

Abstract
Incidences of prostate cancer in most countries are increasing owing to better detection methods; however, prevention with the use of finasteride, a very effective steroid 5α-reductase type II inhibitor, has been met with mixed success. A wide interindividual variation in response exists and is thought to be due to heritable factors. This article summarizes the literature that attempts to elucidate the molecular mechanisms of finasteride in terms of its metabolism, excretion and interaction with endogenous steroid molecules. We describe previously reported genetic variations of steroid-metabolizing genes and their potential association with finasteride efficacy. Based on the literature, we outline directions of research that may contribute to understanding the interindividual variation in finasteride prevention and to the future development of personalized medicine.
Keywords: androgen, CYP3A4, finasteride, haplotype, personalized medicine, pharmacogenetics, prostate cancer, single nucleotide polymorphism, SRD5A2, UGT1A
Prostate cancer is the most common nonskin cancer in men in the western world. Cancer Research UK (CRUK) documented 35,550 new cases in 2006 and 10,239 deaths were attributable to the disease [201]. In 2010, the American Cancer Society (ACS) estimated the occurrence of 217,730 newly diagnosed cases and 32,050 deaths [202]. The exact etiology of prostate cancer remains unknown, although there are numerous risk factors associated with disease susceptibility that have been reviewed elsewhere [1]. It is well recognized that genetic factors and environmental stimuli (and the potential interplay between the two), age, family history, steroid hormone metabolism and race/ethnicity (African–American men are particularly at risk [2]) are all potential risk factors.
Prostate & steroid hormones
Prostate tissue growth and differentiation is regulated by androgen hormones and modulated via the androgen receptor (AR) [3]. Testosterone is irreversibly converted in a nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction reaction to the most physiologically active androgen 5α-dihydrotestosterone (DHT) (FIGURE 1). This is mediated by the androgen-metabolizing enzymes steroid 5α-reductase types I and II in the prostate tissue (encoded by the SRD5A1 and SRD5A2 genes, respectively) [4]. Steroid 5α-reductase type III (encoded by SRD5A3) was more recently discovered to be an enzyme involved in the conversion of testosterone to DHT in hormone-refractory prostate cancer cells [5].
Figure 1. Prostate tissue growth and differentiation.
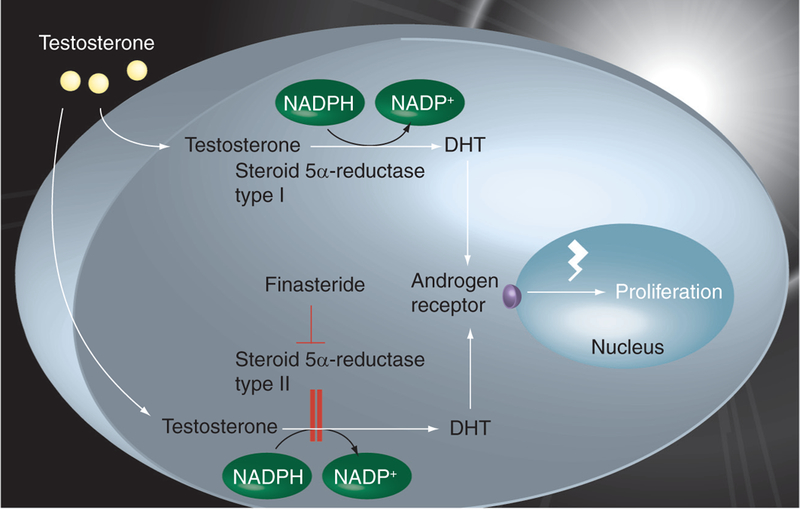
Circulating testosterone diffuses into prostatic cells, where it is irreversibly converted into the more potent androgen DHT via the NADPH-dependent reduction catalyzed by the steroid 5α-reductase type II and, to a lesser extent, by steroid 5α-reductase type I. DHT binds to the androgen receptors located on the nuclear membrane. Two DHT–receptor complexes are formed in the nucleus that bind to specific regions of the DNA – the androgen response elements – with high affinity, enhancing the production of regulatory proteins involved in cell proliferation and prostate-specific antigen. Finasteride, a steroid 5α-reductase type II inhibitor, causes an intra-prostatic DHT decrease, ultimately leading to apoptosis. DHT: 5α-dihydrotestosterone; NADP: Nicotinamide adenine dinucleotide; NADPH: Nicotinamide adenine dinucleotide phosphate. Data taken from [125].
Prostatic androgen metabolism is essential for normal prostate function, although it is thought to play a pivotal role in prostate carcinogenesis, as well [6]. As such, the steroid 5α-reductase type II inhibitor finasteride (Proscar®; Propecia® [Merck, NJ, USA] FIGURE 1), which was approved for benign prostatic hyperplasia (BPH) and alopecia, respectively [203], and the dual steroid 5α-reductase (type I and II) inhibitor dutasteride (Avodart® [GlaxoSmithKline, UK]), approved for BPH [204], mediate their effects by mechanism-based inhibition of steroid 5α-reductase type II by blocking DHT and thereby androgen action through the AR [7]. The steroid 5α-reductase types I and II isoenzymes demonstrate differential expression in various tissues. The steroid 5α-reductase type I is mostly expressed in liver and skin and to a lesser extent in the prostate. Steroid 5α-reductase type II is abundantly expressed in the prostate and minimally expressed in the liver and skin [8]. Steroid 5α-reductase type I is composed of 259 amino acids with a molecular weight of approximately 29 kDa and the type II isoform is composed of 254 amino acids, has a molecular weight of 28 kDa and exhibits increased sensitivity to finasteride [205,206]. Both isoenzymes are pH dependent and optimally active at alkaline and acidic pHs, respectively [205,206].
Dutasteride results in prostate involution and epithelial cell loss via apoptosis [9]. The readministration of androgens induces the prostate to regain normal size and function through rapid proliferation and differentiation of basal epithelial cells [10]. The success of steroid 5α-reductase inhibitors in reducing prostate cancer risk supports the notion that the androgen pathway is necessary for the development of the disease [11–14].
Genetic variants, risk & drug response
Historically, it was proposed as early as 1957 that adverse reactions and interindividual variation in response to drugs was probably due to inheritance [15]. Despite this, the literature documents a limited number of pharmacogenetic-studies [16], and these have predominantly focused on drug-metabolizing enzymes and their transporters [17,18]. However, the importance of pharmacogenetic research in terms of identifying variants associated with dose–response effects is suggested to be more manageable than identifying disease-susceptibility alleles for common, complex diseases [19]. This clearly represents a major challenge to healthcare providers in prescribing chemo-preventive drugs that are beneficial to the patient while minimizing the risk of adverse effects [20–22].
It is thought that the wide interindividual variability of drug efficacy and toxicity can be explained by variations within drug-metabolizing-enzymes [17]. Both finasteride and dutasteride are no exception, as demonstrated by variation of the biochemical and pharmacological properties according to genotype for the target steroid 5α-reductase type II [23–25]. The development of personalized medicine for prostate cancer will ultimately involve studies that investigate the nature of the genetic variability of finasteride- and dutasteride--metabolizing genes and inhibitor action through the steroid 5α-reductase type II enzyme.
In a previous review, it was reported that the existing literature of androgen-metabolizing gene variants and prostate cancer disease susceptibility remains largely inconclusive [26]. The review thoroughly summarized the literature exploring the potential influences of androgens and prostate cancer disease risk. Candidate genes included those that mediate serum testosterone bioavailability, androgen action and metabolism. These included serum sex hormone-binding globulin (SHBG), CYP17A1, the hydroxysteroid dehydrogenase genes HSD3B1, HSD3B2, HSD17B1, HSD17B3, SRD5A2, CYP3A4 and CYP3A5, the uridine diphosphoglucuronosyltransferase (UGT) genes UGT2B15 and UGTB17, AR and the chromosomal locus 8q24. Apart from the highly reproducible but unknown action of the 8q24 loci and its association with prostate cancer risk in different ethnic populations, the lack of reproducibility of other androgen genes probably reflects the genetic heterogeneity of the disease and differences in data interpretation between studies. The authors suggested future studies should focus efforts on determining both disease susceptibility alleles and environmental factors across diverse ethnic populations. In particular, the effects of DHT inactivation, variation of the UGT genes and the 8q24 susceptibility locus were considered to be of particular interest [26].
In general terms, the candidate gene approach has proven most successful thus far, in that genes of biological relevance are associated with drug response [19]. Many positive associations have been identified where genotype–phenotype correlations and disease susceptibility or drug efficacy have been reported with statistically significant p‑values of less than 0.05, but these are not considered clinically relevant until reaching a threshold of p < 10−6 in genome-wide association studies [16].
Studies investigating multiple variants in blocks of inherited variation involve the haplotype-based strategy (statistically correlated single nucleotide polymorphisms [SNPs] inherited together in blocks on one chromosome), which is crucial for capturing the overall genetic diversity of the region of interest rather than analyzing individual SNPs. In most cases, the SNP identified is not the causal SNP but merely one that is associated with a causal variant at a different locus (i.e., a marker) [27]. Because exhaustive genotyping of SNPs within a candidate gene is expensive, technically challenging and rather point-less owing to the presence of ‘redundant SNPs’, another method is to use linkage disequilibrium (the nonrandom association of alleles at two or more loci) and to genotype the informative SNPs (or tagSNPs) based on the pairwise linkage disequilibrium measure [28]. TagSNPs or haplotype-tagging SNPs serve as proxies for those that are not genotyped. However, linkage disequilibrium structures have been shown to demonstrate differences in the distance of measurable linkage disequilibrium between northern European and Nigerian populations [29]. This highlights the impact of linkage disequilibrium diversity across different ethnic populations and the transferability of tagSNPs between populations [30], and must be considered when designing experiments. A disadvantage of using tagSNPs is that rare genetic variants are sometimes missed [27], which supports the view of the importance of implementing the characterization of functional SNPs and haplotypes in the search for alleles that mediate both drug dose–response and disease susceptibility [31].
The importance of haplotype-based studies is highlighted by the fact that most cancers are characterized by multiple genetic variations that are influenced by environmental factors and, as such, form the basis of many common diseases [32–34]. The common disease, common variant hypothesis states that common alleles (i.e., SNPs) in the population with low penetrance are the major genetic determinants of common diseases [35]. An alternative is the common disease, rare variant model, which suggests multiple variants are present at low frequencies but exert larger phenotypic effects [36]. Therefore, the population-attributable risk may be significant [37].
Investigations of the distribution of haplotypes and associations with prostate cancer disease risk, progression and response to finasteride chemoprevention will be particularly useful in population studies. Haplotype-based studies are essential for determining the presence of risk alleles that have the potential to influence the functional expression of the finasteride target, steroid 5α-reductase type II. This has the potential for identifying individuals at increased risk of developing the heterogeneous forms of the disease in terms of latent and aggressive forms of prostate cancer. Such studies may contribute to understanding the interindividual variability associated with finasteride efficacy and toxicity and are necessary for predicting the appropriate dosage of finasteride to achieve therapeutic benefit and administering an effective therapy without delay.
Finasteride: a steroid 5α‑reductase type II inhibitor
The use of finasteride as a chemopreventive for prostate cancer originated from the work performed by Charles B Huggins in the 1930s and 1940s, leading to the discovery of the link between the physiological role of testosterone and prostate growth, for which he was later awarded the Nobel Prize in 1966 [38]. He noted an improvement of symptoms in men with advanced prostate cancer that were either castrated or subjected to estrogen therapy [39]. Later, the discovery of pseudohermaphrodites characterized by steroid 5α-reductase type II deficiency prompted the development of the steroid 5α-reductase type II inhibitor MK‑906 (now termed finasteride), which entered clinical trials and was approved for use in 1992 [40].
Finasteride is well documented as the most effective chemopreventive for reducing prostate cancer risk [11,41–44]. The Prostate Cancer Prevention Trial (PCPT) recruited over 18,000 men aged 55 years or more with normal digital rectal examination and prostate-specific antigen (PSA) levels [11]. Subjects were randomly assigned oral finasteride 5 mg/day or placebo for a total study follow-up period of 7 years. PSA levels and prostate biopsy were performed at the end of the study for all participants. Those who received finasteride demonstrated a 25% reduced incidence of prostate carcinoma (for men with a family history of the disease, the reduction was over 31%). However, adverse effects were reported regarding the prevalence of high-grade tumors with Gleason scores of greater than or equal to 7 in the finasteride arm of the trial in comparison with the control group (p < 0.001) [45]. The PCPT suggested the unfavorable outcome was due to detection bias and so halved PSA cutoff values accordingly in order to improve detection rates for the aggressive form of the disease. Since the PCPT, subsequent studies have shown that finasteride treatment inhibits low-grade prostate cancer, reduces prostate volume and, in doing so, increases the detection rate of high-grade tumors [41]. Moreover, it is hypothesized that finasteride is not so efficient at reducing the size of high-grade in comparison with low-grade tumors and therefore increases the chances of detection [46].
Inhibitory mechanism of finasteride
Finasteride (N-[2-methyl-2-propyl]-3-OxO4-aza-5α-androst-1-ene-17β-carboxamide) is a synthetic 4‑azasteroid steroid 5α-reductase type II inhibitor that is developed by Merck and marketed as Proscar and Propecia, among other names. Originally termed a competitive steroid 5α-reductase type II inhibitor [4], finasteride is now known to demonstrate mechanism-based enzyme inhibition [47]. It has been shown that the reduction of testosterone and inhibition by finasteride reactions are similar, except that the two reactions diverge at the final pathway, in which finasteride exits the reaction by avoiding proton transfer [47]. In short, after binding to the steroid 5α-reductase type II enzyme, hydride transfer from the cofactor NADPH to the ∆1,2-ene double bond present on finasteride occurs. The intermediate product enolate (NADP-dihydrofinasteride) tautomerizes at the active site of the steroid 5α-reductase type II enzyme [47]. Accepted as an alternative substrate, it is reduced to dihydrofinasteride via the enzyme-bound NADP–dihydrofinasteride complex [47].
As a bisubstrate analogue inhibitor, finasteride is one of the most potent enzyme inhibitors [48]. Bisubstrate inhibitors are composed of two conjugated fragments and these are important for binding to different sites of the bisubstrate enzyme [49]. Bisubstrate inhibitors are essentially considered more effective in comparison with single-site inhibitors because of their potential for more interactions with the enzyme [50]. The binding affinity of the enzyme–NADP–dihydro-finasteride complex is directly related to the affinity of the enzyme for NADPH and dihydro-finasteride [47]. Moreover, enzyme kinetics data for free finasteride and enzyme saturated with the cofactor (i.e., NADPH) suggest that conformational changes to the enzyme associated with binding of finasteride may be rate-limiting in the catalytic turnover of both testosterone and finasteride [47].
Finasteride also demonstrates inhibition of the steroid 5α-reductase type I enzyme, although at a slower rate, and may explain why finasteride is a more potent steroid 5α-reductase type II inhibitor [47]. The Michaelis constant [Km] of finasteride is lower than that of testosterone, ranging from 3 to 26 Km and 10 to 30 Km, respectively, thereby inhibiting the production of DHT from testosterone, demonstrating that finasteride has a higher affinity for steroid 5α-reductase type II compared with testosterone [50]. There is no literature at present that determines the binding interactions between finasteride and the steroid 5α-reductase types I and II [51].
Finasteride metabolism
The biotransformation of finasteride consists of two pathways; phase I and phase II metabolism. Phase I metabolism (FIGURE 2) is regulated by the cytochrome P450 enzymes [52]. These enzymes are a subfamily of predominantly hepatic drug metabolizing enzymes (encoded by the CYP3A4, CYP3A5, CYP3A7 and CYP3A43 genes), rendering drugs with an increased water solubility to increase their rate of urinary excretion [53]. These enzymes also account for the bioavailability of a wide range of drugs [54]. The biotransformation of finasteride involves CYP3A4-mediated hydroxylation and oxidation reactions to produce phase I metabolites [55]. Phase I finasteride metabolism involves the conversion of finasteride to ω-hydroxyfinasteride (FIGURE 2) [55] by hydroxylation of the t‑butyril group. CYP3A4 is also involved in the biotransformation of endogenous steroid metabolites, including the oxidation of testosterone [56,57], thereby representing an important pathway in the formation of phase I androgen metabolites in the prostate.
Figure 2. Metabolism of finasteride.

During phase I, finasteride is metabolized to ω-hydroxyfinasteride and finasteride-ω-oic acid, a process mediated by CYP3A4. Phase II metabolism involves glucuronidation of ω-hydroxyfinasteride and finasteride-ω-oic acid metabolites by UGT1A4 and UGT1A3, respectively. Data taken from [65].
Phase II metabolites are catalyzed by glucuronidation (FIGURE 2) [58], a conjugation pathway that mediates the metabolism and excretion of endobiotics and xenobiotics [59]. Glucuronidation is catalyzed by the enzyme superfamily UGT [59]. The endoplasmic reticulum-bound UGT enzymes are documented to be the most important determinants of phase II drug metabolism [60]. Among the numerous drugs and endogenous molecules, steroid hormones are also inactivated and eliminated by glucuronidation [61]. The process involves the transfer of glucuronic acid from UDP-glucuronic acid to the substrate at various sites, such as carboxyl and hydroxyl groups [62,63]. The addition of polar glucuronic acid to the substrate mediates excretion of the lipophilic substrate via urine or bile [64]. It has been reported that androgens are able to effectively switch off their own glucuronidation in prostate cancer cells via AR activation, resulting in a build-up of cellular androgen concentrations that drives the disease process [60,65].
The metabolites of finasteride, ω-hydroxy finasteride and finasteride ω-oic acid, are glucuronidated by UGT1A4 and UGT1A3, respectively [65]. Both enzymes are found in biliary tissue, colon, liver and intestine, as well as the stomach (UGT1A3 only) [66,67]. Phase II glucuronidation involves oxidation of the t‑butyril group and replacement by a carboxylic acid, the end product being finasteride-ω-oic acid (FIGURE 2) [68,69]. This renders the metabolite highly water soluble and, in doing so, promotes inactivation and elimination.
Finasteride bioavailability
The bioavailability of finasteride is yet to be fully characterized. Studies have calculated the percentage bioavailability from 63% [68] to 80% [55]. The peak mean concentration for finasteride is reached approximately 2 h after oral administration (1.7 ± 0.5 h) and complete absorption occurs 6–8 h later [69].
The volume of distribution indicates that the drug dissolves well in tissues, as indicated by the plasma protein binding value of finasteride ranging between 91.3 and 89.8% and the experimentally determined volume of distribution of approximately 76 l [207]. Finasteride has a lipophilic nature (log p = 3.2) and, despite being able to cross the BBB, does not interact with adjacent tissues [70].
Data from a study of six healthy volunteers demonstrated that the dose-predominant routes of excretion for finasteride were urine, in which 39% of the dose was recovered, and feces, in which 57% was recovered [69]. Another study reported that urinary finasteride excretion was essentially unchanged; parent forms of the drug present in the bile were extremely low and were therefore thought to undergo metabolism [68]. The authors performed a subsequent study in which they administered 5 mg/day of finasteride to healthy human subjects and quantified finasteride excretion in vivo using a single-pass perfusion technique, Loc-I-Gut [65]. Bile collection and subsequent measurements of metabolites were quantified [71,72] and the original study was the first to report the identification of two new hydroxyl metabolites [65], one being an intact hydroxyfinasteride glucuronide in addition to the previously characterized ω-hydroxyfinaste-ride and finasteride-ω-oic acid phase I metabolites identified in vitro and in human bile and urine (FIGURE 2) [73]. One of the novel metabolites was identified by the authors to potentially be a ring-closed form of ω-hydroxyfinasteride [65]. The other, the major glucuronide identified in human bile and urine, was thought to be identical to the previously identified metabolite 6α-hydroxyfinasteride [73]. The authors hypothesized that the discovery of the predominant novel metabolite in bile suggests that it may undergo hydrolysis, intestinal reabsorption and enterohepatic circulation, which are thought to lead to increased exposure in the hepatobiliary tract and long terminal plasma half-life [65,74]. The consequences of this in terms of toxicity have yet to be determined [65]. ω-hydroxyfinasteride was glucuronidated in vitro but was not identified in human bile and urine and as such was not considered the principle metabolite in humans, in contrast to previously reported findings [69]. Finasteride-ω-oic acid was also glucuronidated in vitro, but only trace amounts were identified in the bile of some subjects. This glucuronide might be responsible for the toxicity to DNA or proteins by binding to them [75] and, as suggested by Lundahl and colleagues [65], should be investigated further. The discovery of newly identified finasteride phase I and II metabolites is very interesting. The authors hypothesize that there is the possibility of at least one other parallel pathway competing with the formation of finasteride-ω-oic acid. Investigating the genetic basis of finasteride metabolizing genes may determine alternative pathways of finasteride metabolism.
Genetic variation of the finasteride target enzyme: SRD5A2
The importance of the affinity of steroid 5α-reductase type II for NADP and dihydro-finasteride was reported in an early study that suggested conformational changes to the enzyme associated with the binding of finasteride may be rate-limiting in terms of catalytic turn-over of both testosterone and finasteride [47]. A comprehensive study of the biochemical and pharmacogenetic properties of SRD5A2 missense mutations reported interesting and important findings. The effects of each single-point mutation, which naturally occur in humans, compound heterozygotes marking two mutations and haplotypes composed of homozygotes for the common V89L (rs523349) variant plus one rare heterozygous mutation located on one chromosome in cis were determined (TABLE 1) [24]. Firstly, we note the P30L (rs61748121), P48R (rs61748122) and A49T (rs9282858) substitutions demonstrated an increased Km for testoster one (TABLE 1), indicating these residues are necessary for substrate binding. The A49T mutation was previously demonstrated to increase the risk of prostate cancer in African–Americans and Hispanics [76], although these findings were not replicated in other studies [77]. It is possible that the substitution from the nonpolar alanine to the polar threonine creates a structural conformation change to the enzyme and thereby favors substrate binding. Interestingly, the adjacent P48R mutation also demonstrated an increased Km for the substrate. V89L exhibited a low maximum rate of reaction (Vmax) and, therefore, low enzyme activity, suggesting this variation affects the stability of the enzyme. This variant is associated with aggressive prostate cancer risk [78]. P30L, T187M, R227Q and F234L substitutions exhibited an increase in Km for the cofactor (i.e., NADPH), no change in testoster one Km and a decrease in Vmax. This suggests that the wild-type residues are essential for cofactor binding but not enzyme activity. This is a surprising finding for the P30L substitution as it is considered that the NADPH cofactor-binding domain is located primarily at the C‑terminus of the enzyme [24,79,80]. F194L demonstrated an increase in Km for the cofactor and unchanged Vmax, which demonstrates the potential of F194L to be an essential residue involved in mediating binding of NADPH. In contrast to the V89L–A49T compound heterozygotes that displayed a Vmax of an average of the two individual variants, the haplotypes demonstrated an unusually low Vmax (TABLE 1).
Table 1.
Enzyme properties of SRD5A2 missense substitutions.
| Substitution | Testosterone Km (mM) | NADPH Km (mM) | Vmax (nmol/min/mg) |
|---|---|---|---|
| WT (normal range) | 0.9 (0.7–1.0) | 8 (6–14) | 1.9 (1.7–2.2) |
| Single-point variant | |||
| P30L (rs61748121) | 2.1 | 21 | 0.5 |
| P48R (rs61748122) | 2.2 | 12 | 1.2 |
| A49T (rs9282858) | 2.7 | 7 | 9.9 |
| V89L (rs523349) | 0.6 | 8 | 1.1 |
| T187M (rs61748125) | 1.1 | 47 | 0.8 |
| F194L (rs61748126) | 0.7 | 19 | 2.2 |
| R227Q (rs9332964) | 4.6 | 38 | 0.06 |
| F234L (rs9332966) | 1.6 | 21 | 1.4 |
| Compound heterozygote | |||
| V89L–A49T | 1.2 | 8 | 5.5 |
| Haplotype (i.e., double mutant) | |||
| V89L–A49T | 0.8 | 8 | 2.9 |
Km: Michaelis constant; NADPH: Nicotinamide adenine dinucleotide phosphate; Vmax: Maximum rate of reaction; WT: Wild-type.
Reproduced with permission from [23].
Substantial pharmacogenetic variation was observed among the mutants and the three steroid 5α-reductase type II inhibitors [24]. In the case of finasteride in particular, inhibition varied 60‑fold (TABLE 2). The P30L, R227Q and F234L mutations demonstrated the highest inhibition constant (Ki) values, suggesting these mutations have the lowest affinity for finasteride. F194L exhibited the highest affinity for finasteride, followed by P48R. Interestingly, the kinetic properties for these substitutions suggest that these residues are essential for binding of the cofactor and the substrate, respectively. Again, results for the compound heterozygotes were as one would expect, with Ki values averaging the same for both individual SNPs as the biochemical enzymatic data. On the other hand, the haplotypes displayed different pharmacokinetic results to those that were expected. Essentially, V89L–T187M remains unchanged compared with the wild-type; however, V89L–F234L demonstrates a decreased affinity for finasteride. V89L–A49T demonstrates a decrease in Ki and, consequently, the highest affinity for finasteride apart from the single substitutions F194L and P48R.
Table 2.
Pharmacogenetic variation of SRD5A2 variants.
| Substitution | Finasteride (Ki nM) |
|---|---|
| WT | 60 (52–72) |
| Single-point variant | |
| P30L (rs61748121) | 420 |
| P48R (rs61748122) | 22 |
| F194L (rs61748126) | 7 |
| R227Q (rs9332964) | 260 |
| F234L (rs9332966) | 200 |
| Compound heterozygotes | |
| V89L–A49T | 150 |
| V89L–T187M | 89 |
| V89L–F234L | 154 |
| Haplotypes (i.e., double mutants) | |
| V89L–A49T | 48 |
| V89L–T187M | 59 |
| V89L–F234L | 94 |
Ki: Inhibition constant; WT: Wild-type.
Reproduced with permission from [23].
We propose that future studies should investigate the biochemical and pharmacogenetic characterization of steroid 5α-reductase type II haplotypes in large cohorts across diverse ethnic groups in order to determine disease risk and finasteride efficacy at the population level. The study by Makridakis et al. highlights the importance of haplotypes in determining an individual’s risk of developing prostate cancer and response to finasteride chemoprevention [24]. In terms of the PCPT in particular, the identification of risk haplotypes that potentially predict an individual’s likelihood of developing both the latent and aggressive forms of the disease (individuals with low and high Gleason scores, respectively) would improve early detection and chemoprevention strategies. Haplotypes that correlate with the functional expression of the steroid 5α-reductase type II enzyme in particular may aid appropriate dosing of finasteride in response to the overall catalytic turnover of androgen metabolism and thereby DHT bioavailability. Furthermore, elucidation of the steroid 5α-reductase type II enzyme crystal structure and appropriate mutagenesis studies may provide further details of the structure and function of the enzyme and binding interactions with finasteride.
CYP3A variation & phase I finasteride metabolism
Genetic variation in CYP3A genes has generated widespread interest owing to their role in drug metabolism and effects on drug efficacy and toxicity [81]. CYP3A4 activity has been shown to vary by more than than tenfold in vivo [82]. As has been previously noted [81], genetic variability accounts for the observed interindividual variation of CYP3A enzyme activity by as much as 90% [83]. CYP3A4 is the major liver isoform, also found in the intestine [84]. The CYP3A subfamily exhibits high sequence homology (84%), and various isoforms share similar substrate specificity [53]. In particular, CYP3A4 and CYP3A5 exhibit an overlap of substrate specificity. Studies have shown that different drugs bind to various domains of the CYP3A active site [85], thereby producing activation or inhibition of the enzyme [81].
Studies have reported variability in CYP3A4 metabolism that is directly attributable to individual coding SNPs that both increase and decrease the 6β-hydroxylation of testosterone with concurrent increases or decreases of desulfuration of the drug target [86]. It is suggested that because the allele frequencies of the CYP3A4 variants demonstrate relatively modest effects, they are unlikely to be the major cause of interindividual differences mediated by CYP3A4 metabolism [81]. However, the degree of linkage disequilibrium between CYP3A4 isoforms is high and therefore it is likely that these variants are in linkage disequilibrium with functional variants elsewhere on the CYP3A locus [81].
One of the most important CYP3A4 SNPs in the regulatory region has been extensively studied. CYP3A4*1B A-392G (rs2740574) is located immediately upstream of the transcription start site in the nifedipine-specific element, causing decreased CYP3A4 protein activity and thereby decreased oxidative deactivation of testosterone [87]. The frequency of this SNP was investigated in several ethnic groups and was found to be most common in African–Americans (p < 0.0001) [88]. Within the African–American group, almost half of those who presented with prostate cancer were homozygous for the variant allele, whereas this statistic decreased to 28% in healthy African–American volunteers (p < 0.005). Moreover, men who were homozygous for the variant allele were more likely to be diagnosed with high-grade prostate cancer.
CYP3A4*1B (rs2740574) allele frequencies vary greatly between ethnic populations [89]. Population studies have demonstrated a higher frequency of the CYP3A4*1B (rs2740574) variant in an African–American population in comparison with Asian, Caucasian and Hispanic populations (p < 0.0001) [88]. The allele frequencies for this variant are close to zero in the Asian population, 7% in the Caucasian population, 20% in the Hispanics and demonstrate 81% heterozygosity in the African–American population. This trend can be related to the occurrence of prostate cancer; the rate is lowest among the Asian population [88,90,91]. This allele has demonstrated a positive association with the advanced form of prostate cancer in a Caucasian population and an inverse association with risk in the same population with less aggressive disease [91].
Another study reported that most of the variant alleles of the CYP3A4 genotypes investigated showed a protective effect against prostate cancer [92]. CYP3A4*1B (rs2740574) in particular demonstrated an inverse association with low disease aggressiveness, and haplotype analysis revealed an inverse association with low aggressiveness of the disease [92].
CYP3A5 is reported to be a major contributor to the metabolism of many CYP3A-mediated drugs and is highly polymorphic [92]. The CYP3A5*1 (22893A of rs rs776746) allele has been shown to increase the hepatic expression of CYP3A5, even in the presence of a single polymorphic allele [85]. Highly frequent in African–Americans, the CYP3A5*1 variant allele is associated with CYP3A5 expression in the liver and intestine [93]. Studies have shown an inverse association with the less aggressive form of the disease [92]. Collectively, the CYP3A4*1B–CYP3A5*1 haplotype was inversely associated with risk of less aggressive disease. Both variants are known to be in linkage disequilibrium with each other and both are expressed by African–Americans [93]. The CYP3A5*3 (22893G, rs776746) allele produces a cryptic splice site and premature termination of the protein, resulting in loss of CYP3A5 expression [93]. The CYP3A4*1B–CYP3A5*3 haplotype was found to be positively associated with prostate cancer [94]. Collectively, the CYP3A4*1B–CYP3A5*3 haplotype warrants further investigation in large cohorts of diverse ethnic groups.
The literature reveals relatively few CYP3A haplotype studies to date. There are currently 347 human CYP3A4 SNPs deposited in the National Center for Biotechnology Information (NCBI) database [208]. Moreover, many of the positively associated markers are in high link-age disequilibrium with each other, as previously noted [95–98]. Therefore, implementing haplotype-tagged studies in large, diverse ethnic populations that extend across the CYP3A loci is warranted. Haplotype-based investigations, together with the biochemical characterization of functional variants, are essential for understanding the overall impact of the CYP3A loci and finasteride biotransformation. At present, there is no published literature of the effects of CYP3A4 and CYP3A5 genetic variations and finasteride metabolism.
Uridine diphosphoglucuronosyl-transferase variation & phase II finasteride metabolism
The UGT proteins are highly polymorphic and demonstrate tissue specificity and variability between different ethnic/racial populations [99]. Deficiencies in the extent of drug glucuronidation can lead to increased drug toxicity or enhanced substrate bioactivation through toxic metabolite intermediates [100]. The UGT1A locus is characterized by 13 first exons in the amino terminus and five common exons at the carboxy terminus of the protein [63,101]. The unique structure of this locus enables the formation of nine different UGT1A isotypes through exon sharing of each first exon with the common exons [100]. Two of these isoforms, UGT1A3 and UGT1A4, are involved in the glucuronidation of the products resulting from phase I metabolism in vitro, namely finasteride-ω-oic acid and ω-hydroxyfinasteride, respectively [65].
There are approximately 100 SNPs currently listed for each isotype in the NCBI database [208]. The UGT1A3 locus harbors a number of common functional variants in the first exon that affect enzyme efficiency [102,103]. Expression studies have demonstrated an increased Km:Vmax of the enzyme by 121% for the W11R (rs3821242) variant in comparison with the wild-type allele [103]. Studies have shown functional SNPs at the UGT1A4 locus [102]. Exon 1 variants P24T (rs6755571) and L48V (rs2011425) demonstrate reduced enzymatic activity; in particular, P24T reduced testosterone glucuronidation by 50% [102].
Comparisons of the haplotype structure of the UGT1A locus between Asian and Caucasian populations demonstrated differences in haplotype frequencies between ethnic groups [91]. These findings were reportedly attributable to the inclusion of rare nonsynonymous variants. Furthermore, the linkage disequilibrium patterns were also varied as expected between different ethnic populations.
Currently, there is little literature to suggest which UGT protein(s) are involved in the glucuronidation of finasteride [65]. Therefore, it is not known which UGT isozyme is responsible for the glucuronidation of the alternatively hydroxylated metabolite 6α-hydroxyfinasteride, which was the predominant metabolite identified in a recent study [65]. This represents a novel target for future investigations and could potentially explain, at least in part, the interindividual variation in response to finasteride treatment in some individuals.
Other modes of prostate cancer chemoprevention
In addition to finasteride, several modes of chemoprevention have been developed in order to reduce prostate cancer risk [104]. These include selective estrogen-receptor modulators, dietary supplements, nonsteroidal anti-inflammatory drugs (NSAIDs) and statins. Toremifene, one of the selective estrogen-receptor modulators that mediates estrogen-receptor blockade, is currently undergoing clinical trials in men at high risk of developing prostate cancer [105]. Dietary supplements such as selenium supplementation have shown no significant association with prostate cancer risk [106]. The use of NSAIDs and their anti-inflammatory effects mediated through COX-2 inhibition and association with prostate cancer risk is yet to be proven [107], although early studies demonstrated a decreased risk of the disease with NSAID use [108]. More recently, determination of aspirin use in conjunction with other NSAIDs and statins on PSA levels and PSA velocity (rate of change over time) in men diagnosed by biopsy with prostate cancer has been investigated. Subjects were part of an ongoing Phase II clinical trial using selenium to prevent the progression of prostate cancer. Baseline and on-study PSA measurements were evaluated over a period of 3.2 years. The results of the study showed that aspirin use significantly decreased PSA concentrations at baseline. Baseline PSA concentrations were not associated with other NSAIDs and statins. Aspirin use does not lead to a reduction in prostatic volume or disease progression and, as such, the authors suggest that aspirin use lowers PSA but does not actively contribute to the mechanisms of disease progression. They also note that the PSA-lowering effects mediated by aspirin may promote inaccurate PSA measurements and delay necessary treatment [109].
Statins are widely used as a cholesterol-lowering therapy in cardiovascular disease [110,111]. In addition to these properties, statins exert pleiotropic effects, including improvement of endothelial dysfunction, an increase of nitric oxide bio-availability and antioxidant, anti-inflammatory and immunomodulatory effects [112]. Studies have reported the benefits of statin therapy in reducing the risk of prostate cancer [113], although the association between statins and the overall risk of developing the disease is less clear [114]. It is considered more likely that this is due to detection bias of PSA testing among individuals undergoing statin therapy as opposed to nonstatin users [115]. The effects of statin use and their influence in detecting prostate cancer based on PSA levels was demonstrated in a cohort of over 2500 men aged 44 years or older without prostate cancer in the National Health and Nutrition Examination Survey (NHANES) 2001–2004 [116]. The study showed that men receiving statin therapy had slightly lower PSA concentrations compared with those not on statin treatment. The authors suggest that if PSA levels are decreased owing to statin therapy, the specificity of PSA screening in detection rates for asymptomatic prostate cancer are increased. However, the likelihood of detecting advanced prostate tumors is decreased [116]. To control for this potential confound and to determine the extent of actual incidence rates without sampling bias, a cohort of over 80,000 men from the Finnish Prostate Cancer Screening Trial analyzed the effects of statin therapy and overall relative risk of prostate cancer in men screened for the disease, independent of PSA levels between both statin and nonstatin users [117]. The results showed a dose-dependent reduction in overall relative risk in men undergoing statin therapy (independent of PSA lowering by statin therapy).
Steroid 5α-reductase type I enzyme expression demonstrates upregulation during tumor progression to become the dominant steroid 5α-reductase isoform [118]. This could offer another explanation for the reduced efficacy of finasteride in individuals with the aggressive form of the disease. The Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial demonstrated the effectiveness of the dual steroid 5α-reductase inhibitor dutasteride in preventing prostate cancer [12,119]. Over 8000 men between the ages of 50 and 75 years who had undergone a single negative prostate biopsy 6 months previously were recruited to the 4‑year randomized study based on elevated PSA levels and therefore increased risk of developing the disease. Men were assigned dutasteride 0.5 mg/day or placebo on study. The results of the trial demonstrated a 23% overall relative risk reduction of low-grade (Gleason score 5–6) prostate cancer (detectable by biopsy) in the dutasteride arm compared with the control group (p < 0.001) but did not definitively prevent prostate cancer. Overall, prostate volume was significantly decreased by 17.5% in the dutasteride group in comparison with controls over the 4‑year period (p < 0.001). Although no significant difference was observed for years 1 and 2, high-grade prostate cancer (Gleason score 8–10) was significantly increased in the second half of the trial in the dutasteride arm compared with the control group (p = 0.003).
The REDUCE trial investigators hypothesize that the observed increase in the number of high-grade tumors among the dutasteride group points to detection bias. If men in the control group who had low-grade tumors (Gleason score 5–7) detected in the first 2 years had remained in the study, it was considered most likely that at the end of the trial, these tumors would have been upgraded to Gleason scores of 8–10 and thereby no significant difference may have been observed between the two groups. The authors suggest another explanation for their findings, in that it is possible that the observed incidence of high-grade tumors in the dutasteride group was due to dutasteride itself. However, as the authors point out, dutasteride reduces prostate volume and therefore increases the likelihood of tumor detection, although as the results suggest, an overall relative risk reduction of 23% in prostate cancer implies dutasteride-mediated tumor shrinkage. These results exclude PSA and digital rectal examination bias owing to the careful study design. Of the adverse side effects reported, the study found an unexpectedly higher incidence of cardiac failure in the dutasteride group in comparison with the control group, affecting 30 out of 4105 subjects (0.7%) in the dutasteride group compared with 0.4% in the placebo arm of the trial (p = 0.03) [41].
The Combination of Avodart and Tamsolusin (CombAT) study was performed over 4 years and involved 4844 men older than 50 years who were diagnosed with BPH [120]. The results demonstrated that the cardiac failure event proportion was highest in the combination group (0.9%) compared with the dutasteride group (0.2%). This event was more likely to occur in men being treated with a combination therapy involving dutasteride and α-blockers. Despite this, the authors conclude that dutasteride should be considered to be a treatment option for men at increased risk of prostate cancer [119].
The influence of steroid 5α-reductase type II genetic variations and dutasteride response needs to be noted. Pharmacogenetic analyses of the efficacy of dutasteride and steroid 5α-reductase type II inhibition has shown that the Ki varies 35‑fold among the SNPs studied. This is a narrower variation range compared with the 60-fold variation demonstrated with finasteride inhibition [23]. Moreover, a lower Ki of 17 nM was defined, ranging from 15 to 20 nM for the wild-type enzyme compared with 60 nM described for finasteride. In addition, the study reveals a lower Ki for the majority of SNPs studied, except for the F194L variant, which varies threefold between finasteride (7 nM) and dutasteride (21 nM) [23].
A subsequent study investigated the pharmacological variation for both finasteride and dutasteride at 10- and 30‑min reaction times [25]. The observed decreased Ki values for both inhibitors demonstrate that they are slow, time-dependent inhibitors. The data presented in this study show a decrease in the Ki for most variants, except A49T, R227Q and F234L, for which Ki increased approximately twofold. In addition, the Ki values for P30L, F194L, R227Q and F234L were found to be above the normal range of the wild-type enzyme [25]. It is also interesting to note that for the steroid 5α-reductase type II A49T variant, the enzyme exhibits increased affinity for dutasteride in comparison with finasteride at longer reaction times of 30 min, despite the twofold increased Ki and 1.5-fold decreased Ki, respectively. The Ki remains below the wild-type range in the case of dutasteride [25].
In 2009, the American Urological Association (AUA) and the American Society of Clinical Oncology (ASCO) published a practice guideline for healthcare professionals that outlined recommendations for the administration of steroid 5α-reductase inhibitors as a prostate cancer chemopreventive therapy [41]. The PCPT and REDUCE trials demonstrated the efficacy of finasteride and dutasteride in reducing the risk of developing prostate cancer in high-risk populations [11,12,119]. Despite the benefits, the panel emphasized the use of these inhibitors as a chemopreventive therapy to reduce the risk of developing prostate cancer, although such therapies are not considered curative [41]. In addition, the effects of these drugs during clinical trials after 4 years for dutasteride and 7 years for finasteride remain unknown. Consequently, although the use of steroid 5α-reductase inhibitors have proven beneficial as a chemopreventive therapy for high-risk populations, information on the risk of increased grade tumors [45], the uncertainty of effects beyond the trials period and sexual- and endocrine-related side effects, such as decreased libido and ejaculate volume and gynecomastia, should be discussed with the patient when prescribed by the practitioner. Although the latter side effects tend to disappear over time, they are still significant. In addition, the benefits of such therapy should be outweighed against the risk of developing high-grade tumors as observed in the PCPT, events possibly owing to detection bias. This results in the increased chance of the detection of high-grade cancers in a smaller prostate; thus, it appears unlikely that finasteride would be the cause [41]. Finally, the long-term effects and the influence on mortality and life expectancy of chemopreventive therapy have not been studied beyond a 7‑year period. For these reasons, the panel recommends that steroid 5α-reductase inhibitors as a primary chemopreventive should not be administered to the patient for more than 7 years until further safety information is compiled. Therefore, it is recommended that a steroid 5α-reductase inhibitor regimen be prescribed to nonsexually active patients at high-risk of developing prostate cancer [41]. The decision to undergo this chemo-preventive therapy should be determined on the basis of histological information [41].
Conclusion & future perspective
The search for prostate cancer disease susceptibility alleles has yielded positive associations at multiple loci [121–124]. Identification of alleles across potentially multiple genes that predict phenotypic responses to finasteride chemoprevention is yet to be determined. There have been recent advances in the knowledge of finasteride metabolism [65] that have identified novel phase I and II metabolites, suggesting that an alternative pathway competing for the formation of finasteride-ω-oic acid exists [65]. The effects of this in terms of finasteride bioavailability and toxicity warrants further investigation. Overall, the extent of genetic variation of the enzymes involved in the regulation of both previously identified and novel finasteride metabolites and endogenous steroid activity needs to be established.
The SRD5A2 data show distinct biochemical and pharmacogenetic properties for the variants under study [24]. In particular, the V89L–A49T haplotype (TABLE 2) demonstrated the highest affinity for finasteride compared with the other haplotypes and almost all of the other variants studied. We believe the A49T mutation is involved in substrate binding. Based on the literature we have reviewed, we propose that there is another binding site on the steroid 5α-reductase type II enzyme that mediates finasteride binding and the subsequent inhibition reactions, which is similar to but distinct from the natural testosterone ligand binding. If correct, this hypothesis supports the views of others who suggest that there is a parallel pathway competing for the formation of finasteride-ω-oic acid [65]. Studies of steroid 5α-reductase type II crystal structure are essential to elucidate the mechanism-induced inhibition by finasteride. Furthermore, as finasteride is a bisubstrate inhibitor, genetic variation of the enzyme (steroid 5α-reductase type II) may result in structural conformational changes that affect the catalysis of both testosterone and finasteride; therefore, studies to address this question have been proposed.
CYP3A4 is the major enzyme involved in the biotransformation of phase I finasteride metabolites [55] and is also implicated in endogenous testosterone metabolism [56,57]. The reported associations between CYP3A4 genotypes/haplotypes and prostate cancer supports the role of the CYP3A4 gene as a plausible candidate for investigating dose–response effects of finasteride. Furthermore, we propose that future studies should focus on CYP3A4 and CYP3A5 genes. At present, there is no literature that has investigated CYP3A4 and CYP3A5 variations and potential effects on the metabolism of finasteride.
Finasteride-ω-oic acid glucuronides, the end product of finasteride metabolism, were identified in trace amounts in bile in vivo [65]. Glucuronidation is mediated by UGT1A3 in vitro [65]. However, studies to investigate the UGT proteins involved in the glucuronidation of finasteride metabolites are needed. Similarly, one of the novel glucuronides, possibly a ring-closed form of ω-hydroxyfinasteride, is potentially glucuronidated by UGT1A4. Both UGT isotypes have been implicated in drug glucuronidation activities and steroid metabolism and raise questions regarding drug efficacy and toxicity, but these issues have yet to be determined in studies of finasteride metabolism and prostate cancer risk. The other novel hydroxy metabolite is thought to be identical in structure to 6α-hydroxyfinasteride [65]. If such an alternative pathway of finasteride metabolism exists, it is essential to determine which UGT isozyme is responsible for the glucuronidation of 6α-hydroxyfinasteride.
We propose that future studies should characterize the biochemical and pharmacogenetic profiles of individual SNPs and implement extended haplotype-based studies of CYP3A4/5, UGT1A3/4 and SRD5A2 genes across diverse ethnic populations. This will determine the differences (if any exist) of the haplotypic structure between populations (especially high-risk populations such as African–Americans). This is crucial for determining the potential of haplotypes to confer both positive and less favorable responses to finasteride chemoprevention and thereby minimize the delay of alternative chemoprevention strategies, such as the dual steroid 5α-reductase inhibitor dutasteride.
In summary, studies of genes involved in both finasteride metabolism and its target steroid 5α-reductase type II may potentiate the search for the development of a personalized medicine approach to identifying prostate cancer disease risk, progression and response to finasteride chemoprevention. These investigations may include resequencing of genes in order to determine the true extent of overall genetic variation and haplotype-tagging strategies to capture the maximum genetic diversity of individuals and their response to finasteride in population studies. Furthermore, the biochemical characterization and pharmacogenetic analyses of functional SNPs and haplotypes of candidate genes is essential for confirming an alternative pathway(s) for finasteride metabolism and possibly accounting for the huge interindividual variation observed. This would enable investigators to identify variants that influence both positive and negative interactions with finasteride and thereby an individual’s likely of response to finasteride chemoprevention.
Author summary.
Prostate cancer
-
▪
Prostate cancer is the most common nonskin cancer in men in the western world.
The prostate & steroid hormones
-
▪
Testosterone is irreversibly converted to the most physiologically active androgen dihydrotestosterone by the androgen-metabolizing enzymes steroid 5α-reductase types I and II.
-
▪
Androgen metabolism occurs within the prostate gland and is essential for normal prostate function, although is thought to play a pivotal role in prostate carcinogenesis.
Chemoprevention
-
▪
Finasteride, a steroid 5α-reductase type II inhibitor, reduces prostate cancer risk; however, a large interindividual variation in responses to finasteride treatment exists. Such variation is thought to be due at least in part to genetic variation.
-
▪
The Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial demonstrated the efficacy of dutasteride in reducing prostate cancer risk by 23%. However, genetic variants of the steroid 5α-reductase type II enzyme may influence pharmacological responses to therapy.
-
▪
A higher prescription dose is required for finasteride chemoprevention therapy compared with dutasteride in order to achieve similar prostate cancer risk reductions. This may be relevant in terms of drug toxicity.
Finasteride metabolism
-
▪
The recent identification of the predominant and novel hydroxy metabolite in human bile and urine suggests the possibility that an alternative pathway competing for the formation of finasteride-ω-oic acid exists.
CYP3A & finasteride biotransformation
-
▪
CYP3A4 mediates the hydroxylation and oxidation reactions of phase I finasteride metabolites. The CYP3A4 locus is highly polymorphic and is in high linkage disequilibrium with the functional variants of CYP3A5.
-
▪
Future studies aimed at the characterization of the CYP3A4–CYP3A5 locus and effects of finasteride phase I biotransformation are warranted.
Uridine diphosphoglucuronosyltransferases & finasteride glucuronidation
-
▪
It has been shown that the UGT1A locus mediates the glucuronidation of finasteride metabolites in vitro. However, a lack of literature exists in terms of determining the uridine diphosphoglucuronosyltransferases involved in finasteride metabolism and the impact of genetic variants and finasteride glucuronidation. Furthermore, it is not known which uridine diphosphoglucuronosyltransferase isoform is involved in the glucuronidation of the novel finasteride metabolite, 6α-hydroxyfinasteride.
Steroid 5α-reductase type II
-
▪
The biochemical and pharmacogenetic properties of SRD5A2 variants demonstrate a wide variation of affinities for substrate, cofactor, enzyme and inhibitor. It is possible that an alternative substrate-binding site, in addition to the testosterone ligand-binding site, exists, owing to genetic variations that promote structural conformational changes to the enzyme.
Guidelines on the use of steroid 5α-reductase inhibitors
-
▪
Chemopreventive therapies do not eliminate the risk of developing prostate cancer, but reduce the likelihood of developing the disease in high-risk groups on long-term therapy. However, the long-term effects of therapy currently remain unknown.
Future perspective
-
▪
The characterization of finasteride-metabolizing genetic variants and the impact of finasteride metabolism clearly warrant further investigation.
-
▪
Elucidation of the crystal structure of steroid 5α-reductase type II may help to reveal the drug interactions and functional properties of this enzyme.
-
▪
The careful molecular characterization of genetic variants, in particular haplotypes, involved in the metabolism process may highlight novel pathways involving finasteride metabolism and explain, at least in part, the interindividual responses to finasteride treatment.
Acknowledgments
Financial
This work was supported in part by National Cancer Institute (NCI) grant PO1 CA108964 (project 1) to Juergen KV Reichardt who is also a Medical Foundation Fellow at the University of Sydney. This work was also supported by the Intramural Research Program of the NIH, NCI, Division of Cancer Epidemiology and Genetics, USA.
Footnotes
competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Haas GP, Sakr WA: Epidemiology of prostate cancer. CA Cancer J. Clin 47, 273–287 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Sakr WA, Grignon DJ, Haas GP, Heilbrun LK, Pontes JE, Crissman JD: Age and racial distibution of prostatic intraepithelial neoplasia. Eur. Urol 30, 138–144 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Jänne OA, Palvimo JJ, Kallio P, Mehto M: Androgen receptor and mechanisms of androgen action. Ann. Med 25, 83–89 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Andersson S, Russell DW: Structural and biochemical properties of cloned and expressed human and rat steroid 5α-reductases. Proc. Natl Acad. Sci 87, 3640–3644 (1990).▪ Describes both of the steroid 5α-reductase isoforms (types I and II).
- 5.Tamura K, Furihata M, Tsunoda T et al. : Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res 67, 5117–5125 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Hsing AW, Reichardt JK, Stanczyk FZ: Hormones and prostate cancer: current perspectives and future directions. Prostate 52, 213–235 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Zhu YS, Imperato-McGinley JL: 5α-reductase isozymes and androgen actions in the prostate. Ann. NY Acad. Sci 1155, 43–56 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Nickel JC: Comparison of clinical trials with finasteride and dutasteride. Rev. Urol 6(Suppl. 9), S31–S39 (2004). [PMC free article] [PubMed] [Google Scholar]
- 9.Lazier CB, Thomas LN, Douglas RC, Vessey JP, Rittmaster RS: Dutasteride, the dual 5α-reductase inhibitor, inhibits androgen action and promotes cell death in the LNCaP prostate cancer cell line. Prostate 58, 130–144 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Bruchovsky N, Lesser B, Van Doorn E, Craven S: Hormonal effects on cell proliferation in rat prostate. Vitam. Horm 33, 61–102 (1975). [DOI] [PubMed] [Google Scholar]
- 11.Thompson IM, Phyllis JG, Tangen CM et al. : The influence of finasteride on the development of prostate cancer. N. Engl. J. Med 349, 215–224 (2003).▪▪ Describes the Prostate Cancer Prevention Trial (PCPT), which demonstrated the effectiveness of the steroid 5α-reductase inhibitor finasteride in reducing prostate cancer risk.
- 12.Andriole GL, Roehrborn C, Schulman C, Slawin KM, Somerville M, Rittmaster RS: Effect of dutasteride on the detection of prostate cancer in men with benign prostatic hyperplasia. Urology 64, 537–541 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Shahani R, Fleshner NE, Zlotta AR: Pharmacotherapy for prostate cancer: the role of hormonal treatment. Discov. Med 7, 118–124 (2007). [PubMed] [Google Scholar]
- 14.Walsh PC: Chemoprevention of prostate cancer. N. Engl. J. Med 362, 1237–1238 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Motulsky AG: Drug reaction enzymes, and biochemical genetics. J. Am. Med. Assoc 165, 835–837 (1957). [DOI] [PubMed] [Google Scholar]
- 16.Nebert DW: From genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab. Rev 40, 187–224 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldstein DB: The genetics of human drug response. Phil. Trans. R. Soc. B 360, 1571–1572 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tate SK, Depondt C, Sisodiya SM et al. : Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc. Natl Acad. Sci. USA 102, 5507–5512 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein DB, Tate SK, Sisodiya SM: Pharmacogenetics goes genomic. Nat. Rev. Genet 4, 937–947 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Surendiran A, Pradhan SC, Adithan C: Role of pharmacogenomics in drug discovery and development. Indian J. Pharmacol 40, 137–143 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walko CM, McLeod H: Pharmacogenomic progress in individualised dosing of key drugs for cancer patients. Nat. Clin. Prac. Oncol 6, 153–162 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Zhou SF, Di YM, Chan E et al. : Clinical pharmacogenetics and potential application in personalized medicine. Curr. Drug Metab 8, 738–784 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Makridakis NM, di Salle E, Reichardt JKV: Biochemical and pharmacogenetic dissection of human steroid 5α-reductase type II. Pharmacogenetics 10, 407–413 (2000).▪▪ Characterizes the biochemical and pharmacogenetic properties of SRD5A2 variants with steroid 5α-reductase type II inhibitors.
- 24.Makridakis N, Akalu A, Reichardt JKV: Identification and characterisation of somatic steroid 5α-reductase (SRD5A2) mutations in human prostate cancer tissue. Oncogene 23, 7399–7405 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Makridakis N, Reichardt JKV: Pharmacogenetic analysis of human steroid 5α‑reductase type II: comparison of finasteride and dutasteride. J. Mol. Endocrinol 34, 617–623 (2005).▪▪ Characterizes the pharmacogenetic variation of dutasteride and finasteride. Demonstrates dutasteride to be a more effective 5α-reductase type II inhibitor than finasteride.
- 26.Chu LW, Reichardt JK, Hsing AW: Androgens and the molecular epidemiology of prostate cancer. Curr. Opin. Endocrinol. Diabetes Obes 15, 261–270 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Goldstein DB, Weale ME: Linkage disequilibrium holds the key. Curr. Biol 11, R576–R579 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Johnson GC, Esposito L, Barratt BJ et al. : Haplotype tagging for the identification of common disease genes. Nat. Genet 29, 233–237 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Reich DE, Cargill M, Bolk M et al. : Linkage disequilibrium in the human genome. Nature 411, 199–204 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Gu CC, Yu K, Ketkar S, Templeton AR, Rao DC: On transferability of genome-wide tag SNPs. Genet. Epidemiol 32, 89–97 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Mehrain-Shai R, Reichardt JKV: A renaissance of ‘biochemical genetics’? SNPs, haplotypes, function, and complex diseases. Mol. Gen. Metab 83, 47–50 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Vineis P, Khan AE, Vlaanderen J, Vermeulen R: The impact of new research technologies on our understanding of environmental causes of disease: the concept of clinical vulnerability. Environ. Health 8, 54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma BBY, Hui EP, Mok TSK: Population-based differences in treatment outcome following anticancer drug therapies. Lancet Oncol 11, 75–84 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Cascorbi I: Genetic basis of toxic reactions to drugs and chemicals. Toxicol. Lett 162, 16–28 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Reich DE, Lander ES: On the allelic spectrum of human disease. Trends Genet 17, 502–510 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Campbell H, Manolio T: Commentary: rare alleles, modest genetic effects and the need for collaboration. Int. J. Epidemiol 36(2), 445–448 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB: Rare variants create synthetic genome-wide associations. PLoS Biol 8, 1–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huggins C, Steven R: The effect of castration on benign hypertrophy of the prostate in man. J. Urol 43, 705 (1940). [DOI] [PubMed] [Google Scholar]
- 39.Marks LS: 5α-reductase: history and clinical importance. Rev. Urol 6, S11–S21 (2004). [PMC free article] [PubMed] [Google Scholar]
- 40.Gormley GJ, Stoner E, Bruskewitz RC et al. : The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N. Engl. J. Med 327, 1185–1191 (1992). [DOI] [PubMed] [Google Scholar]
- 41.Kramer BS, Hagerty KL, Justman S et al. : Use of 5-α-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 clinical practice guideline. J. Clin. Oncol 27, 1502–1516 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephenson AJ, Abouassaly R, Klein EA: Chemoprevention of prostate cancer. Urol. Clin. North Am 37, 11–21 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Strope SA, Andriole GL: Update on chemoprevention for prostate cancer. Curr. Opin. Urol 20(3), 194–197 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Vickers AJ, Savage CJ, Lilja H: Finasteride to prevent prostate cancer: should all men or only a high-risk subgroup be treated? J. Clin. Oncol 28, 1112–1116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynolds T: Prostate cancer prevention trial yields positive results, but with a few cautions. J. Natl Cancer Inst 95, 1030–1031 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Crawford ED, Andriole GL, Marberger M, Rittmaster RS: Reduction on the risk of prostate cancer: future directions after the prostate cancer prevention trial. Urology 75, 502–510 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Bull HG, Garcia-Calvo M, Andersson S et al. : Mechanism-based inhibition of human steroid 5α-reductase by finasteride: enzyme-catalyzed formation of NADP– dihydrofinasteride, a potent bisubstrate analog inhibitor. J. Am. Chem. Soc 118, 2359–2365 (1996). [Google Scholar]
- 48.Schloss JV: Significance of slow-binding enzyme inhibition and its relationship to reaction-intermediate analogues. Accounts Chem. Res 21, 348–353 (1988). [Google Scholar]
- 49.Lavogina D, Enkvist E, Uri A: Bisubstrate inhibitors of protein kinases: from principles to practical applications. Chem. Med. Chem 5, 23–34 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Faller B, Farley D, Nick H: Finasteride: a slow-binding 5α-reductase inhibitor. Biochemistry 32, 5705–5710 (1993). [DOI] [PubMed] [Google Scholar]
- 51.Drury JE, Di Costanzo L, Penning TM, Christianson DW: Inhibition of human steroid 5β-reductase (AKR1D1) by finasteride and structure of the enzyme–inhibitor complex. J. Biol. Chem 284(30), 19786–19790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrighton SA, Stevens JC: The human hepatic cytochromes P450 involved in drug metabolism. Crit. Rev. Toxicol 22, 1–21 (1992). [DOI] [PubMed] [Google Scholar]
- 53.Nebert DW, Russell DW: Clinical importance of the cytochromes P450. Lancet 360, 1155–1162 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Perera MA: The missing linkage: what pharmacogenetic associations are left to find in CYP3A? Expert Opin. Drug Metab. Toxicol 6, 17–28 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Huskey SW, Dean DC, Miller RR, Rasmusson GH, Chiu SH: Identification of human cytochrome P450 isozymes responsible for the in vitro oxidative metabolism of finasteride. Drug Metab. Dispos 23, 1126–1135 (1995). [PubMed] [Google Scholar]
- 56.Waxman DJ, Attisano C, Guengerich FP, Lapenson DP: Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 β-hydroxylase cytochrome P-450 enzyme. Arch. Biochem. Biophys 263, 424–436 (1988). [DOI] [PubMed] [Google Scholar]
- 57.Sata F, Sapone A, Elizondo G et al. : CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: evidence for an allelic variant with altered catalytic activity. Clin. Pharmacol. Ther 67, 48–56 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Smith RL, Williams RT: Implication of the conjugation of drugs and other exogenous compounds. In: Glucuronic Acid: Free and Combined: Chemistry, Biochemistry, Pharmacology and Medicine Dutton GJ (Ed.). Academic Press, NY, USA, 58–69 (1966). [Google Scholar]
- 59.Wells PG, Mackenzie PI, Chowdury JR et al. : Glucuronidation and the UDP-glucuronosyltransferases in health and disease. Drug Metab. Dispos 32, 281–290 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Nagar S, Remmel RP: Uridine diphosphoglucuronosyltransferase pharmacogenetics and cancer. Oncogene 25, 1659–1672 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Barbier O, Belaner A: Inactivation of androgens by UDP-glucuronosyltransferases in the human prostate. Best Pract. Res. Clin. Endocrinol. Metab 22, 259–270 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Ritter JK: Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem. Biol. Interact 129, 171–193 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Guillemette C, Lévesque E, Harvey M, Bellemare J, Menard V: UGT genomic diversity: beyond gene duplication. Drug Metab. Rev 42, 24–44 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Operaña TN, Tukey RH: Oligomerization of the UDP-glucuronosyltransferase 1A proteins. J. Biol. Chem 282, 4821–4829 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Lundahl A, Lennernas H, Knutson L et al. : Identification of finasteride metabolites in human bile and urine by high-performance liquid chromatography/tandem mass spectrometry. Drug Metab. Dispos 37, 2008–2017 (2009).▪▪ Identifies novel hydroxy phase I and II finasteride metabolites.
- 66.Soars MG, Burchell B, Riley RJ: In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J. Pharmacol. Exp. Ther 301, 382–390 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Tukey RH, Strassburg CP: Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol 40, 581–616 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Lundahl A, Hedeland M, Bondesson U, Knutson L, Lennernäs H: The effect of St. John’s wort on the pharmacokinetics, metabolism and biliary excretion of finasteride and its metabolites in healthy men. Eur. J. Pharm. Sci 36, 433–443 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Carlin JR, Hoglund P, Eriksson LO, Christofalo P, Gregoire SL, Taylor AM: Disposition and pharmacokinetics of 14C finasteride after oral administration in humans. Drug Metab. Dispos 20, 148–155 (1992). [PubMed] [Google Scholar]
- 70.Loftsson T, Hreinsdóttir D: Determination of aqueous solubility by heating and equilibration: a technical note. AAPS PharmSciTech 7, E1–E4 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Bergman E, Forsell P, Tevell A et al. : Biliary secretion of rosuvastatin and bile acids in humans during the absorption phase. Eur. J. Pharm. Sci 29, 205–214 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Persson EM, Nilsson RG, Hansson GL, Löfgren LJ, Lidbäck F, Knutson L: A clinical single-pass perfusion investigation of the dynamic in vivo secretory response to a dietary meal in human proximal small intestine. Phar. Res 23, 742–751 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Ishii Y, Mukoyama H, Ohtawa M: In vivo biotransformation of finasteride in rat hepatic microsomes. Drug Metab. Dispos 22, 79–84 (1994). [PubMed] [Google Scholar]
- 74.Liu MJ, Pollack GM: Pharmacokinetics and pharmacodynamics of valproate analogs in rats. II. Pharmacokinetics of octanoic acid, cyclohexanecarboxylic acid, and 1-methyl-1-cyclohexanecarboxylic acid. Biopharm. Drug Dispos 14, 325–339 (1993). [DOI] [PubMed] [Google Scholar]
- 75.Shipkova M, Armstrong VW, Oellerich M, Wieland E: Acyl glucuronide drug metabolites: toxicological and analytical implications. Ther. Drug Monit 25, 1–16 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Makridakis NM, Ross RK, Pike MC et al. : Association of missense substitution in SRD5A2 gene with prostate cancer in African–American and Hispanic men in Los Angeles, USA. Lancet 354, 975–978 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Latil AG, Azzouzi R, Cancel GS et al. : Prostate carcinoma risk and allelic variants of genes involved in androgen biosynthesis and metabolism pathways. Cancer 92, 1130–1137 (2001). [DOI] [PubMed] [Google Scholar]
- 78.Cussenot O, Azzouzi AR, Nicolaiew N et al. : Low-activity V89L variant in SRD5A2 is associated with aggressive prostate cancer risk: an explanation for the adverse effects observed in chemoprevention trials using 5-α-reductase inhibitors. Eur. Urol 52, 1082–1089 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Wang M, Bhattacharyya AK, Taylor MF, Tai HH, Collins DC: Site-directed mutagenesis studies of the NADPH-binding domain of rat steroid 5α-reductase (isozyme-1) I: analysis of aromatic and hydroxylated amino acid residues. Steroids 64, 356–362 (1999). [DOI] [PubMed] [Google Scholar]
- 80.Ramos L, Chávez B, Vilchis F: Cloning and differential expression of steroid 5‑a‑reductase type I (SRD5A1) and type II (SRD5A2) from the Harderian glands of hamsters. Gen. Comp. Endocrinol 166, 388–395 (2010). [DOI] [PubMed] [Google Scholar]
- 81.Lamba JK, Lin YS, Schuetz EG, Thummel KE: Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Delivery Rev 54, 1271–1294 (2002). [DOI] [PubMed] [Google Scholar]
- 82.Schmidt R, Baumann F, Hanschmann H, Geissler F, Preiss R: Gender difference in ifosfamide metabolism by human liver microsomes. Eur. J. Drug Metab. Pharmacokinet 26, 193–200 (2001). [DOI] [PubMed] [Google Scholar]
- 83.Ozdemir V, Kalowa W, Tang BK et al. : Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method. Pharmacogenetics 10, 373–388 (2000). [DOI] [PubMed] [Google Scholar]
- 84.Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB: Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human bowel enterocytes. J. Clin. Invest 90, 1871–1878 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kenworthy KE, Bloomer JC, Clarke SE, Houston JB: CYP3A4 drug interactions: correlation of 10 in vitro probe substrates. Br. J. Clin. Pharmacol 48, 716–727 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dai D, Tang J, Rose R et al. : Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J. Pharmacol. Exp. Ther 299, 825–831 (2001). [PubMed] [Google Scholar]
- 87.Rebbeck TR, Jaffe JM, Walker AH, Wein AJ, Malkowicz SB: Modification of clinical presentation of prostate tumors by a novel genetic variant in CYP3A4. J. Natl Cancer Inst 90, 1225–1229 (1998). [DOI] [PubMed] [Google Scholar]
- 88.Paris PL, Kupelian PA, Hall JM, Williams TL, Levin H, Klein EA: Association between a CYP3A4 genetic variant and clinical presentation in African– American prostate cancer patients. Cancer Epidemiol. Biomarkers Prev 8, 901–905 (1999). [PubMed] [Google Scholar]
- 89.Zeigler-Johnson CM, Walker AH, Mancke B et al. : Ethnic differences in the frequency of prostate cancer susceptibility alleles at SRD5A2 and CYP3A4. Hum. Hered 54, 13–21 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Bangsi D, Zhou JY, Sun Y, Patel NP, Darga LL, Heilbrun LK: Impact of a genetic variant in CYP3A4 on risk and clinical presentation of prostate cancer among white and African–American men. Urol. Oncol 24, 21–27 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Thomas SS, Li SS, Lampe JW, Potter JD, Bigler J: Genetic variability, haplotypes, and htSNPs for exons 1 at the human UGT1A locus. Hum. Mut 27, 717–730 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Loukola A, Chadha M, Penn SG et al. : Comprehensive evaluation of the association between prostate cancer and genotypes/ haplotypes in CYP17A1, CYP3A4, and SRD5A2. Eur. J. Hum. Genet 12, 321–332 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Keuhl P, Zhang J, Lin Y et al. : Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet 27, 383–391 (2001). [DOI] [PubMed] [Google Scholar]
- 94.Plummer SJ, Conti DV, Paris PL, Curran AP, Casey G, Witte JS: CYP3A4 and CYP3A5 genotypes, haplotypes, and risk of prostate cancer. Cancer Epidemiol. Biomarkers Prev 12, 928–932 (2003). [PubMed] [Google Scholar]
- 95.Sinues B, Vicente J, Fanlo A et al. : CYP3A5*3 and CYP3A4*1B allele distribution and genotype combinations: differences between Spaniards and Central Americans. Ther. Drug Monit 29, 412–416 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Bosch TM, Doodeman VD, Smits PHM, Meijerman I, Schellens JHM, Beijnen JH: Pharmacogenetic screening for polymorphisms in drug-metabolizing enzymes and drug transporters in a Dutch population. Mol. Diag. Ther 10, 175–185 (2006). [DOI] [PubMed] [Google Scholar]
- 97.Thompson EE, Kuttab-Boulos H, Yang L, Roe BA, Di Renzo A: Sequence diversity and haplotype structure at the human CYP3A cluster. Pharmacogenomics J 6, 105–114 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Dally H, Bartsch H, Jäger B et al. : Genotype relationships in the CYP3A locus in Canadians. Cancer Lett 207, 95–99 (2004). [DOI] [PubMed] [Google Scholar]
- 99.Strassburg CP, Kalthoff S, Ehmer U: Variability and function of family 1 uridine-5´-diphosphate glucuronosyltransferases (UGT1A). Crit. Rev. Clin. Lab. Sci 45, 485–530 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Wells PG, Mackenzie PI, Chowdhury JR et al. : Glucuronidation and the UDP-glucuronosyltransferases in health and disease. Drug Metab. Dispos 32, 281–290 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Gong QH, Cho JW, Huang T et al. : Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenetics 11, 357–368 (2001). [DOI] [PubMed] [Google Scholar]
- 102.Ehmer U, Vogel A, Schutte JK, Krone B, Manns MP, Strassburg CP: Variation of hepatic glucuronidation: novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology 39, 970–977 (2004). [DOI] [PubMed] [Google Scholar]
- 103.Iwai M, Maruo Y, Ito M, Yamamoto K, Sato H, Takeuchi Y: Six novel UDP glucuronosyltransferase (UGT1A3) polymorphisms with varying activity. J. Hum. Genet 49, 123–128 (2004). [DOI] [PubMed] [Google Scholar]
- 104.Rittmaster RS, Fleshner NE, Thompson IM: Pharmacological approaches to reducing the risk of prostate cancer. Eur. Urol 55, 1064–1107(2009). [DOI] [PubMed] [Google Scholar]
- 105.Price D, Stein B, Sieber P: Toremifene for the prevention of prostate cancer in men with high grade prostatic intraepithelial neoplasia: results of a double-blind, placebo controlled, Phase IIb clinical trial. J. Urol 176, 965–970 (2006). [DOI] [PubMed] [Google Scholar]
- 106.Lippman SM, Klein EA, Goodman PJ et al. : Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J. Am. Med. Assoc 301, 39–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jafari S, Etminan M, Afshar K: Nonsteroidal anti-inflammatory drugs and prostate cancer: a systematic review of the literature and meta-analysis. Can. Urol. Assoc. J 3, 323–330 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hussain T, Gupta S, Mukhtar H: Cyclooxygenase-2 and prostate carcinogenesis. Cancer Lett 191, 125–135 (2003). [DOI] [PubMed] [Google Scholar]
- 109.Algotar AM, Thompson PA, Ranger-Moore J et al. : Effect of aspirin, other NSAIDs, statins on PSA and PSA velocity. Prostate 70(8), 883–888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brown G, Albers JJ, Fisher LD et al. : Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N. Engl. J. Med 323, 1289–1298 (1990). [DOI] [PubMed] [Google Scholar]
- 111.Kane JP, Malloy MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ: Regression of coronary atherosclerosis during treatment of familial hypercholesterolemia with combined drug regimes. JAMA 264, 3007–3012 (1990). [PubMed] [Google Scholar]
- 112.Jasinska M, Owczarek J, Orszulak‑Michalak D: Statins: a new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol. Rep 59, 483–499 (2007). [PubMed] [Google Scholar]
- 113.Friedman GD, Flick ED, Udaltsova N, Chan J, Quesenberry CP, Habel LA: Screening statins for possible carcinogenic risk: up to 9 years of follow-up of 361,859 recipients. Pharmacoepidemiol. Drug Saf 17, 27–36 (2008). [DOI] [PubMed] [Google Scholar]
- 114.Murtola TJ, Visakorpi T, Lahtela J, Syvälä H, Tammela TLJ: Statins and prostate cancer prevention: where are we now and future directions. Nat. Clin. Pract. Urol 5, 376–387 (2008). [DOI] [PubMed] [Google Scholar]
- 115.Brookhart MA, Patrick AR, Dormuth C et al. : Adherence to lipid-lowering therapy and the use of preventive health services: an investigation of the healthy user effect. Am. J. Epidemiol 166, 348–354 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Mondul AM, Selvin E, De Marzo AM, Freedland SJ, Platz EA: Statin drugs, serum cholesterol, and prostate-specific antigen in the National Health and Nutrition Examination Survey 2001–2004. Cancer Causes Control 21, 61–68 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murtola TJ, Tammela TLJ, Määttänen L et al. : Prostate cancer and PSA among statin users in the Finnish Prostate Cancer Screening Trial. Int. J. Cancer 127(7), 1650–1659 (2010). [DOI] [PubMed] [Google Scholar]
- 118.Hofland J, van Weerden WM, Dits NF et al. : Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res 70, 1256–1264 (2010). [DOI] [PubMed] [Google Scholar]
- 119.Andriole GL, Bostwick DG, Brawley OW et al. : Effect of dutasteride on the risk of prostate cancer. N. Engl. J. Med 362, 1192–1202 (2010).▪▪ Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial, demonstrating the effectiveness of dutasteride in reducing the risk of prostate cancer.
- 120.Roehrborn CG, Siami P, Barkin J et al. : The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4 year results from the CombAT study. Eur. Urol 57, 123–131 (2010). [DOI] [PubMed] [Google Scholar]
- 121.Eeles RA, Kote-Jarai Z, Al Olama AA et al. : Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet 41, 1116–1121 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hooker S, Hernandez W, Chen H et al. : Replication of prostate cancer risk loci on 8q24, 11q13, 17q12, 19q33, and XpII in African– Americans. Prostate 70, 270–275 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Xu J, Kibel AS, Hu JJ et al. : Prostate cancer risk associated loci in African–Americans. Cancer Epidemiol. Biomarkers Prev 18, 2145–2149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kote-Jarai Z, Easton DF, Stanford JL et al. : Multiple novel prostate cancer predisposition loci confirmed by an international study: the PRACTICAL constortium. Cancer Epidemiol. Biomarkers Prev 17, 2052–2061 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Carson C, Rittmaster R: The role of dihydrotestosterone in benign prostatic hyperplasia. Urology 61(Suppl. 4A), 2–7 (2003). [DOI] [PubMed] [Google Scholar]
Websites
- 201.Cancer Research UK homepage. www.cancerresearchuk.org.
- 202.American Cancer Society: Cancer facts and figures 2010. www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026238.pdf.
- 203.MedlinePlus drug information for finasteride. www.nlm.nih.gov/medlineplus/druginfo/meds/a698016.html.
- 204.Prescribing information for Avodart®. www.accessdata.fda.gov/drugsatfda_docs/label/2008/021319s015lbl.pdf.
- 205.GeneCards: SRD5A1 gene information. www.genecards.org/cgi-bin/carddisp.pl?gene=Srd5a1.
- 206.GeneCards: SRD5A2 gene information. www.genecards.org/cgi-bin/carddisp.pl?gene=Srd5a2.
- 207.Finasteride: drug information provided by Lexi-Comp. www.merck.com/mmpe/lexicomp/finasteride.html.
- 208.National Center for Biotechnology Information single nucleotide polymorphisms database. www.ncbi.nlm.nih.gov/snp.