

Abstract
Glioblastomas (GBMs) are malignant brain tumors that can outstrip nutrient supplies due to rapid growth. Cyclooxygenase-2 (COX-2) has been linked to GBMs and may contribute to their aggressive phenotypes. Amino acid starvation results in COX-2 mRNA and protein induction in multiple human glioma cell lines in a process requiring p38 mitogen-activated protein kinase (p38-MAPK) and the Sp1 transcription factor. Increased vascular endothelial growth factor expression results from starvation-dependent COX-2 induction. These data suggest that COX-2 induction with amino acid deprivation may be part of the adaptation of glioma cells to these conditions and potentially alter cellular response to anti-neoplastic therapy.
Keywords: Glioblastomas, Amino acid deprivation, COX-2, p38-MAPK, VEGF
Introduction
Cyclooxygenase (COX) is the rate-limiting enzyme in the biosynthesis of prostaglandins (for review, see (1)). Two isoforms of the COX enzyme, COX-1 and COX-2, have been identified. The COX-1 isoform synthesizes prostaglandins that are required for normal physiologic function like gastrointestinal cytoprotection and platelet activity. Inhibition of COX-1 may have an important role in non-steroidal anti-inflammatory drug (NSAID)-induced toxicity in humans, such as gastric ulcer formation. The second isoform, COX-2, is not detectable in most normal tissues; however, it is induced at sites of inflammation by cytokines, growth factors, tumor promoters, and other agents. Expression of cyclooxygenase-2 (COX-2) has been linked to many cancers and may contribute to malignant phenotypes, including enhanced proliferation, angiogenesis, and resistance to cytotoxic therapies (2–7).
Amino acids (AAs) are the building blocks of proteins and can also serve as intermediates in metabolism. Biosynthetic pathways are available for the production of many AAs. However, certain essential AAs cannot be synthesized within the cell and therefore must be externally acquired. Changes in the cellular milieu may potentially limit the availability of AAs, particularly the essential ones, which may have significant implications for basic cellular processes. Overall, cellular responses to nutrient deprivation such as autophagy have been extensively studied and may have implications for cancer growth and treatment response (for reviews, see (8–10)). AA deprivation does represent a form of metabolic stress, which can lead to activation of certain signaling pathways including the mitogen-activated protein kinase (MAPKs). MAPKs, which include extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 subfamilies, are important regulatory proteins through which various extracellular stimuli and stresses can be transduced into intracellular events (for reviews, see (11, 12))
Glioblastomas (GBMs) are the most common malignant primary brain tumors in adults. Despite advances in the treatment of GBMs, these tumors remain highly aggressive and relatively resistant to treatment with chemotherapy and radiotherapy (13, 14). COX-2 appears to be an important factor in the malignancy of GBMs with previous studies demonstrating the association of higher COX-2 expression with higher grade and more mitotically-active gliomas (15, 16). Furthermore, increased expression of COX-2 correlates with worse survival in this disease (16). In addition, significant hypoxia is often present in GBMs (17) and the mechanisms responsible for driving this hypoxia can similarly limit delivery of nutrients including amino acids. While hypoxia is thought to contribute to therapeutic resistance in GBMs (18), less is known regarding the role of amino acid starvation on response to cytotoxic therapies. Najim et al. did find that methionine restriction reduces sensitivity of a glioma cell to a variety of chemotherapies (19). Finally, amino acid homeostasis is potentially of importance to the pathogenesis of GBMs especially since specific amino acid transporters are elevated in these tumors as evidenced by the utility of amino acid PET tracers such as 11C-methionine for imaging GBMs (20, 21).
Due to rapid growth, GBMs can quickly outstrip their vascular supply resulting in significant hypoxia and nutrient deprivation necessitating an adaptive metabolic response by tumor cells (22–24). We sought to simulate nutrient deprivation with an in vitro model by culturing human glioma cells under conditions of AA starvation to evaluate potential adaptive responses to this cellular stress. In this study, we show that the expression of COX-2 is strongly induced by AA deprivation in multiple human glioma cell lines, further explore the mechanistic basis for starvation-dependent induction of COX-2, and identify a downstream effect of this COX-2 induction on vascular endothelial growth factor (VEGF) expression.
Materials and Methods
Cell lines, culture conditions, and transfections.
Glioma cells lines (U87MG, SF767) were chosen for use in these studies due to their frequent use as representative glioma models and previous characterization of COX-2 response to other signals (7, 25). These cells were cultured in high-glucose DMEM supplemented with 10% fetal calf serum, sodium pyruvate (1 mM), and penicillin (100 units/mL)/streptomycin (100 μg/mL) (Sigma-Aldrich, St. Louis, MO) unless otherwise indicated. Transient transfection of small interfering RNAs (siRNAs) or plasmids was accomplished using LipofectAMINE 2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s recommendation. siRNA against p38-MAPK and Sp1 (Santa Cruz Biotechnology, Santa Cruz, CA) was obtained commercially. siRNA (sequence: 5’-aaagacugguauuucaucugccugc-3’) against COX-2 was custom synthesized (Invitrogen). The Stealth RNAi medium GC duplex negative control or a custom scrambled siRNA (sequence: 5’-ugaucuggaugucaagacauaacuc-3’) (Invitrogen) was used to control for sequence independent effects of introducing short RNA duplexes into cells. The COX-2 promoter reporter construct containing the human COX-2 promoter from bases −1122 to +27 relative to the transcriptional start site inserted upstream of the firefly luciferase gene (COX2/P1-luciferase) has previously been described (7).
Amino acid deprivation and inhibitor treatments.
Cells were serum starved overnight prior to culturing in Gln/Lys/Arg or Met/Cys deficient media for the indicated times. DMSO stocks of inhibitors against the following enzymes, p38-MAPK (SB202190), MAP/ERK kinase (U0126), c-Jun-NH2-kinase (JNK; SP600125) (Calbiochem, San Diego, CA), and COX-2 (celecoxib; LC Laboratories, Woburn, MA) were made and working concentrations for a particular inhibitor are noted in the figure legends with vehicle only (DMSO) used as a negative control. The Sp1 transcription factor inhibitor mithramycin (Sigma) was also made as stock solutions in DMSO and used at concentrations noted for each experiment.
Western blotting and antibodies used.
All western blots were done according to standard procedures. Blots were probed with antibodies against human COX-2 (Cayman Chemical, Ann Arbor, MI), p38-MAPK/phospho- p38-MAPK (Cell Signaling Technology, Beverly, MA), JNK/phospho-JNK (Cell Signaling), Sp1 (Santa Cruz), and LC3 (Novus, Littleton, CO). As loading controls, blots were also probed with antibodies against EIF5α (Santa Cruz). Blots were detected using anti-rabbit or mouse IgG secondary antibody conjugated with horseradish peroxidase and chemiluminescent substrate according to standard procedures.
Quantitative real-time PCR.
COX-2 mRNA levels was assessed by quantitative real-time PCR. Briefly, total RNA was isolated in TRIzol and cDNA synthesized according to standard protocols. The real-time PCR reaction was performed using the TaqMan primer and probe sets for PTGS2 (COX-2; Hs00153133_m1), VEGFA (VEGF; Hs00900055_m1) and PPIA (cyclophilin A; Hs04194521_s1) on an ABI 7500 unit (Life Technologies, Grand Island, NY). Real-time PCR assays were performed in 20 μL reaction with standard reagents under conditions as follows: 50oC x 2 min, 95oC x 10 min, 40 cycles of 95oC x 15 sec and 60oC x 1 min. COX-2 levels were determined relative to cyclophilin levels as a normalization control.
Luciferase reporter assay.
The COX2/P1-luciferase reporter construct was transiently transfected into U87 and SF767 glioma cells using LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s recommendations. Two days post-transfection, cells were serum starved overnight and then subjected to AA starvation for 4 hours without or with SB202190 or mithramycin. Cells were harvested and luciferase activity measured according to standard procedure as described previously (26, 27). Luciferase activities were normalized to total protein concentration in each lysate.
Results
AA deprivation induces expression of COX-2.
We first noted in metabolic labeling experiments that culturing glioma cells in media lacking methionine/cysteine leads to significant induction of COX-2. Therefore, we sought to assess this response under more controlled conditions. Two human glioma cell lines (U87 and SF767) are cultured under conditions of AA deprivation with media lacking either glutamine/arginine/lysine (Gln/Arg/Lys) or methionine/cysteine (Met/Cys) and assessed for expression of COX-2. After culturing under these conditions of AA starvation, COX-2 expression increases significantly at the level of protein as assessed by immunoblot analysis (Figure 1A-B). COX-2 is induced within two hours and appears to reach its peak at 4–6 hours after exposure to starvation conditions. Of note, similar responses are seen with starvation using either Gln/Arg/Lys or Met/Cys deficient medias except that COX-2 induction is slightly more pronounced and seen somewhat sooner with Met/Cys deprivation prompting us to use Met/Cys deficient conditions on subsequent experiments (and will be referred to synonymously as AA starvation). Next, RNA expression of COX-2 is assessed by quantitative real-time PCR and shows similar increases in response to AA starvation (Figure 1C). Thus, nutrient starvation by AA depletion results in COX-2 induction at both the RNA and protein levels in malignant glioma cells.
Figure 1.
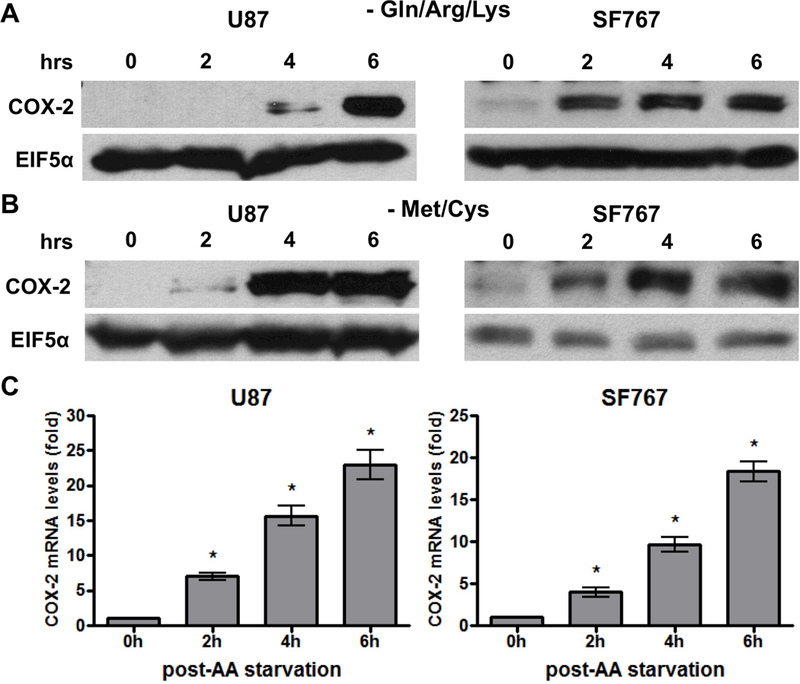
AA starvation induces COX-2 expression. Indicated cells were cultured in media lacking (A) glutamine/arginine/lysine (Gln/Arg/Lys) or (B/C) methionine/cysteine (Met/Cys). Cells were subjected to starvation conditions for various times and (A/B) probed for COX-2 protein expression on immunoblots or (C) COX-2 mRNA expression by real-time PCR. EIF5α and cyclophilin A served as normalization controls for the immunoblots and real-time PCR assays, respectively. Blots are representative of three independent experiments. Graphs show average value of three independent experiments with error bar representing ± one SEM. * indicate statistically significant (p < 0.05) difference by student’s t-test compared with initial time point.
Signaling through p38-MAPK is required for AA deprivation-induced COX-2 expression.
We next determined which signaling pathways are needed for COX-2 induction in response to AA starvation. Nutrient starvation is expected to trigger stress signals prompting us to evaluate the role of the p38-MAPK, MEK/ERK and JNK/SAPK pathways on COX-2 induction. These pathways are pharmacologically inhibited in two different glioma cell lines and the p38-MAPK inhibitor SB202190 is found to consistently suppress AA deprivation-dependent COX-2 response to the greatest extent (Figure 2A-B). Interestingly, reduced COX-2 induction is also seen with JNK inhibition using SP600125 in U87 cells while our other assessed cell line (SF767) showed no such dependence in JNK activity (Figure 2A-B). Consistent with these findings, AA starvation induces p38-MAPK phosphorylation in both U87 and SF767 cells (Figure 3A) and JNK phosphorylation in U87 cells (Figure 3B) indicative of pathway activation. To further confirm the role of p38-MAPK on AA deprivation-dependent COX-2 induction, a p38-MAPK–specific siRNA is introduced into the glioma cells to knockdown p38-MAPK expression. In each case, p38-MAPK expression is substantially reduced (>70%) and results in a corresponding decrease in COX-2 induction both at the protein and RNA levels after AA starvation (Figure 4A-B). Overall, these results strongly suggest that the p38-MAPK pathway is needed for AA deprivation-induced COX-2 expression in malignant glioma cells.
Figure 2.
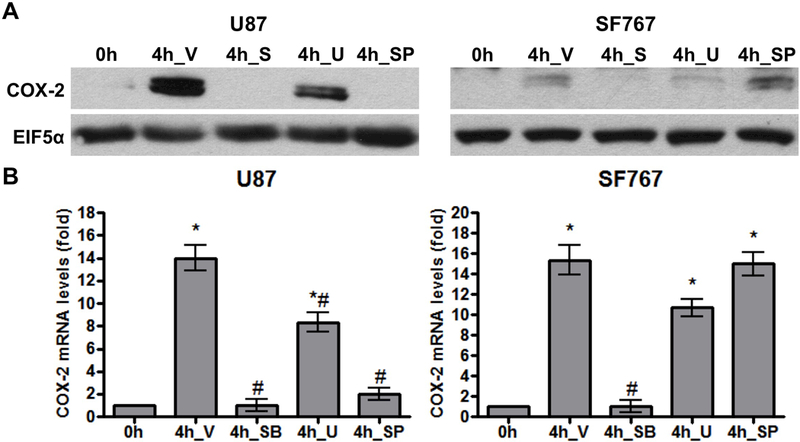
AA starvation-dependent COX-2 induction is inhibited by p38-MAPK inhibition. Indicated cells were pretreated for one hour with DMSO (V, vehicle), SB202190 (SB, 20 μM, p38-MAPK inhibitor), U0126 (U, 10 μM, MEK inhibitor), or SP600125 (SP, 30 μM, JNK inhibitor) and then AA starved for 4 hours. COX-2 (A) protein and (B) mRNA expression were assessed. EIF5α and cyclophilin A again served as normalization controls. Blots are representative of three independent experiments. Graphs show average value of three independent experiments with error bar representing ± one SEM. * and # indicate statistically significant (p < 0.05) difference by t-test compared with the initial and V-treated 4 hour time points, respectively.
Figure 3.
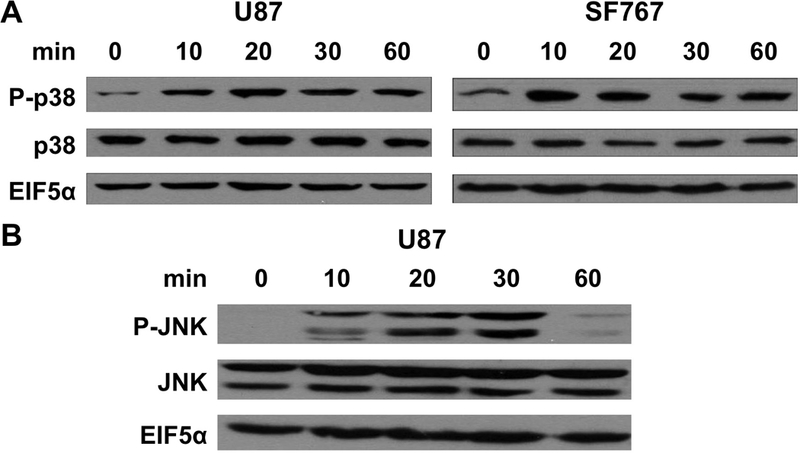
Activation of p38-MAPK and JNK by AA starvation. Indicated cells were harvested at the indicated times after AA starvation and assessed for expression of (A) p38-MAPK and phosphorylated p38-MAPK (P-p38) or (B) JNK and phosphorylated JNK (P-JNK) by immunoblot analysis. EIF5α again served as normalization controls. Blots are representative of three independent experiments.
Figure 4.
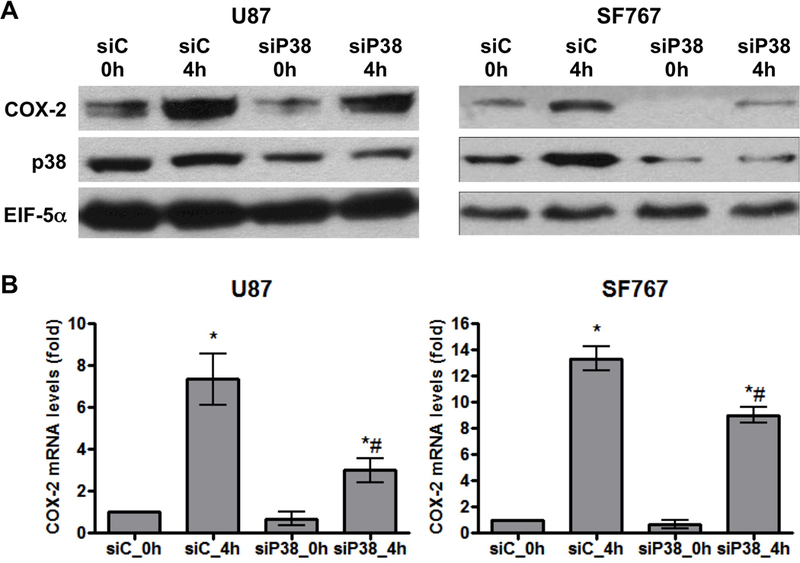
Knockdown of p38-MAPK reduces AA starvation-induced COX-2 expression. Indicated cells were subjected to AA starvation for 4 hours two days after transient transfection with siRNA against control (siC) or p38-MAPK (siP38). COX-2 (A) protein and (B) mRNA expression were assessed. EIF5α and cyclophilin A again served as normalization controls. Blots are representative of three independent experiments. Graphs show the average value of three independent experiments with error bar representing ± one SEM. * and # indicate statistically significant (p < 0.05) difference by t-test compared with the corresponding initial and siC-transfected 4 hour time points, respectively.
Sp1 is involved in AA deprivation-induced COX-2 expression.
Base on our previous work demonstrating the involvement of the Sp1 family of transcription factors downstream of p38-MAPK on EGFR-dependent induction of COX-2 (7, 28, 29), we speculate that Sp1 is also involved in AA deprivation-induced COX-2 expression. To test this possibility, Sp1 is pharmacologically inhibited with mithramycin and assessed for effect on COX-2 expression after AA starvation. In each case, mithramycin is capable of significantly suppressing induction of COX-2 at both the mRNA and protein levels in U87 and SF767 cells in response to AA deprivation (Figure 5A-B). These cells are then assessed after siRNA knockdown of Sp1, which could reduce expression of Sp1 by more than 90% (Figure 6A). In each case after Sp1 knockdown, COX-2 induction in response to AA starvation is significantly attenuated (Figure 6A-B). Finally, a COX-2 promoter reporter construct (COX2/P1-luciferase) is transiently introduced into U87 and SF767 cells (Figure 7A) and assessed for luciferase activity under AA starvation condition in the absence or presence of SB202190 and mithramycin. In this experiment, luciferase activity is induced by AA deprivation and attenuated by p38-MAPK and Sp1 inhibition (Figure 7B-C). These results suggest that AA starvation-dependent COX-2 induction involves a transcriptional mechanism requiring Sp1.
Figure 5.
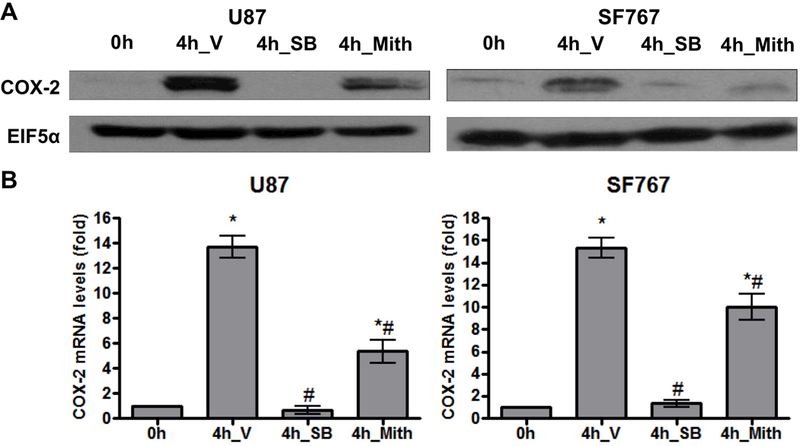
AA starvation-dependent COX-2 induction is reduced with Sp1 inhibition. Indicated cells were pretreated for one hour with V, SB (20 μM), or mithramycin (Mith, 0.5 μM, Sp1 inhibitor) and then AA starved for 4 hours. COX-2 (A) protein and (B) mRNA expression were assessed. EIF5α and cyclophilin A again served as normalization controls. Blots are representative of three independent experiments. Graphs show the average value of three independent experiments with error bar representing ± one SEM. * and # indicate statistically significant (p < 0.05) difference by t-test compared with the initial and V-treated 4 hour time points, respectively.
Figure 6.
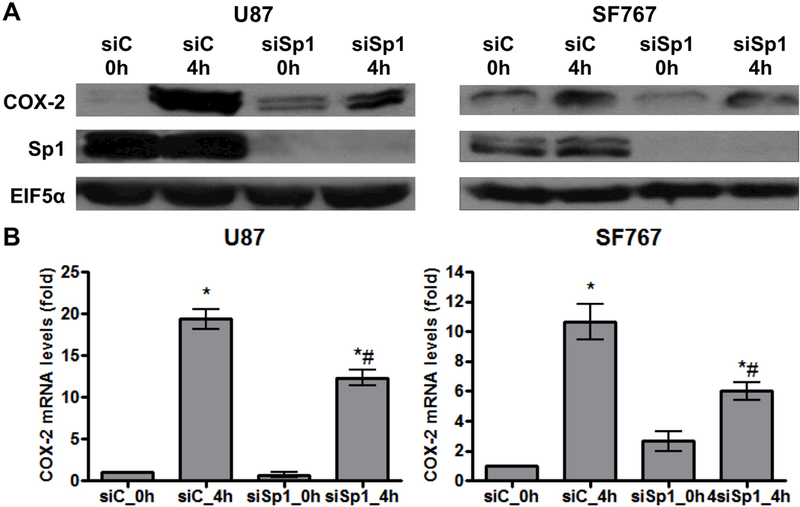
Knockdown of Sp1 reduces AA starvation-induced COX-2 expression. Indicated cells were subjected to AA starvation for 4 hours two days after transient transfection with siRNA against control (siC) or Sp1 (siSp1). COX-2 (A) protein and (B) mRNA expression were assessed. EIF5α and cyclophilin A again served as normalization controls. Blots are representative of three independent experiments. Graphs show the average value of three independent experiments with error bar representing ± one SEM. * and # indicate statistically significant (p < 0.05) difference by t-test compared with the corresponding initial and siC-transfected 4 hour time points, respectively.
Figure 7.
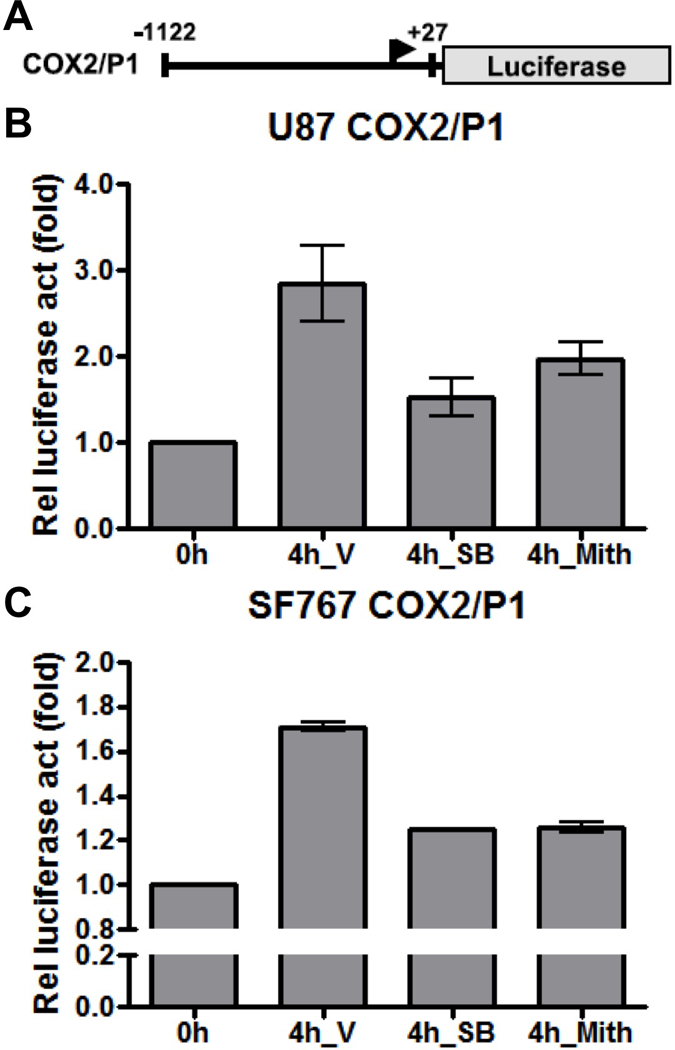
AA starvation induces COX-2 promoter activity. Indicated cells were subjected to AA starvation for 4 hours two days after transient transfection with the COX2/P1-luciferase reporter plasmid. (A) The COX-2 promoter expression cassette is shown. (B) U87 or (C) SF767 cells were assessed for luciferase expression with values expressed as fold induction relative to the initial time point. Cells were treated with V, SB (20 μM) or Mith (0.5 μM), as indicated. Graphs show the average value of two independent experiments with error bar representing ± one SEM.
AA deprivation-induced VEGF expression requires COX-2.
Because cells undergoing starvation would naturally try to adapt by enhancing nutrient acquisition through stimulating angiogenesis and we have previously found that increased COX-2 activity results in VEGF induction (7), we hypothesized that VEGF would also be induced by AA deprivation in glioma cells in a process dependent on COX-2 induction. To test this hypothesis, COX-2 expression was knocked down in U87 cells and then subjected to AA deprivation. Under these conditions, AA starvation results in substantial induction of COX-2 mRNA in the control cells with very significant knockdown of COX-2 expression after introduction of siRNA against COX-2 (Figure 8A). Knockdown of COX-2 protein expression (>90%) was also confirmed by immunoblot analysis (data not shown). Similarly, VEGF mRNA expression was induced 3–4 fold at baseline with AA starvation in control cells while COX-2 knockdown also significantly attenuated this starvation-induced VEGF expression (Figure 8B). Significant relative reduction of VEGF expression after COX-2 knockdown was in the 60–75% range (Figure 8C). These results suggest that COX-2 induction after AA deprivation enhances VEGF expression and stimulates angiogenesis as part of the adaptive response to starvation.
Figure 8.
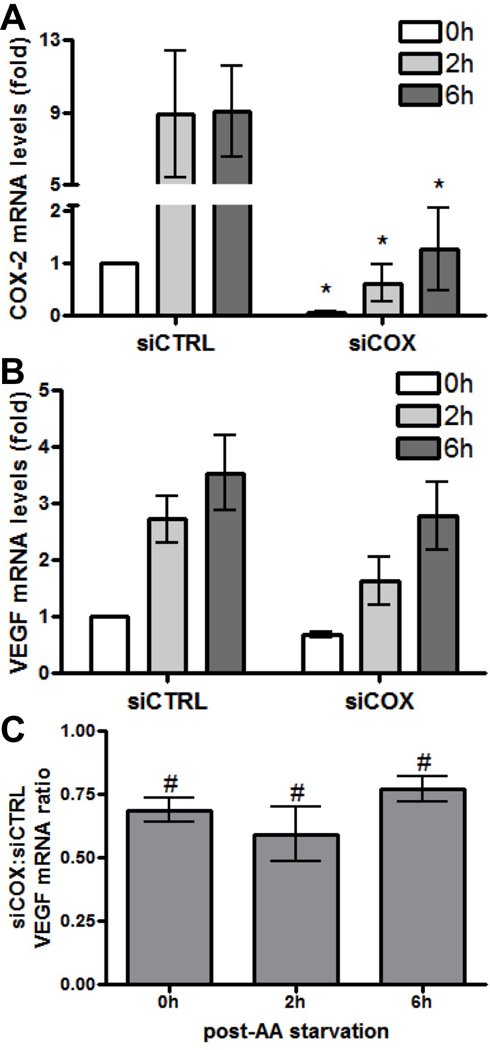
AA starvation-induced VEGF expression is dependent on COX-2 expression. U87 cells were subjected to AA starvation for the indicated times after transient transfection with siRNA against control (siCTRL) or COX-2 (siCOX). COX-2 and VEGF mRNA levels were determined at each time point by real-time PCR. (A) COX-2 and (B) VEGF mRNA expression at 0, 2 and 6 hours post-starvation (0h, 2h and 6h) in siCTRL and siCOX cells are shown relative to values from siCTRL cells that have not undergone starvation (0h, arbitrarily set at a value of 1). * indicate statistically significant (p < 0.05) differences by paired t-test compared with the corresponding siCTRL time points. (C) Relative reduction in VEGF mRNA expression is shown expressed as a ratio of values from siCOX to siCTRL cells. # indicate statistically significant reduction in VEGF expression by paired one-tailed t-test at each time point. Graphs show the average value of four independent experiments with error bar representing ± one SEM.
Discussion
Our data show that AA deprivation, as a form of metabolic stress, increases the expression of COX-2 in human glioma cell lines. The p38-MAPK pathway has previously been shown to mediate COX-2 induction in response to growth factor stimulation in glioma cells (7, 28, 29). Similarly, the p38-MAPK pathway appears to be particularly important in COX-2 induction in response to AA starvation although signaling through JNK is also a strong factor in one of the assessed cell lines (U87MG). Like with growth factor-mediated COX-2 induction, the Sp1 transcription factor also appears to be required in this process. Therefore, our work links AA deprivation and COX-2 expression and defines a molecular mechanism for this association.
Because of unchecked growth, malignancies including glioblastoma brain tumors can quickly outstrip their vascular supply and be potentially subjected to conditions of nutrient deprivation. To continue to thrive under these conditions, these cells need to generate appropriate adaptive responses. We find that COX-2 expression is induced after AA deprivation. COX-2 is overexpressed in a variety of malignancies and has been linked to aggressive phenotypes, including enhanced proliferation, angiogenesis, suppression of cell apoptosis, and resistance to cytotoxic therapies (2–7, 30). Given this range of potential activities, we speculate that its induction may be an important part of the adaptive response that these cells need to undergo to survive such conditions. We and others have previously found that increased COX-2 activity induces VEGF and results in enhanced angiogenesis (7, 27, 31, 32). In fact, we have now demonstrated that a consequence of the adaptive response of COX-2 induction in the face of AA starvation is enhanced VEGF expression. Transcriptional activation of COX-2 has not been previously linked to amino acid deprivation. Therefore, our report is the first to describe this finding and provide a molecular mechanism linking AA deprivation through p38-MAPK/Sp1 activation to transcriptional activation of COX-2 that results in VEGF induction and angiogenesis.
While the p38-MAPK pathway appears to be the primary one involved in regulation of COX-2 expression in response to AA starvation, we also find that the MEK/ERK pathway has a partial role in our assessed cells while the JNK/SAPK pathway has a very strong role in one of our assessed cell lines (U87). A rapid and transient activation of ERK1/2 and, to a minor extent, of JNK1 has been found to occur in fibroblasts incubated in a saline solution deprived for all amino acid providing precedence for the activation of these signaling pathways in response to AA deprivation (33). Since the MEK/ERK pathway appears to only have a minor role and the JNK/SAPK did not have consistent effects in both of our assessed cell lines in AA starvation-dependent COX-2 expression, we did not pursue identifying additional mechanistic details involved in these pathways. Zhang et al. did find that the JNK1/c-JUN/AP-1 pathway was important in TPA-induced COX-2 expression (34). Given the strong effect that inhibition of JNK/SAPK had on COX-2 expression in the U87 cells after AA starvation, it is certainly possible that this is also an important pathway in a subset of malignant glioma cells. Thus, identifying biomarker(s) that can indicate whether JNK/SAPK play a strong role in AA starvation-induced COX-2 expression will be of importance and can potentially be the subject of future investigations. Overall, inhibition of the p38-MAPK pathway along with JNK/SAPK, in selected GBMs, may be part of an ultimate strategy for targeting AA starvation-dependent COX-2 induction.
The pathogenesis of GBMs may be impacted by AA deprivation in multiple ways. As we found, AA starvation-dependent COX-2 induction may directly increase VEGF expression, which leads to microvascular proliferation and enhances tumor viability. Besides increasing angiogenesis with VEGF expression, we speculate that COX-2 induction is contributing to the adaptation of our malignant glioma cells to AA starvation in other ways. Autophagy is known to be one of the major cellular responses to nutrient deprivation. Under these conditions, a cell needs to take “drastic” measures to ensure survival by digesting portions of itself to sustain metabolism. While autophagy can be associated with increased cell death, it is also often involved in responding to metabolic stress and promoting cellular survival (for review, see (35)). In the context of malignancies, it is certainly conceivable that adaptive responses to nutrient deprivation such as autophagy may alter a tumor’s overall responsiveness to anti-neoplastic therapies. Kang et al. have previously shown that COX-2 inhibition with celecoxib in GBM cell lines results in autophagy (36). This suggests that COX-2 induction may potentially act to limit starvation-dependent autophagy. It is conceivable that COX-2 is playing a role in the tight regulation of such autophagic processes so that this adaption to starvation does not overshoot resulting in greater cell death. Exploration of these mechanisms, while intriguing, is beyond the scope of this paper.
Malignant glial neoplasms are notoriously resistant to cytotoxic therapies (37–40). Since regions of necrosis and hypoxia, which are likely to also be nutrient deprived, are very common with these tumors (41–43), we speculate that these adaptive responses, including COX-2 induction, may contribute to some of the resistance to therapy we see with these tumors. Like with other malignancies, increased expression of COX-2 is associated with higher grade gliomas and worse overall prognosis (15, 16). This has prompted investigations into COX-2 inhibition as anti-neoplastic therapy in various malignancies including GBMs (44–48). We speculate that COX-2 inhibition may be a particularly effective anti-neoplastic therapy in such nutrient-deprived tumors.
In summary, we have identified that p38-MAPK to Sp1 signaling pathway as the predominant mechanism contributing to AA deprivation-dependent induction of COX-2 transcription. In addition, COX-2 induction, in this setting, is an important contributor to the adaptation to nutrient deprivation with potential implications for response to cytotoxic therapies.
Acknowledgements
Work supported in part by grant (to H.G.S.) from the Southeast Brain Tumor Foundation and fellowship (to Z.L.) from the National Clinical Key Specialist Foundation to Anesthesiology Department of First Hospital of Jilin University. A Cancer Center grant (to Emory University) from the National Cancer Institute (P30-CA138292) also provided partial support for this work.
Footnotes
Declaration of Interest
The authors report no conflicts of interest.
References
- 1.Smith WL; DeWitt DL; Garavito RM Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem 2000. 69, 145–82. [DOI] [PubMed] [Google Scholar]
- 2.Dandekar DS; Lokeshwar BL Inhibition of cyclooxygenase (COX)-2 expression by Tet-inducible COX-2 antisense cDNA in hormone-refractory prostate cancer significantly slows tumor growth and improves efficacy of chemotherapeutic drugs. Clin Cancer Res 2004. 10, 8037–47. [DOI] [PubMed] [Google Scholar]
- 3.Kishi K; Petersen S; Petersen C; Hunter N; Mason K; Masferrer JL; Tofilon PJ; Milas L Preferential enhancement of tumor radioresponse by a cyclooxygenase-2 inhibitor. Cancer Res 2000. 60, 1326–31. [PubMed] [Google Scholar]
- 4.Oshima M; Dinchuk JE; Kargman SL; Oshima H; Hancock B; Kwong E; Trzaskos JM; Evans JF; Taketo MM Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996. 87, 803–9. [DOI] [PubMed] [Google Scholar]
- 5.Tsujii M; Kawano S; DuBois RN Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A 1997. 94, 3336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsujii M; Kawano S; Tsuji S; Sawaoka H; Hori M; DuBois RN Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998. 93, 705–16. [DOI] [PubMed] [Google Scholar]
- 7.Xu K; Shu HK EGFR activation results in enhanced cyclooxygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res 2007. 67, 6121–9. [DOI] [PubMed] [Google Scholar]
- 8.Jin S; White E Role of autophagy in cancer: management of metabolic stress. Autophagy 2007. 3, 28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kondo Y; Kondo S Autophagy and cancer therapy. Autophagy 2006. 2, 85–90. [DOI] [PubMed] [Google Scholar]
- 10.Lozy F; Karantza V Autophagy and cancer cell metabolism. Semin Cell Dev Biol 2012. 23, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishina H; Wada T; Katada T Physiological roles of SAPK/JNK signaling pathway. J Biochem 2004. 136, 123–6. [DOI] [PubMed] [Google Scholar]
- 12.Roux PP; Blenis J ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 2004. 68, 320–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stupp R; Hegi ME; Mason WP; van den Bent MJ; Taphoorn MJ; Janzer RC; Ludwin SK; Allgeier A; Fisher B; Belanger K; Hau P; Brandes AA; Gijtenbeek J; Marosi C; Vecht CJ; Mokhtari K; Wesseling P; Villa S; Eisenhauer E; Gorlia T; Weller M; Lacombe D; Cairncross JG; Mirimanoff RO Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009. 10, 459–66. [DOI] [PubMed] [Google Scholar]
- 14.Stupp R; Mason WP; van den Bent MJ; Weller M; Fisher B; Taphoorn MJ; Belanger K; Brandes AA; Marosi C; Bogdahn U; Curschmann J; Janzer RC; Ludwin SK; Gorlia T; Allgeier A; Lacombe D; Cairncross JG; Eisenhauer E; Mirimanoff RO Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005. 352, 987–96. [DOI] [PubMed] [Google Scholar]
- 15.Joki T; Heese O; Nikas DC; Bello L; Zhang J; Kraeft SK; Seyfried NT; Abe T; Chen LB; Carroll RS; Black PM Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res 2000. 60, 4926–31. [PubMed] [Google Scholar]
- 16.Shono T; Tofilon PJ; Bruner JM; Owolabi O; Lang FF Cyclooxygenase-2 expression in human gliomas: prognostic significance and molecular correlations. Cancer Res 2001. 61, 4375–81. [PubMed] [Google Scholar]
- 17.Evans SM; Judy KD; Dunphy I; Jenkins WT; Nelson PT; Collins R; Wileyto EP; Jenkins K; Hahn SM; Stevens CW; Judkins AR; Phillips P; Geoerger B; Koch CJ Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Res 2004. 64, 1886–92. [DOI] [PubMed] [Google Scholar]
- 18.Yang L; Lin C; Wang L; Guo H; Wang X Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp Cell Res 2012. 318, 2417–26. [DOI] [PubMed] [Google Scholar]
- 19.Najim N; Podmore ID; McGown A; Estlin EJ Methionine restriction reduces the chemosensitivity of central nervous system tumour cell lines. Anticancer Res 2009. 29, 3103–8. [PubMed] [Google Scholar]
- 20.Ogawa T; Shishido F; Kanno I; Inugami A; Fujita H; Murakami M; Shimosegawa E; Ito H; Hatazawa J; Okudera T; et al. Cerebral glioma: evaluation with methionine PET. Radiology 1993. 186, 45–53. [DOI] [PubMed] [Google Scholar]
- 21.Sasaki M; Kuwabara Y; Yoshida T; Nakagawa M; Fukumura T; Mihara F; Morioka T; Fukui M; Masuda K A comparative study of thallium-201 SPET, carbon-11 methionine PET and fluorine-18 fluorodeoxyglucose PET for the differentiation of astrocytic tumours. Eur J Nucl Med 1998. 25, 1261–9. [DOI] [PubMed] [Google Scholar]
- 22.Damert A; Machein M; Breier G; Fujita MQ; Hanahan D; Risau W; Plate KH Up-regulation of vascular endothelial growth factor expression in a rat glioma is conferred by two distinct hypoxia-driven mechanisms. Cancer Res 1997. 57, 3860–4. [PubMed] [Google Scholar]
- 23.Tenan M; Fulci G; Albertoni M; Diserens AC; Hamou MF; El Atifi-Borel M; Feige JJ; Pepper MS; Van Meir EG Thrombospondin-1 is downregulated by anoxia and suppresses tumorigenicity of human glioblastoma cells. J Exp Med 2000. 191, 1789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu H; Yang JM; Jin S; Zhang H; Hait WN Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res 2006. 66, 3015–23. [DOI] [PubMed] [Google Scholar]
- 25.Kim SY; Ahn BH; Min KJ; Lee YH; Joe EH; Min DS Phospholipase D isozymes mediate epigallocatechin gallate-induced cyclooxygenase-2 expression in astrocyte cells. J Biol Chem 2004. 279, 38125–33. [DOI] [PubMed] [Google Scholar]
- 26.Abbott KL; Loss JR 2nd; Robida AM; Murphy TJ Evidence that Galpha(q)-coupled receptor-induced interleukin-6 mRNA in vascular smooth muscle cells involves the nuclear factor of activated T cells. Mol Pharmacol 2000. 58, 946–53. [DOI] [PubMed] [Google Scholar]
- 27.Xu K; Gao H; Shu HK Celecoxib can induce vascular endothelial growth factor expression and tumor angiogenesis. Mol Cancer Ther 2011. 10, 138–47. [DOI] [PubMed] [Google Scholar]
- 28.Xu K; Chang CM; Gao H; Shu HK Epidermal growth factor-dependent cyclooxygenase-2 induction in gliomas requires protein kinase C-delta. Oncogene 2009. 28, 1410–20. [DOI] [PubMed] [Google Scholar]
- 29.Xu K; Shu HK Transcription factor interactions mediate EGF-dependent COX-2 expression. Mol Cancer Res 2013. 11, 875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu K; Wang L; Shu HK COX-2 overexpression increases malignant potential of human glioma cells through Id1. Oncotarget 2014. 5, 1241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murono S; Inoue H; Tanabe T; Joab I; Yoshizaki T; Furukawa M; Pagano JS Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc Natl Acad Sci U S A 2001. 98, 6905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seno H; Oshima M; Ishikawa TO; Oshima H; Takaku K; Chiba T; Narumiya S; Taketo MM Cyclooxygenase 2- and prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc(Delta716) mouse intestinal polyps. Cancer Res 2002. 62, 506–11. [PubMed] [Google Scholar]
- 33.Franchi-Gazzola R; Visigalli R; Bussolati O; Dall’Asta V; Gazzola GC Adaptive increase of amino acid transport system A requires ERK1/2 activation. J Biol Chem 1999. 274, 28922–8. [DOI] [PubMed] [Google Scholar]
- 34.Zhang D; Li J; Song L; Ouyang W; Gao J; Huang C A JNK1/AP-1-dependent, COX-2 induction is implicated in 12-O-tetradecanoylphorbol-13-acetate-induced cell transformation through regulating cell cycle progression. Mol Cancer Res 2008. 6, 165–74. [DOI] [PubMed] [Google Scholar]
- 35.Mohammad RM; Muqbil I; Lowe L; Yedjou C; Hsu HY; Lin LT; Siegelin MD; Fimognari C; Kumar NB; Dou QP; Yang H; Samadi AK; Russo GL; Spagnuolo C; Ray SK; Chakrabarti M; Morre JD; Coley HM; Honoki K; Fujii H; Georgakilas AG; Amedei A; Niccolai E; Amin A; Ashraf SS; Helferich WG; Yang X; Boosani CS; Guha G; Bhakta D; Ciriolo MR; Aquilano K; Chen S; Mohammed SI; Keith WN; Bilsland A; Halicka D; Nowsheen S; Azmi AS Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol 2015. 35 Suppl, S78–S103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang KB; Zhu C; Yong SK; Gao Q; Wong MC Enhanced sensitivity of celecoxib in human glioblastoma cells: Induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol Cancer 2009. 8, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shu HK; Kim MM; Chen P; Furman F; Julin CM; Israel MA The intrinsic radioresistance of glioblastoma-derived cell lines is associated with a failure of p53 to induce p21(BAX) expression. Proc Natl Acad Sci U S A 1998. 95, 14453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baumann M; DuBois W; Pu A; Freeman J; Suit HD Response of xenografts of human malignant gliomas and squamous cell carcinomas to fractionated irradiation. Int J Radiat Oncol Biol Phys 1992. 23, 803–9. [DOI] [PubMed] [Google Scholar]
- 39.Ruan S; Okcu MF; Ren JP; Chiao P; Andreeff M; Levin V; Zhang W Overexpressed WAF1/Cip1 renders glioblastoma cells resistant to chemotherapy agents 1,3-bis(2-chloroethyl)-1-nitrosourea and cisplatin. Cancer Res 1998. 58, 1538–43. [PubMed] [Google Scholar]
- 40.Shapiro JR; Pu PY; Mohamed AN; Galicich JH; Ebrahim SA; Shapiro WR Chromosome number and carmustine sensitivity in human gliomas. Cancer 1993. 71, 4007–21. [DOI] [PubMed] [Google Scholar]
- 41.Nelson JS; Tsukada Y; Schoenfeld D; Fulling K; Lamarche J; Peress N Necrosis as a prognostic criterion in malignant supratentorial, astrocytic gliomas. Cancer 1983. 52, 550–4. [DOI] [PubMed] [Google Scholar]
- 42.Shweiki D; Itin A; Soffer D; Keshet E Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992. 359, 843–5. [DOI] [PubMed] [Google Scholar]
- 43.Brat DJ; Castellano-Sanchez AA; Hunter SB; Pecot M; Cohen C; Hammond EH; Devi SN; Kaur B; Van Meir EG Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 2004. 64, 920–7. [DOI] [PubMed] [Google Scholar]
- 44.Edelman MJ; Watson D; Wang X; Morrison C; Kratzke RA; Jewell S; Hodgson L; Mauer AM; Gajra A; Masters GA; Bedor M; Vokes EE; Green MJ Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2 expression is a positive predictive factor for celecoxib + chemotherapy--Cancer and Leukemia Group B Trial 30203. J Clin Oncol 2008. 26, 848–55. [DOI] [PubMed] [Google Scholar]
- 45.Ng K; Meyerhardt JA; Chan AT; Sato K; Chan JA; Niedzwiecki D; Saltz LB; Mayer RJ; Benson AB 3rd; Schaefer PL; Whittom R; Hantel A; Goldberg RM; Venook AP; Ogino S; Giovannucci EL; Fuchs CS Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J Natl Cancer Inst 2015. 107, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips RK; Wallace MH; Lynch PM; Hawk E; Gordon GB; Saunders BP; Wakabayashi N; Shen Y; Zimmerman S; Godio L; Rodrigues-Bigas M; Su LK; Sherman J; Kelloff G; Levin B; Steinbach G A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002. 50, 857–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grossman SA; Olson J; Batchelor T; Peereboom D; Lesser G; Desideri S; Ye X; Hammour T; Supko JG Effect of phenytoin on celecoxib pharmacokinetics in patients with glioblastoma. Neuro Oncol 2008. 10, 190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reardon DA; Quinn JA; Vredenburgh J; Rich JN; Gururangan S; Badruddoja M; Herndon JE 2nd; Dowell JM; Friedman AH; Friedman HS Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer 2005. 103, 329–38. [DOI] [PubMed] [Google Scholar]