retrovirus, the envelope protein of which mediates oncogenic transformation. Proc Natl Acad Sci U S A. 2001;98(8):4443–4448. PubMed PMID: 11296287; PubMed Central PMCID:PMC31854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zavala G, Pretto C, Chow YH, et al. Relevance of Akt phosphorylation in cell transformation induced by Jaagsiekte sheep retrovirus. Virology. 2003;312(1):95–105. PubMed PMID: 12890624. [DOI] [PubMed] [Google Scholar]
- [41].Johnson C, Sanders K, Fan H. Jaagsiekte sheep retrovirus transformation in Madin-Darby canine kidney epithelial cell three-dimensional culture. J Virol. 2010;84(10):5379–5390. PubMed PMID: 20219922; PubMed Central PMCID:PMC2863794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu SL, Miller AD. Transformation of madin-darby canine kidney epithelial cells by sheep retrovirus envelope proteins. J Virol. 2005;79(2):927–933. PubMed PMID: 15613321; PubMed Central PMCID:PMC538587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bonifati S, Daly MB, St Gelais C, et al. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology. 2016;495:92–100. PubMed PMID: 27183329; PubMed Central PMCID:PMC4912869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ross TM, Minella AC, Fang ZY, et al. Mutational analysis of human T-cell leukemia virus type 2 Tax. J Virol. 1997;71(11):8912–8917. PubMed PMID: 9343258; PubMed Central PMCID:PMC192364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang F, St Gelais C, de Silva S, et al. Phosphorylation of mouse SAMHD1 regulates its restriction of human immunodeficiency virus type 1 infection, but not murine leukemia virus infection. Virology. 2016;487:273–284. PubMed PMID: 26580513; PubMed Central PMCID:PMC4679491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wu L. SAMHD1 knockout mice: modeling retrovirus restriction in vivo. Retrovirology. 2013;10:142 PubMed PMID: 24257155; PubMed Central PMCID:PMC3842632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].St Gelais C, de Silva S, Hach JC, et al. Identification of cellular proteins interacting with the retroviral restriction factor SAMHD1. J Virol. 2014;88(10):5834–5844. PubMed PMID: 24623419; PubMed Central PMCID:PMC4019113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Buzovetsky O, Tang C, Knecht KM, et al. The SAM domain of mouse SAMHD1 is critical for its activation and regulation. Nat Commun. 2018;9(1):411 PubMed PMID: 29379009; PubMed Central PMCID:PMC5788916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang R, Bloch N, Nguyen LA, et al. SAMHD1 restricts HIV-1 replication and regulates interferon production in mouse myeloid cells. PLoS One. 2014;9(2):e89558 PubMed PMID: 24586870; PubMed Central PMCID:PMC3929709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lahouassa H, Daddacha W, Hofmann H, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012;13(3):223–228. PubMed PMID: 22327569; PubMed Central PMCID:PMC3771401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127(1):53–61. PubMed PMID: 26660429; PubMed Central PMCID:PMC4705610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schneider C, Oellerich T, Baldauf HM, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med. 2017;23(2):250–255. PubMed PMID: 27991919. [DOI] [PubMed] [Google Scholar]
- [53].Herold N, Rudd SG, Ljungblad L, et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat Med. 2017;23(2):256–263. PubMed PMID: 28067901. [DOI] [PubMed] [Google Scholar]
- [54].Herold N, Rudd SG, Sanjiv K, et al. With me or against me: tumor suppressor and drug resistance activities of SAMHD1. Exp Hematol. 2017;52:32–39. PubMed PMID: 28502830. [DOI] [PubMed] [Google Scholar]