

Abstract
Red blood cell (RBC) traits provide insight into a wide range of physiological states and exhibit moderate to high heritability, making them excellent candidates for genetic studies to inform underlying biologic mechanisms. Previous RBC trait genome-wide association studies were performed primarily in European- or Asian-ancestry populations, missing opportunities to inform understanding of RBC genetic architecture in diverse populations and reduce intervals surrounding putative functional SNPs through fine-mapping. Here, we report the first fine-mapping of six correlated (Pearson’s r range: |0.04 – 0.92|) RBC traits in up to 19,036 African Americans and 19,562 Hispanics/Latinos participants of the Population Architecture using Genomics and Epidemiology (PAGE) consortium. Trans-ethnic meta-analysis of race/ethnic- and study-specific estimates for approximately 11,000 SNPs flanking 13 previously identified association signals as well as 150,000 additional array-wide SNPs was performed using inverse-variance meta-analysis after adjusting for study and clinical covariates. Approximately half of previously reported index SNP-RBC trait associations generalized to the trans-ethnic study population (p<1.7×10−4); previously unreported independent association signals within the ABO region reinforce the potential for multiple functional variants affecting the same locus. Trans-ethnic fine-mapping did not reveal additional signals at the HFE locus independent of the known functional variants. Finally, we identified a potential novel association in the Hispanic/Latino study population at the HECTD4/RPL6 locus for RBC count (p=1.9×10−7). The identification of a previously unknown association, generalization of a large proportion of known association signals, and refinement of known association signals all exemplify the benefits of genetic studies in diverse populations.
Keywords: Genomics, fine-mapping, generalization, RBC traits, trans-ethnic meta-analysis
INTRODUCTION
Red blood cell (RBC) trait measurements are used to characterize the physiology of RBCs in both clinical and research settings, are captured by a complete blood count panel, and include the primary traits hematocrit (HCT), hemoglobin (HGB), and RBC count. Accompanying HCT, HGB, and RBC count are three derived traits—mean corpuscular hemoglobin (MCH), MCH concentration (MCHC), and mean corpuscular volume (MCV)—which can be used in combination with primary traits to evaluate RBC development and maintenance. Together, primary and derived RBC trait deficiencies (e.g., abnormally low HGB or excessive RBC count) cause circulatory diseases such as thalassemia, polycythemia, and genetic or nonhereditary anemias1–5. Population-specific HBB causal alleles for recessive diseases such as sickle-cell anemia and β-thalassemia have also been associated with protection against malaria and myocardial infarction, respectively, in the heterozygous state.6–8 Additionally, RBC traits have been associated with stroke, cardiovascular disease (CVD) in populations with chronic kidney disease, and all-cause mortality.9–12 RBC traits are therefore of substantial public health and clinical importance, yet their underlying pathophysiological mechanisms remain incompletely characterized.
As RBC traits exhibit moderate to high heritability (40-90%), population-based genetic analysis of these phenotypes can help identify causal alleles for and inform the underlying biology of RBC-related disorders.3; 13; 14 To date, over 80 independent association signals with one or more RBC traits have been reported, primarily in studies of European- or Asian-ancestry populations15–24. One genome-wide association study (GWAS) performed in over 16,000 African Americans identified 12 genome-wide-significant loci previously reported in European-ancestry or Japanese populations, indicating a shared role for common variants at RBC trait association signals16. However, fine-mapping of RBC trait associations identified in GWAS has had limited success narrowing broad GWAS signals to prioritize functional candidates due to large linkage disequilibrium (LD) blocks or characterizing variants that are rare or monomorphic in Europeans or Asians, as has been demonstrated for platelet count25; 26. Narrowing and fine-mapping of previously identified association signals may be improved by performing analyses in ancestrally diverse populations with multi-continental admixture, including African Americans and Hispanics/Latinos16; 17; 27.
Here, we evaluated 32 index SNP-RBC trait associations in 11 fine-mapped Metabochip regions, previously identified in populations of European-, Japanese-, and South Asian descent (SPTA1, BCL11A, HFE, ABO, HK1, SH2B3/ATXN2, LIPC, PPCDC, NUTF2, NEUROD2, and TMPRSS6) for evidence of generalization and locus refinement in African American and Hispanic/Latino participants of the Population Architecture using Genomics and Epidemiology (PAGE) consortium28. Additionally, we evaluated all SNPs genotyped on the Metabochip for associations not previously described in any of the six RBC traits. These efforts will help address gaps in understanding the genetic underpinnings of RBC traits.
MATERIALS AND METHODS
Study Populations
The PAGE consortium is a National Human Genome Research Institute funded effort to examine the epidemiologic architecture of genetic variants associated with human diseases and traits across diverse populations29. The following PAGE I studies contributed to this manuscript (Supplemental Materials and Methods): the Atherosclerosis Risk in Communities Study (ARIC),30 the Coronary Artery Risk Disease in Young Adults study (CARDIA),31 the Cardiovascular Health Study (CHS),32 the Hispanic Community Health Study/Study of Latinos (HCHS/SOL),33 and the Women’s Health Initiative (WHI)34. The Icahn Mt. Sinai School of Medicine (MSSM) contributed both African American and Hispanic/Latino study populations separately from PAGE I29. The Institutional Review Board at all participating institutions approved the study protocol and all participants gave written consent.
Genotype platforms
The Metabochip was a custom Illumina iSELECT array that contained approximately 195,000 SNPs and was designed to support large scale follow up of putative associations for cardiovascular and metabolic traits28. Further information on genotyping and quality control is provided in the supplemental material. We defined an index SNP as a SNP reported in the GWAS catalog prior to October 1, 2016, as having a genome-wide significant association (5×10−8) with at least one of the six RBC traits we evaluated. Index SNPs that were not directly genotyped on the Metabochip were represented by proxies, defined as SNPs in high (r2 ≥ 0.80) with the GWAS index SNP in the ancestral population in which the association was first reported. For one index SNP, rs671 (ALDH2), no proxy was available because this variant is specific to populations of East Asian ancestry. A total of 74% of participants were directly genotyped on the Illumina custom Metabochip array; genotypes for the remaining participants were imputed from the Affymetrix 6.0 panel35. After QC and study-population-specific effective heterozygosity criteria were applied, 163,929 SNPs were available for analysis in African Americans and 159,467 SNPs were available for analysis in Hispanics/Latinos.
Statistical Analysis
We performed four types of analysis: (1) generalization, whereby we examined 32 index SNP associations across six RBC traits; (2) fine-mapping of association signals that generalized in (1); (3) testing for independent association signals for any RBC trait within one of the 11 densely genotyped regions; and (4) discovery of previously unreported associations with any RBC trait in all remaining Metabochip regions. Only SNPs meeting an effective heterozygosity of 35 were used within each race/ethnic study population; 4,814 SNPs were excluded in Hispanics/Latinos but included for African Americans, whereas 9,431 were excluded for African Americans but included for Hispanics/Latinos. We examined a maximum of 8,082 SNPs in African Americans and 7,991 SNPs in Hispanics/Latinos (9,201 SNPs total) within one of 11 regions densely genotyped on the Illumina Metabochip for all non-discovery analyses.
To interpret fine-mapping results, LD was calculated in 500kb sliding windows using PLINK (http://pngu.mgh.harvard.edu/purcell/plink) and African American (ARIC data), Hispanic/Latino (HCHS/SOL data), and trans-ethnic panels (randomly sampled ARIC and HCHS/SOL participant data in proportion to the racial/ethnic-specific sample population sizes)36. In addition, Metabochip LD and frequency information (but not individual-level information) was provided by the Malmö Diet and Cancer Study on 2,143 control participants from a Swedish population to facilitate LD and MAF comparisons between PAGE African American and Hispanic/Latino populations and populations of European ancestry37. We used NCBI build 36 positions for regional association plots. Recombination rates were estimated from the combined HapMap phase II data.
A weighted version of generalized estimating equations (GEE; HCHS/SOL) accommodating the HCHS/SOL sampling design, relatedness, and household structure was implemented in SUGEN38. Race/ethnic-stratified linear regression was performed for all other studies (Atherosclerosis Risk in Communities [ARIC], Coronary Artery Risk Development in Young Adults [CARDIA], Cardiovascular Health Study [CHS], Icahn Mt. Sinai School of Medicine BioMe Biobank [MSSM], and WHI) using PLINK36. We evaluate the association between each quantitative RBC trait (see Supplement for RBC trait measurement methods and calculations for derived equations, Table S2) and a maximum of 9,201 SNPs (racial/ethnic- and study-specific effective heterozygosity >35, present in more than one study in either African Americans or Hispanics/Latinos) from 11 previously identified RBC trait loci. An additive genetic model was assumed including age, sex, study center/region, and ten ancestry principal components. Racial/ethnic-stratified and trans-ethnic study-specific association results were combined via inverse variance meta-analysis as implemented in METAL39. Genomic inflation factors were not calculated as the design of the Metabochip purposefully emphasizes potential functional candidates, leading to expected early departure from a uniform p-value distribution.
Generalization
We defined an “association signal” as a set of SNPs genotyped in a Metabochip fine-mapped region and exhibiting linkage disequilibrium (r2≥0.2 in the Malmö Diet and Cancer Study) with a previously reported genome-wide significant SNP for one or more RBC traits. For two or more previously reported genome-wide-significant SNPs to be considered within the same association signal in our study, those variants had to be in LD.
We next defined an “index SNP” as the most significant previously reported SNP within an association signal for each RBC trait. In instances for which multiple SNPs were published as the most significant SNP within a particular association signal for the same trait, we selected the SNP with the lowest reported p-value as the index SNP. The index SNP within an association signal may vary by trait due to differences in sample size, measurement error, and allelic heterogeneity among other possible reasons related to genetic architecture of the traits. Therefore, we evaluated the most significant SNP reported for each association signal-trait combination rather than selecting one index SNP to evaluate in all traits for which that association signal was previously reported as genome-wide-significant, even though some of the index SNPs likely tag the same genetic association across multiple RBC traits. For example, the SH2B3/ATXN2 association signal has been reported for multiple RBC traits with the most significant SNP differing by trait, meaning the index SNP for RBC count is rs3184504 whereas the index SNP for hematocrit is rs11065987. These two SNPs are in LD and likely represent the same association signal. Furthermore, while several RBC trait associations examined in this paper were first reported in Japanese populations, those associations have since been generalized to European populations. European LD blocks are typically larger than for African or admixed-ancestry haplotypes, therefore we used European LD to conservatively define loci when analyzing potential independent associations in fine-mapped regions containing previously reported RBC trait GWAS associations.
We then evaluated whether association signals identified in populations of European, Japanese, or South Asian ancestry generalized to African American and Hispanic/Latino populations. Approximately 44% of all previously reported genome-wide-significant RBC trait SNPs, but only 13% of reported association signals (defined above based on European LD, identified as of September 2016), were located in fine-mapped regions on the Metabochip15–24. The generalization significance criterion was then defined as α = 1.7 × 10−4, a Bonferroni-corrected threshold calculated using 294 tag SNPs in African Americans (r2 ≥0.80; determined using African American LD from the ARIC Study) that captured all SNPs correlated with the index SNPs representing 32 index SNP-trait associations as identified in the Malmö Diet Study population.
Fine-Mapping Generalized Associations
We evaluated association-signal narrowing across ancestral backgrounds by comparing the number of SNPs in high LD with the trans-ethnic lead SNP, as well as the width of the region covered by the high-LD SNPs (Table 2, Figures 1, S2). LD for African Americans was calculated using ARIC study participants; LD for Hispanics/Latinos was calculated using HCHS/SOL study participants.
Table 2.
Narrowing of generalized fine-mapped RBC trait association signals using LD with lead SNPs.
| Trait | Locus | Trans-ethnic lead SNP | Trans-ethnic LD SNPs (n) | Trans-ethnic range (kb) | Malmo LD SNPs (n) | Malmo range (kb) | African American LD SNPs (n) | African American range (kb) | Hispanic/Latino LD SNPs (n) | Hispanic/Latino range (kb) | Locus Refinement (kb) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HGB | HFE (1) | rs1799945 | 4 | 37.2 | 11 | 150 | 5 | 46.2 | 5 | 37.2 | 113 |
| MCH | HFE (1) | rs1799945 | 4 | 37.2 | 11 | 150 | 5 | 46.2 | 5 | 37.2 | 113 |
| MCV | HFE (1) | rs1799945 | 4 | 37.2 | 11 | 150 | 5 | 46.2 | 5 | 37.2 | 113 |
| MCH | HFE (2) | rs55925606 | 7 | 280 | 5 | 255 | 7 | 280 | 5 | 220 | – |
| HCTa | ABO (1) | rs635634 | 4 | 5.1 | 5 | 13.1 | 3 | 13.1 | 5 | 13.1 | 8 |
| HGB | ABO (1) | rs495828 | 2 | 0.7 | 5 | 13.1 | 1 | 0.7 | 4 | 13.1 | 12 |
| HGBa | ABO (2) | rs10901252 | 3 | 3.4 | 18 | 79.3 | 5 | 4.6 | 5 | 14.2 | 76 |
| MCHCa | ABO (2) | rs8176722 | 0 | – | 15 | 21.2 | 0 | – | 0 | – | 21 |
| HCT | HK1 | rs72805692 | 0 | – | 1 | 4.6 | 1 | 4.6 | 0 | – | 5 |
| HGB | HK1 | rs72805692 | 0 | – | 1 | 4.6 | 1 | 4.6 | 0 | – | 5 |
| HCTa | SH2B3/ATXN2 | rs10774625 | 10 | 1022 | 3 | 123 | 5 | 188 | 5 | 188 | – |
| HGBa | SH2B3/ATXN2 | rs10774631 | 18 | 173.7 | 41 | 181 | 18 | 176 | 19 | 174 | 7 |
| HCT | TMPRSS6 | rs855791 | 0 | – | 1 | 6.7 | 0 | – | 0 | – | 7 |
| HGB | TMPRSS6 | rs855791 | 0 | – | 1 | 6.7 | 0 | – | 0 | – | 7 |
| MCH | TMPRSS6 | rs855791 | 0 | – | 1 | 6.7 | 0 | – | 0 | – | 7 |
| MCHC | TMPRSS6 | rs4820268 | 0 | – | 1 | 6.7 | 0 | – | 0 | – | 7 |
| MCV | TMPRSS6 | rs855791 | 0 | – | 1 | 6.7 | 0 | – | 0 | – | 7 |
SNPs reported as in LD if MAF > 0.01 in the study population and r2>0.8 with the lead SNP.
Previously reported association did not generalize when only study populations reporting all six RBC traits were considered. African American LD represented by the ARIC study population; Hispanic/Latino LD represented by the HCHS/SOL study population. PAGE LD was calculated using relevant proportions of African Americans and Hispanic/Latinos to represent trans-ethnic study population (see Methods).
Figure 1.
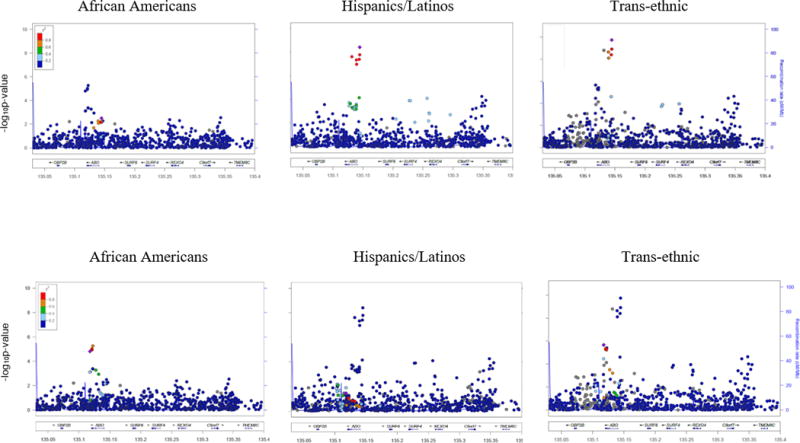
a. HGB association signal at ABO (1) generalizes to Hispanics/Latinos but not African Americans.
b. HGB association signal at ABO (2) generalizes to African Americans but not Hispanics/Latinos.
Independent and Discovery SNP Identification
To identify independent SNPs influencing RBC traits, we identified all SNPs at the 11 RBC trait loci that were uncorrelated with the index SNPs (r2 < 0.20 in the Malmö Diet and Cancer Study). Sequential conditional analyses were then performed by adjusting for significant racial/ethnic-specific lead SNPs. If a statistically significant association was identified, defined as 0.05 divided by the number of SNPs in African Americans with MAF ≥ 0.01 that were uncorrelated with the index SNPs (n=8,907; α = 5.61 × 10−6), the SNP was identified as independent and added to the adjustment set. Sequential conditional analysis was repeated until no significant SNPs were identified. We evaluated all remaining SNPs for discovery in African Americans or Hispanics/Latinos using a Metabochip-wide significant threshold of 0.05/155,022 (the number of SNPs available for evaluation after exclusion of SNPs evaluated for generalization), or α = 3.23 × 10−7, and only considered SNPs with an effective heterozygosity >35 in more than one cohort study population per race/ethnicity.
Bioinformatic Characterization of RBC Trait loci
For each of the significant RBC trait SNPs (i.e., any lead SNP that generalized in one or both race/ethnic populations or the trans-ethnic meta-analysis; or any novel SNP identified in either race/ethnic population), all SNPs in LD (r2 ≥ 0.8) were identified in the appropriate 1000 Genomes reference superpopulations (AFR [Africans] for African Americans and AMR [Admixed Americans] for Hispanic/Latinos) for functional annotation. Using HaploRegV240, all variants in each LD block were characterized with putative functional roles including: conservation; promoter and/or enhancer epigenetic markers, derived from the Roadmap Epigenomics Project41 and ENCODE;42 DNAse hypersensitive sites; and transcription factor binding motifs calculated as a library of position weight matrices.43–45 Evidence of functional activity considered promoter and enhancer regions based on histone modification patterns in k562 erythroleukemia cells in the Roadmap Epigenome Project; DNase hypersensitive sites in ENCODE tissues including erythroblasts and erythroid leukemia cell lines; and transcription factor binding evidence in k562 erythroid leukemia cells. Evidence of cis-eQTL status was performed using Blueprint for relevant blood tissues46. All functional elements that varied by cell type were restricted to RBC-relevant tissues (Tables S12a, S12b).
In order to evaluate the relevance of trans-ethnic PAGE lead SNPs across tissue types, we compared the eQTL status of index SNPs in both blood-relevant and other tissues (Table S13). We looked up significant eQTLs for each index SNP (p<1E-06) in whole blood in GTEx, which provides data on a wide array of tissues; and two blood-specific eQTL databases: the blood eQTL browser; and the NESDA NTR Conditional eQTL catalog47–49. Only GTEx tissues which showed an association with at least one SNP are reported. We further reported clinical relevance of the trans-ethnic lead SNPs as described in the literature50; 51.
RESULTS
We analyzed six correlated RBC traits (Pearson’s correlation coefficient range: -0.29 to 0.92 in Hispanic Community Health Study/Study of Latinos (HCHS/SOL) participants; Tables S1, S2) in a maximum of 19,036 African American and 19,562 Hispanic/Latino participants from six studies participating in the PAGE consortium (Table S3). Females were over-represented among both African Americans (83%) and Hispanic/Latinos (70%). The HCHS/SOL (n=11,675) and Women’s Health Initiative (WHI, n=17,363, of which 12,022 are African American) studies contributed the largest proportion of Hispanic/Latino (60%) and African American (63%) participants, respectively.
Generalization and fine-mapping of 11 densely genotyped Metabochip regions
Generalization
First, we examined 11 regions densely genotyped on the Illumina Metabochip and harboring one or more variants previously associated at genome-wide significance (p<5×10−8) with at least one RBC trait (Table S4). All but two of the 11 regions contained one association signal, with the HFE and ABO regions each containing two association signals (see Methods). Of these 13 association signals, eight were previously associated with two or more RBC traits and two were previously associated with four traits, for a total of 32 index SNP-trait associations (Tables 1, S5, S6).
Table 1.
Association results examining evidence of generalization to a PAGE trans-ethnic population for 13 RCB trait association signals.
| Trait (N, AfAm) (N, Hisp) |
Association Signal | Published index
SNP |
Trans-ethnic Lead SNPb
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trans-ethnic Population
|
African Americans |
Hispanics/Latinos |
|||||||||||||
| Index SNP | Pop** | CA | 1000G CAF Range | SNP | CA | Beta (SE) | p-value | CAF | Index LD (r2) | p-value | CAF | Index LD (r2) | p-value | ||
| HCT
(19,036) (19,562)0 |
HFE (2) | rs1800562 | EU | A | 0.00 – 0.05 | rs78273613 | A | −0.17 (0.078) | 0.03 | 0.98 | 0.67 | 0.21 | 0.86 | 0.39 | 0.08 |
| ABO (1)* | rs495828 | JP | T | 0.13 – 0.22 | rs635634 | T | −0.21 (0.038) | 5.1×10−8 | 0.11 | 0.75 | 0.01 | 0.15 | 0.86 | 5.9×10−7 | |
| HK1* | rs16926246 | EU | C | 0.84 – 1.00 | rs72805692 | A | −0.37 (0.061) | 8.2×10−10 | 0.98 | 0.08 | 2.7×10−3 | 0.93 | 0.52 | 8.0×10−8 | |
| SH2B3/ATXN2* | rs11065987 | EU | G | 0.01 – 0.42 | rs10774625 | A | 0.15 (0.034) | 1.7×10−5 | 0.09 | 0.83 | 3.3×10−3 | 0.29 | 0.86 | 1.1×10−3 | |
| TMPRSS6* | rs2143450f | EU | A | 0.43 – 0.70 | rs855791 | A | −0.22 (0.029) | 8.8×10−14 | 0.16 | 0.15 | 5.4×10−3 | 0.44 | 0.73 | 8.1×10−13 | |
|
| |||||||||||||||
| HGB
(19,036) (19,562) |
HFE (1)* | rs198846 | EU, SA | A | 0.03 – 0.16 | rs1799945 | C | −0.10 (0.017) | 1.8×10−9 | 0.97 | 0.24 | 8.6×10−6 | 0.88 | 0.82 | 9.6×10−6 |
| HFE (2) | rs1800562 | EU | A | 0.00 – 0.05 | rs55925606 | A | −0.13 (0.036) | 5.4×10−4 | 0.99 | 0.90 | 0.05 | 0.98 | 0.84 | 4.3×10−3 | |
| ABO (1)* | rs495828 | JP | T | 0.13 – 0.22 | rs495828 | T | −0.08 (0.012) | 1.8×10−10 | 0.14 | 1† | 3.2×10−3 | 0.17 | 1† | 3.8×10−9 | |
| ABO (2)* | rs7853989f | EU, SA | T | 0.06 – 0.20 | rs10901252 | C | 0.06 (0.014) | 2.2×10−6 | 0.16 | 0.94 | 1.5×10−5 | 0.07 | 0.97 | 0.04 | |
| HK1* | rs16926246 | EU | C | 0.84 – 1.00 | rs72805692 | A | −0.13 (0.021) | 1.4×10−10 | 0.98 | 0.08 | 8.1×10−3 | 0.93 | 0.52 | 4.6×10−9 | |
| rs10159477 | EU, SA | A | 0.00 – 0.14 | 0.10 | 0.56 | ||||||||||
| SH2B3/ATXN2* | rs11065987 | EU | A | 0.58 – 0.99 | rs10774631 | A | −0.05 (0.011) | 1.9×10−6 | 0.22 | 0.02 | 1.9×10−4 | 0.19 | 0.06 | 2.7×10−3 | |
| rs3184504 | EU, SA | T | 0.01 – 0.47 | 0.03 | 0.07 | ||||||||||
| LIPC | rs1532085 | EU, SA | A | 0.37 – 0.52 | rs2414577 | T | 0.02 (0.009) | 0.08 | 0.60 | 0.24 | 0.99 | 0.61 | 0.74 | 0.01 | |
| TMPRSS6* | rs855791 | EU | A | 0.13 – 0.56 | rs855791 | A | −0.11 (0.010) | 1.3×10−25 | 0.16 | 1† | 2.1×10−5 | 0.44 | 1† | 6.1×10−23 | |
|
| |||||||||||||||
| RBC
(6,389) (14,460) |
BCL11A | rs2540913f | EU, SA | T | 0.43 – 0.76 | rs17027944 | A | −0.0026 (0.0013) | 0.05 | 0.30 | 0.34 | 0.08 | 0.20 | 0.17 | 0.01 |
| ABO (1) | rs495828 | JP | T | 0.13 – 0.22 | rs635634 | T | −0.0027 (0.0017) | 0.11 | 0.10 | 0.75 | 0.22 | 0.15 | 0.86 | 0.18 | |
| rs579459 | EU, SA | T | 0.78 – 0.87 | 0.74 | 0.87 | ||||||||||
| SH2B3/ATXN2 | rs3184504 | EU, SA | T | 0.01 – 0.47 | rs10849944 | T | 0.0039 (0.0012) | 9.9×10−4 | 0.50 | 0.09 | 0.22 | 0.66 | 0.17 | 9.9×10−4 | |
| NUTF2 | rs2271294 | EU, SA | A | 0.26 – 0.97 | rs73612222 | A | −0.0036 (0.0016) | 0.02 | 0.87 | 0.02 | 2.8×10−3 | 0.84 | 0.34 | 0.46 | |
| NEUROD2 | rs8182252f | EU, SA | T | 0.16 – 0.32 | rs14050 | T | −0.0026 (0.0012) | 0.03 | 0.46 | 0.12 | 0.09 | 0.57 | 0.30 | 0.35 | |
|
| |||||||||||||||
| MCH
(6,386) (14,343) |
BCL11A | rs13027161f | EU, SA | T | 0.43 – 0.76 | rs2058703 | A | 0.0022 (0.0009) | 0.01 | 0.68 | 0.15 | 0.05 | 0.71 | 0.22 | 0.08 |
| HFE (1)* | rs198846 | EU, SA | A | 0.03 – 0.16 | rs1799945 | C | −0.0088 (0.0014) | 6.7×10−10 | 0.97 | 0.24 | 0.09 | 0.89 | 0.82 | 1.8×10−9 | |
| HFE (2)* | rs1800562 | NR | A | 0.00 – 0.05 | rs55925606 | A | −0.0126 (0.0032) | 8.8×10−5 | 0.99 | 0.90 | 0.04 | 0.98 | 0.84 | 8.7×10−4 | |
| rs1408272 | EU | T | 0.95 – 1.00 | 0.97 | 0.77 | ||||||||||
| rs17342717 | NR | T | 0.00 – 0.08 | 0.73 | 0.44 | ||||||||||
| TMPRSS6* | rs855791 | EU, SA | A | 0.13 – 0.56 | rs855791 | A | −0.0096 (0.0008) | 1.0×10−30 | 0.15 | 1† | 2.6×10−3 | 0.43 | 1† | 6.8×10−27 | |
| rs4820268 | EU, NR | A | 0.43 – 0.70 | 0.15 | 0.73 | ||||||||||
| rs2143450 | EU | A | 0.43 – 0.70 | 0.15 | 0.73 | ||||||||||
|
| |||||||||||||||
| MCHC
(19,027) (19,553) |
SPTA1 | rs857684f | EU, SA | C | 0.11 – 0.44 | rs863931 | A | 0.0009 (0.0003) | 1.5×10−3 | 0.33 | 0.04 | 6.7×10−3 | 0.40 | 0.24 | 0.19 |
| rs857721f | EU | C | 0.11 – 0.44 | 0.04 | 0.24 | ||||||||||
| ABO (2)* | rs8176746 | JP | T | 0.06 – 0.20 | rs8176722 | A | 0.0018 (0.0005) | 1.7×10−4 | 0.14 | 0.76 | 2.3×10−4 | 0.09 | 0.76 | 0.14 | |
| TMPRSS6* | rs855791 | EU, SA | A | 0.13 – 0.56 | rs4820268 | A | 0.0021 (0.0003) | 1.9×10−11 | 0.73 | 0.15 | 0.04 | 0.54 | 0.73 | 1.4×10−11 | |
| rs4820268 | NR | A | 0.43 – 0.70 | 1† | 1† | ||||||||||
|
| |||||||||||||||
| MCV
(6,397) (14,411) |
SPTA1 | rs3737515 | EU | C | 0.21 – 0.30 | rs952094 | A | −0.0008 (0.0007) | 0.20 | 0.58 | 0.33 | 0.04 | 0.47 | 0.21 | 0.87 |
| BCL11A | rs243070f | EU, SA | A | 0.43 – 0.84 | rs17402905 | T | −0.0018 (0.0010) | 0.06 | 0.86 | 0.30 | 0.37 | 0.84 | 0.20 | 0.12 | |
| rs2540917f | EU | T | 0.43 – 0.76 | 0.30 | 0.20 | ||||||||||
| HFE (1)* | rs198846 | EU, SA | A | 0.03 – 0.16 | rs1799945 | C | −0.0052 (0.0012) | 2.6×10−5 | 0.97 | 0.24 | 0.04 | 0.89 | 0.82 | 3.0×10−4 | |
| HFE (2) | rs1800562 | NR | A | 0.00 – 0.05 | rs74662487 | A | −0.0060 (0.0016) | 2.7×10−4 | 0.97 | <0.01 | 0.02 | 0.94 | <0.01 | 2.8×10−3 | |
| rs1408272 | EU, SA | T | 0.95 – 1.00 | 0.25 | 0.10 | ||||||||||
| HK1 | rs16926246 | EU, SA | T | 0.00 – 0.14 | rs72805692 | A | −0.0042 (0.0016) | 9.7×10−3 | 0.98 | 0.08 | 0.07 | 0.93 | 0.52 | 0.04 | |
| PPCDC | rs8028632 | EU, SA | T | 0.41 – 0.77 | rs79269642 | T | −0.0028 (0.0013) | 0.02 | 0.94 | 0.02 | 0.37 | 0.90 | 0.11 | 0.04 | |
| TMPRSS6* | rs855791 | EU, SA | A | 0.13 – 0.56 | rs855791 | A | −0.0069 (0.0007) | 1.0×10−20 | 0.15 | 1† | 5.8×10−3 | 0.43 | 1† | 2.4×10−20 | |
| rs4820268 | EU, NR | A | 0.43 – 0.70 | 0.15 | 0.73 | ||||||||||
Allele frequencies obtained from HaploReg, v4.1, for 1000 Genomes Phase 3 global populations: AFR = African, AMR = American, ASN = Asian, and EUR = European. Alleles presented on the positive strand.
Restricted to SNPs with effective heterozygosity > 30.
Index SNP not included on the Metabochip; proxy SNP substituted (see Table S5).
Index SNP and lead SNP are the same. AfAm, African American; Hisp, Hispanic; CAF, coded allele frequency; SNP, single nucleotide polymorphism;
Independent signals that generalized to the trans-ethnic PAGE population at a significance threshold of α = 1.70×10−4.
Pop = published GWAS study population for the first report of the index SNP. EU = European; JP = Japanese; SA = South Asian; NR = Not Reported: Kullo, et al (2010) used electronic medical record data including patients from six potential race/ethnicity categories, but did not report the frequency of subpopulations or adjust for race/ethnicity in their linear regression analysis.
Seventeen of the 32 index SNP-trait associations (53%) generalized at p<1.7×10−4 to the trans-ethnic study population (Tables 1, S7, S8), of which six trans-ethnic lead SNPs were identical to the previously reported index SNP. Of the remaining 11 generalized associations, nine trans-ethnic lead SNP p-values exceeded the index SNP p-values by at least an order of magnitude (Tables 1, S10). Effect sizes for both generalized and non-generalized association signals for index SNPs and trans-ethnic lead SNPs were consistent with previously reported estimates (Table S6)17.
The first HFE association signal (index SNP: rs198846) generalized with the same trans-ethnic lead SNP (rs1799945, the functional H63D hemochromatosis variant) to all three previously reported traits—HGB, MCH, and MCV. Furthermore, both ABO association signals (Figure 1) and the SH2B3/ATXN2 association signal generalized to all traits except RBC count. Notably, RBC count was the only trait for which none of the index SNP generalized in the trans-ethnic population; it also was the trait with the smallest sample size (46% of the maximum number of participants). Association signals for SPTA1, BCL11A, LIPC, NUTF2, PPCDC, and NEUROD2 did not generalize. Six non-generalized index SNP-trait associations could not be evaluated for directional consistency because a proxy SNP was used in generalization analyses or the effect size was not reported in the initial publication (Table S9). For the remaining eight non-generalized index SNP-trait associations with sufficient information to evaluate directional consistency, six were directionally consistent in the trans-ethnic population. Additionally, when compared to SNP-trait associations from a previously published RBC trait GWAS, seven of 11 PAGE lead SNPs exceeded the generalization significance threshold in 24,167 participants of the CHARGE consortium (Table S14)17.
In race/ethnicity-specific meta-analyses (Tables 2, S6-S8), 9% (n=3) of index SNP-trait associations generalized to African Americans and generalization was limited to HGB. Conversely, 38% (n=12) of index SNP-trait associations generalized to Hispanics/Latinos, including the SH2B3/ATXN2 association with RBC count, representing the only instance where evidence of generalization for the RBC trait was detected. Of note, HCT, HGB, and MCHC were reported for similar numbers of Hispanics/Latino and African American participants in our study population, whereas MCH, MCV, and RBC count were reported for more than twice as many Hispanics/Latinos as African Americans, potentially contributing to the disparity in generalization by race/ethnicity.
Novel independent signals in 11 fine-mapped regions
We next evaluated the 11 fine-mapped regions to identify significant variants independent of published association signals by examining all SNPs that were uncorrelated with any of the index SNPs (see Methods). We identified no independent associations in previously reported regions for any of the six RBC traits (significance threshold: p<1.3×10−5).
Fine-Mapping
To fine-map association signals that generalized, we then evaluated the LD structure in the trans-ethnic study population and by race/ethnicity (Figures 1 & S2, Table 2). The median reduction in interval width was 75%, likely due to the large reduction in association signals for which the index variant was functional but fell within a large LD block in Europeans. The first HFE association signal showed consistent evidence of narrowing across three traits (113kb decrement), with the same trans-ethnic lead SNP for all traits (the causal H63D variant rs1799945, CAF = 0.97 in African Americans, CAF = 0.88 in Hispanics/Latinos). Both ABO association signals (the latter of which has the determining variant for blood type B, rs8176746, as the published index SNP) fine-mapped to a limited number of SNPs in narrow LD blocks in the trans-ethnic study population, with the trans-ethnic lead SNP varying by trait. Rs855791 is a known functional coding variant in TMPRSS6, therefore we do not consider this signal to be narrowed.
Discovery
Next, all SNPs outside the 11 previously identified RBC trait-associated regions were evaluated for evidence of discovery associations (p < 3.03×10−7, see Methods). No SNP association exceeded Metabochip-wide significance in the total trans-ethnic study population or among African Americans for any RBC trait. However, one previously unreported association met Metabochip-wide significance in Hispanics/Latinos: rs76350043 at HECTD4/RPL6 (chr 12q24.13, p=2.5×10−7) for RBC count (Table S11). This SNP was also nominally significant for HCT and HGB (p < 0.05).
In Silico Bioinformatics Analysis
All SNPs highly correlated (r2≥0.9 in relevant AFR or AMR 1000 Genomes Phase I ancestral populations) with trans-ethnic lead SNPs from generalization analysis were examined using publicly available functional prediction data for erythrocytes or erythroblastoid cell lines, as well as pathogenicity prediction (Tables S12a, S12b)52. With the exception of the well-established TMPRSS6 missense variant rs855791 (generalized to HCT, HGB, MCH, MCHC, and MCV), all trans-ethnic lead variants and their LD proxies were noncoding variants. Lead SNPs and their LD proxies most commonly exhibited potential regulatory effects including disruption of RBC-relevant transcription factor consensus sequences and sites exhibiting DNase I activity. Several SNPs at generalized loci exhibited promise for molecular characterization, including rs198851, the HCT lead SNP in Hispanics/Latinos at the first HFE association signal. In k562 erythroid leukemia cells, rs198851 exhibits both DNase and enhancer activity, is an eQTL for TRIM38, and is located within an RNA Polymerase II ChIP-seq peak. With the exception of one association signal (TMPRSS6 in both African Americans and Hispanics/Latinos), all independent signals contained at least one SNP with evidence for a regulatory function or cis-eQTL activity in relevant blood cell types (Tables S12a, S12b).
We also evaluated tissue specificity of significant eQTLs (p<1E-06) for each published index SNP or trans-ethnic lead SNP for all generalized association signals, as well as the putative clinical relevance of each SNP when information was available (Table S13)47–51. EQTL results show varied evidence of tissue expression effects. Lead or index variants for SH2B3/ATXN2 and TMPRSS6 fine-mapped regions demonstrated no significant association with any gene expression in any tissue type. In contrast, SNPs within the second ABO association signal showed evidence of broad ABO expression across 30 tissue types. SNPs in the first ABO association signal show evidence of expression for multiple genes, but across fewer tissues than the second signal. Lead and index SNPs within the second HFE association signal were only associated with expression of genes other than HFE across a broad array of GTEx tissues; no lead or index SNPs for the second HFE association signal exhibited eQTL activity for the HFE gene in any tissue type. The first HFE association signal showed some evidence of tissue specificity in gene expression profiles—both the index SNP and trans-ethnic lead SNP exhibited cis-eQTLs for either HFE or several other genes, but overlap by tissue type was uncommon in this association signal.
DISCUSSION
In this study we performed generalization, fine-mapping, and discovery analysis of six RBC traits in a population of over 38,000 African American and Hispanic/Latino PAGE participants. We demonstrated that genetic regions influencing RBC traits identified in European- and Asian-ancestry populations are also applicable to African American and Hispanic/Latino populations. The merits of incorporating multi-ethnic study populations in genomic studies were also displayed via locus refinement and identification of a previously unreported RBC trait association that warrants validation in future studies.
In the eleven fine-mapped regions we evaluated, over half of known index SNP-trait associations generalized to the trans-ethnic study population across all six RBC traits, indicating that the effects of known RBC loci are likely shared across ancestral populations. Additionally, ten of 17 generalized associations (59%) met or exceeded the more stringent genome-wide significance threshold of 5×10−8 in the trans-ethnic study population. Although some association signals showed variation in lead SNP, the trans-ethnic lead SNPs almost always matched across traits when we restricted to participants with all traits measured (results not shown). The higher proportion of generalized associations in Hispanics/Latinos compared to African Americans suggests that results in Hispanic/Latino populations may contribute disproportionately to the larger trans-ethnic findings. This was not surprising given the Metabochip design that was enriched for European ancestral content as well as Hispanic/Latino genetic architecture, which shares more features with European-ancestry or Asian-ancestry individuals than does African American architecture53. Additionally, the SNPs designated as proxies for index SNPs discovered in European- or Japanese-ancestry individuals were almost always in much lower LD in African Americans than Hispanics/Latinos, suggesting that previously reported index SNPs are not highly effective for characterizing the genetic architecture of RBC traits in African Americans.
We also detected several instances where trans-ethnic lead SNPs showed considerably stronger evidence of association with RBC traits in our study population than previously reported GWAS index SNPs identified in primarily European or East Asian populations. By examining visualizations of generalized association signals, we further identified several cases in which the lead SNP in LD with the European index SNP was not the most significant SNP in the region, indicating differences. These findings are consistent with recent work in large trans-ethnic populations, which demonstrated considerable effect heterogeneity by genetic ancestry in GWAS index SNPs reported in studies of predominantly European populations; considerably less evidence of heterogeneity was detected when examining index SNPs identified in multi-ethnic populations54. Of particular relevance are recent demonstrations of inappropriate designation of variants which are rare in European populations as pathogenic when they are in fact common in other ancestral groups55. GWAS inclusive of diverse populations can improve the accuracy of identifying functional variants, but fine-mapping is particularly well-suited to this type of exercise. As interest in genetic risk scores for RBC traits increases, e.g., to predict adverse outcomes in pregnancy, cardiovascular and neurologic diseases, and mortality, studies examining generalization of reported loci to global populations will become even more important, particularly in the era of precision medicine22; 56–59.
Over the past decade, GWAS have identified hundreds of loci associated with RBC traits, but these findings incompletely account for the population-level variability attributable to additive genetic effects. A possible explanation for this missing heritability is that all genes expressed within RBC-relevant tissues play a role in RBC trait biology, but their identification may require infinite statistical power60. A recent review described the suite of genes affecting complex traits as including both “core” genes (i.e., those with tissue-specific effects crucial to one or few complex traits) and “peripheral” genes (i.e., those with broad expression profiles playing a role in many traits)61. Distinguishing core from peripheral genes may inform canonical pathways for RBC traits, provide mechanistic insight into biology, and inform targets for pharmaceutical intervention60; 61. Importantly, this designation occurs on a spectrum, with some genes not clearly predisposed to one class over the other. For example, hexokinase 1 (HK1) is highly and ubiquitously expressed, and several GWAS have identified associations within 200kb of HK1 for psychiatric phenotypes, autoimmune disorders, and blood metabolite levels17; 62. However, HK1 was also the only generalized association signal in our study with evidence of a blood-specific eQTL, which localized to a narrow segment of intron four representing GWAS study populations of multiple ancestries. The approximately 10kb segment contains multiple regulatory elements (e.g., DNase hypersensitivity regions and histone methylation marks), but GWAS findings to-date for this region remain restricted to RBC traits, and RBC trait index SNPs within 500kb of this region remain restricted to this narrow genomic fragment. These results reinforce the concept that tissue-specific regulators may play an important role for individual complex traits in broadly expressed genes. In light of this information and other complex-trait GWAS findings, tissue-specific expression data and genomic information will be particularly relevant when considering candidate variants for functional studies.
Large-scale genetic evaluation of correlated traits is challenging, particularly when evaluating multiple populations and traits with variable sample sizes. Importantly, novel statistical methods scalable to GWAS that leverage correlation among phenotypes for novel locus discovery have been reported56–59; 63. Such approaches seem particularly well suited for RBC traits, given evidence of a shared genetic architecture and the number of GWAS associations which have been reported in multiple RBC traits17; 22. Regarding fine-mapping, extensions of correlated-phenotype methods were recently described and have similarly shown promise for reducing sets of SNPs for functional evaluation over single trait methods. However, no studies to date have leveraged such innovations for discovery or fine-mapping of RBC traits.
Finally, we identified a potential novel association at the HECTD4/RPL6 locus for RBC count. The HECTD4/RPL6 locus has been previously associated with MCV (which exhibits modest correlation with RBC count and HGB), blood pressure, coronary heart disease, and multiple metabolic traits64–68. Additionally, coding mutations within several members of the ribosomal protein gene family have been causally associated with Diamond-Blackfan anemia, making an association with RBC count plausible69; 70. This association signal fell within a sparsely genotyped region on the Metabochip, and hence could not be further evaluated via fine-mapping. Evidence of association with multiple non-RBC traits should motivate larger efforts to understand whether the mechanisms underlying these associations are shared across traits or whether, for instance, tissue-specific effects relevant to each trait are represented by the same signal. Multi-ethnic fine-mapping to narrow the association signal for molecular characterization likely represents an ideal first-step, as functional variants in this region have not been described for other associated traits.
This study faced several limitations which deserve consideration. First, phenotype availability differed by study and smaller sample sizes for MCH, MCV, and RBC count likely reduced power, specifically among the African American study population. Second, five of eleven fine-mapped regions we evaluated (SPTA1, BCL11A, HK1, LIPC, and TMPRSS6) were mapped narrowly (<100kb) with few SNPs, potentially providing insufficient coverage of African American or Hispanic/Latino genetic content to perform comprehensive fine-mapping and generalization analyses. Sparse coverage in the SPTA1 and BCL11A regions could also contribute to lack of generalization at these loci, at which the respective genes have established, functional roles in RBC development and maintenance71–73. With regard to bioinformatic characterization for functional candidate SNP evaluation, eQTL analysis—while insufficient as the sole determinant of tissue specificity—is an important component for ascertaining functional status of candidate variants. Finally, the Metabochip design emphasized regions identified for cardiometabolic traits, so overlap with RBC-trait associations was coincidental; we therefore could not examine generalization or fine-mapping in several well established RBC trait associations, including HBS1L/MYB, LUC7L/ITGF3, HBA1/2, and HBB17; 18; 21; 74.
CONCLUSION
Population-based GWAS emphasize discovery, and are often the first step toward elucidating the genetic architecture underlying complex quantitative traits like RBC traits. Fine-mapping previously reported associations—particularly associations identified in genetically homogeneous populations, including European- and East Asian-ancestry populations—provides additional information about known association signals and can lead to narrowing of broad association signals to reduce the burden for bioinformatics and molecular functional analysis. Additional characterization of genetic associations contributing to population-level variability of RBC traits through large-scale sequencing and methods exploiting the correlation of RBC traits may further illuminate biological pathways for these complex quantitative traits.
Supplementary Material
Acknowledgments
(a) The Population Architecture Using Genomics and Epidemiology (PAGE) program is funded by the National Human Genome Research Institute (NHGRI), supported by U01HG004803 (CALiCo), U01HG004790 (WHI), and U01HG004801 (Coordinating Center), and their respective NHGRI ARRA supplements. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The complete list of PAGE members can be found at http://www.pagestudy.org.
(b) The data and materials included in this report result from a collaboration between the following studies:
Funding support for the “Epidemiology of putative genetic variants: The Women’s Health Initiative” study is provided through the NHGRI PAGE program (U01HG004790 and its NHGRI ARRA supplement). The WHI program is funded by the National Heart, Lung, and Blood Institute; NIH; and U.S. Department of Health and Human Services through contracts N01WH22110, 24152, 32100-2, 32105-6, 32108-9, 32111-13, 32115, 32118-32119, 32122, 42107-26, 42129-32, and 44221. The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whiscience.org/publications/WHI_investigators_shortlist.pdf.
Funding support for the Genetic Epidemiology of Causal Variants Across the Life Course (CALiCo) program was provided through the NHGRI PAGE program (U01HG004803 and its NHGRI ARRA supplement). The following studies contributed to this manuscript and are funded by the following agencies: The Atherosclerosis Risk in Communities (ARIC) Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, N01-HC-55022. The Cardiovascular Health Study (CHS) is supported by contracts HHSN268201200036C, HHSN268200800007C, N01 HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants HL080295 and HL087652 from the National Heart, Lung, and Blood Institute (NHLBI), with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org/PI.htm. CHS GWAS DNA handling and genotyping at Cedars-Sinai Medical Center was supported in part by the National Center for Research Resources, grant UL1RR033176, and is now at the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124; in addition the National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
Assistance with phenotype harmonization, SNP selection and annotation, data cleaning, data management, integration and dissemination, and general study coordination was provided by the PAGE Coordinating Center (U01HG004801-01 and its NHGRI ARRA supplement). The National Institutes of Mental Health also contributes to the support for the Coordinating Center.
The PAGE consortium thanks the staff and participants of all PAGE studies for their important contributions.
Works Cited
- 1.Taliaferro WH, Huck JG. The Inheritance of Sickle-Cell Anaemia in Man. Genetics. 1923;8:594–598. doi: 10.1093/genetics/8.6.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williamson GR, Crawford R. Fatal Mediterranean (Cooley’s) anemia. New Orleans Med Surg J. 1945;98:280–284. [PubMed] [Google Scholar]
- 3.Whitfield JB, Martin NG. Genetic and environmental influences on the size and number of cells in the blood. Genet Epidemiol. 1985;2:133–144. doi: 10.1002/gepi.1370020204. [DOI] [PubMed] [Google Scholar]
- 4.Lamson PD. The Processes Taking Place in the Body by Which the Number of Erythrocytes Per Unit Volume of Blood is Increased in Acute Experimental Polycythaemia. Proc Natl Acad Sci U S A. 1916;2:365–369. doi: 10.1073/pnas.2.7.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neel JV, Valentine WN. Further Studies on the Genetics of Thalassemia. Genetics. 1947;32:38–63. doi: 10.1093/genetics/32.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chami N, Lettre G. Lessons and Implications from Genome-Wide Association Studies (GWAS) Findings of Blood Cell Phenotypes. Genes. 2014;5:51–64. doi: 10.3390/genes5010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hashemi M, Shirzadi E, Talaei Z, Moghadas L, Shaygannia I, Yavari M, Amiri N, Taheri H, Montazeri H, Shamsolkottabi H. Effect of heterozygous beta-thalassaemia trait on coronary atherosclerosis via coronary artery disease risk factors: a preliminary study. Cardiovascular journal of Africa. 2007;18:165–168. [PMC free article] [PubMed] [Google Scholar]
- 8.Wang CH, Schilling RF. Myocardial infarction and thalassemia trait: an example of heterozygote advantage. American journal of hematology. 1995;49:73–75. doi: 10.1002/ajh.2830490112. [DOI] [PubMed] [Google Scholar]
- 9.Franczuk P, Kaczorowski M, Kucharska K, Franczuk J, Josiak K, Zimoch W, Kosowski M, Reczuch K, Majda J, Banasiak W, et al. Could an analysis of mean corpuscular volume help to improve a risk stratification in non-anemic patients with acute myocardial infarction? Cardiol J. 2015 doi: 10.5603/CJ.a2015.0031. [DOI] [PubMed] [Google Scholar]
- 10.Panwar B, Judd SE, Warnock DG, McClellan WM, Booth JN, 3rd, Muntner P, Gutierrez OM. Hemoglobin Concentration and Risk of Incident Stroke in Community-Living Adults. Stroke. 2016;47:2017–2024. doi: 10.1161/STROKEAHA.116.013077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barlas RS, Honney K, Loke YK, McCall SJ, Bettencourt-Silva JH, Clark AB, Bowles KM, Metcalf AK, Mamas MA, Potter JF, et al. Impact of Hemoglobin Levels and Anemia on Mortality in Acute Stroke: Analysis of UK Regional Registry Data, Systematic Review, and Meta-Analysis. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.003019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solak Y, Yilmaz MI, Saglam M, Demirbas S, Verim S, Unal HU, Gaipov A, Oguz Y, Kayrak M, Caglar K, et al. Mean corpuscular volume is associated with endothelial dysfunction and predicts composite cardiovascular events in patients with chronic kidney disease. Nephrology (Carlton) 2013;18:728–735. doi: 10.1111/nep.12130. [DOI] [PubMed] [Google Scholar]
- 13.Evans DM, Frazer IH, Martin NG. Genetic and environmental causes of variation in basal levels of blood cells. Twin research: the official journal of the International Society for Twin Studies. 1999;2:250–257. doi: 10.1375/136905299320565735. [DOI] [PubMed] [Google Scholar]
- 14.Wright FA, Sullivan PF, Brooks AI, Zou F, Sun W, Xia K, Madar V, Jansen R, Chung W, Zhou YH, et al. Heritability and genomics of gene expression in peripheral blood. Nature genetics. 2014;46:430–437. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambers JC, Zhang W, Li Y, Sehmi J, Wass MN, Zabaneh D, Hoggart C, Bayele H, McCarthy MI, Peltonen L, et al. Genome-wide association study identifies variants in TMPRSS6 associated with hemoglobin levels. Nature genetics. 2009;41:1170–1172. doi: 10.1038/ng.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, Tang H, Qayyum R, Schick UM, Nalls MA, Handsaker R, Li J, Lu Y, Yanek LR, Keating B, et al. Genome-wide association analysis of red blood cell traits in African Americans: the COGENT Network. Human molecular genetics. 2013;22:2529–2538. doi: 10.1093/hmg/ddt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesh SK, Zakai NA, van Rooij FJ, Soranzo N, Smith AV, Nalls MA, Chen MH, Kottgen A, Glazer NL, Dehghan A, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nature genetics. 2009;41:1191–1198. doi: 10.1038/ng.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, Kamatani N. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nature genetics. 2010;42:210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
- 19.Kullo IJ, Ding K, Jouni H, Smith CY, Chute CG. A genome-wide association study of red blood cell traits using the electronic medical record. PloS one. 2010;5 doi: 10.1371/journal.pone.0013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Glessner JT, Zhang H, Hou C, Wei Z, Bradfield JP, Mentch FD, Guo Y, Kim C, Xia Q, et al. GWAS of blood cell traits identifies novel associated loci and epistatic interactions in Caucasian and African-American children. Human molecular genetics. 2013;22:1457–1464. doi: 10.1093/hmg/dds534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soranzo N, Spector TD, Mangino M, Kuhnel B, Rendon A, Teumer A, Willenborg C, Wright B, Chen L, Li M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nature genetics. 2009;41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Harst P, Zhang W, Mateo Leach I, Rendon A, Verweij N, Sehmi J, Paul DS, Elling U, Allayee H, Li X, et al. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012;492:369–375. doi: 10.1038/nature11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira MA, Hottenga JJ, Warrington NM, Medland SE, Willemsen G, Lawrence RW, Gordon S, de Geus EJ, Henders AK, Smit JH, et al. Sequence variants in three loci influence monocyte counts and erythrocyte volume. Am J Hum Genet. 2009;85:745–749. doi: 10.1016/j.ajhg.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Q, Kathiresan S, Lin JP, Tofler GH, O’Donnell CJ. Genome-wide association and linkage analyses of hemostatic factors and hematological phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S12. doi: 10.1186/1471-2350-8-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, Yu F, Gibbs RA, Genomes P, Bustamante CD. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 2011;108:11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schick UM, Jain D, Hodonsky CJ, Morrison JV, Davis JP, Brown L, Sofer T, Conomos MP, Schurmann C, McHugh CP, et al. Genome-wide Association Study of Platelet Count Identifies Ancestry-Specific Loci in Hispanic/Latino Americans. Am J Hum Genet. 2016;98:229–242. doi: 10.1016/j.ajhg.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Human molecular genetics. 2008;17:R156–165. doi: 10.1093/hmg/ddn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, Burtt NP, Fuchsberger C, Li Y, Erdmann J, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matise TC, Ambite JL, Buyske S, Carlson CS, Cole SA, Crawford DC, Haiman CA, Heiss G, Kooperberg C, Marchand LL, et al. The Next PAGE in understanding complex traits: design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. American journal of epidemiology. 2011;174:849–859. doi: 10.1093/aje/kwr160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. American journal of epidemiology. 1989;129:687–702. [PubMed] [Google Scholar]
- 31.Friedman GD, Cutter GR, Donahue RP, Hughes GH, Hulley SB, Jacobs DR, Jr, Liu K, Savage PJ. CARDIA: study design, recruitment, and some characteristics of the examined subjects. Journal of clinical epidemiology. 1988;41:1105–1116. doi: 10.1016/0895-4356(88)90080-7. [DOI] [PubMed] [Google Scholar]
- 32.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 33.Daviglus ML, Talavera GA, Aviles-Santa ML, Allison M, Cai J, Criqui MH, Gellman M, Giachello AL, Gouskova N, Kaplan RC, et al. Prevalence of major cardiovascular risk factors and cardiovascular diseases among Hispanic/Latino individuals of diverse backgrounds in the United States. JAMA. 2012;308:1775–1784. doi: 10.1001/jama.2012.14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.WHI Study Group. Design of the Women’s Health Initiative clinical trial and observational study. The Women’s Health Initiative Study Group. Controlled clinical trials. 1998;19:61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Li Y, Browning SR, Browning BL, Slater AJ, Kong X, Aponte JL, Mooser VE, Chissoe SL, Whittaker JC, et al. Performance of genotype imputation for rare variants identified in exons and flanking regions of genes. PloS one. 2011;6:e24945. doi: 10.1371/journal.pone.0024945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minisymposium: The Malmo Diet and Cancer Study. Design, biological bank and biomarker programme. 23 October 1991, Malmo, Sweden. J Intern Med. 1993;233:39–79. doi: 10.1111/j.1365-2796.1993.tb00645.x. [DOI] [PubMed] [Google Scholar]
- 38.Lin DY, Tao R, Kalsbeek WD, Zeng D, Gonzalez F, 2nd, Fernandez-Rhodes L, Graff M, Koch GG, North KE, Heiss G. Genetic association analysis under complex survey sampling: the Hispanic Community Health Study/Study of Latinos. Am J Hum Genet. 2014;95:675–688. doi: 10.1016/j.ajhg.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic epidemiology. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roadmap Epigenomics, C. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Pena-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, 3rd, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature biotechnology. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, Bock C, Boehm B, Campo E, Caricasole A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nature biotechnology. 2012;30:224–226. doi: 10.1038/nbt.2153. [DOI] [PubMed] [Google Scholar]
- 47.Consortium, E.P. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jansen R, Hottenga JJ, Nivard MG, Abdellaoui A, Laport B, de Geus EJ, Wright FA, Penninx B, Boomsma DI. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Human molecular genetics. 2017;26:1444–1451. doi: 10.1093/hmg/ddx043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Koebbe BC, Nielsen C, Hirst M, Farnham P, et al. The Human Epigenome Browser at Washington University. Nat Methods. 2011;8:989–990. doi: 10.1038/nmeth.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Research. 2016;44:D877–D881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conomos MP, Reiner AP, Weir BS, Thornton TA. Model-free Estimation of Recent Genetic Relatedness. Am J Hum Genet. 2016;98:127–148. doi: 10.1016/j.ajhg.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, Highland HM, Patel YM, S EP, Avery CL, Belbin GM, Bien SA, Cheng I, Hodonsky CJ, Huckins LM, Jeffs J, Justice AE, Kocarnik JM, Lin BM, Lu YK, Nelson SC, Park SL, Preuss M, Richard MA, Schurmann C, S VW, Vahi K, Vishnu A, Verbanck M, Walker R, Young KL, Zubair N, Ambite JL, Boerwinkle E, Bottinger EP, Bustamante CD, Caberto C, Conomos MP, Deelman E, Do R, D K, Fernandez-Rhodes L, Fornage M, Heiss G, Hindorff LA, Jackson RD, James R, Laurie CA, Laurie CC, Li Y, Lin DY, Nadkarni G, Pankow J, Pooler LC, Reiner AP, Romm J, S C, Sheng X, Stahl E, Stram DO, Thornton TA, Wassel CL, Wilkens LR, Yoneyama S, Buyske S, Haiman C, Kooperberg C, LeMarchand L, Loos RJF, Matise TC, North KE, Peters U, Kenny EE, Carlson CS. Genetic Diversity Turns a New PAGE in Our Understanding of Complex Traits. 2017 (In review) [Google Scholar]
- 55.Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med. 2016;375:655–665. doi: 10.1056/NEJMsa1507092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim J, Bai Y, Pan W. An Adaptive Association Test for Multiple Phenotypes with GWAS Summary Statistics. Genet Epidemiol. 2015;39:651–663. doi: 10.1002/gepi.21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan W, Kim J, Zhang Y, Shen X, Wei P. A powerful and adaptive association test for rare variants. Genetics. 2014;197:1081–1095. doi: 10.1534/genetics.114.165035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pasaniuc B, Price AL. Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet. 2016 doi: 10.1038/nrg.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kichaev G, Roytman M, Johnson R, Eskin E, Lindstrom S, Kraft P, Pasaniuc B. Improved methods for multi-trait fine mapping of pleiotropic risk loci. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btw615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakravarti A, Turner TN. Revealing rate-limiting steps in complex disease biology: The crucial importance of studying rare, extreme-phenotype families. Bioessays. 2016;38:578–586. doi: 10.1002/bies.201500203. [DOI] [PubMed] [Google Scholar]
- 61.Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rawofi L, Edwards M, Krithika S, Le P, Cha D, Yang Z, Ma Y, Wang J, Su B, Jin L, et al. Genome-wide association study of pigmentary traits (skin and iris color) in individuals of East Asian ancestry. PeerJ. 2017;5:e3951. doi: 10.7717/peerj.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei P, Cao Y, Zhang Y, Xu Z, Kwak IY, Boerwinkle E, Pan W. On Robust Association Testing for Quantitative Traits and Rare Variants. G3 (Bethesda) 2016;6:3941–3950. doi: 10.1534/g3.116.035485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016;167:1415–1429 e1419. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, Kelly TN, Saleheen D, Lehne B, Mateo Leach I, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nature genetics. 2015;47:1282–1293. doi: 10.1038/ng.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Rooij FJ, Qayyum R, Smith AV, Zhou Y, Trompet S, Tanaka T, Keller MF, Chang LC, Schmidt H, Yang ML, et al. Genome-wide Trans-ethnic Meta-analysis Identifies Seven Genetic Loci Influencing Erythrocyte Traits and a Role for RBPMS in Erythropoiesis. Am J Hum Genet. 2017;100:51–63. doi: 10.1016/j.ajhg.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, Tay WT, Chen CH, Zhang Y, Yamamoto K, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nature genetics. 2011;43:531–538. doi: 10.1038/ng.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ligthart S, Vaez A, Hsu YH, Inflammation Working Group of the, C.C. Pmi Wg XCP, LifeLines Cohort, S. Stolk R, Uitterlinden AG, Hofman A, Alizadeh BZ, et al. Bivariate genome-wide association study identifies novel pleiotropic loci for lipids and inflammation. BMC Genomics. 2016;17:443. doi: 10.1186/s12864-016-2712-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Petrtylova K, Mihal V, Stary J, Cerna Z, Jabali Y, Pospisilova D. Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum Mutat. 2009;30:321–327. doi: 10.1002/humu.20874. [DOI] [PubMed] [Google Scholar]
- 70.Konno Y, Toki T, Tandai S, Xu G, Wang R, Terui K, Ohga S, Hara T, Hama A, Kojima S, et al. Mutations in the ribosomal protein genes in Japanese patients with Diamond-Blackfan anemia. Haematologica. 2010;95:1293–1299. doi: 10.3324/haematol.2009.020826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bauer DE, Orkin SH. Hemoglobin switching’s surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr Opin Genet Dev. 2015;33:62–70. doi: 10.1016/j.gde.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008;141:367–375. doi: 10.1111/j.1365-2141.2008.07091.x. [DOI] [PubMed] [Google Scholar]
- 73.Mankelow TJ, Satchwell TJ, Burton NM. Refined views of multi-protein complexes in the erythrocyte membrane. Blood Cells Mol Dis. 2012;49:1–10. doi: 10.1016/j.bcmd.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hodonsky CJ, Jain D, Schick UM, Morrison JV, Brown L, McHugh CP, Schurmann C, Chen DD, Liu YM, Auer PL, et al. Genome-wide association study of red blood cell traits in Hispanics/Latinos: The Hispanic Community Health Study/Study of Latinos. PLoS Genet. 2017;13:e1006760. doi: 10.1371/journal.pgen.1006760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.