

Abstract
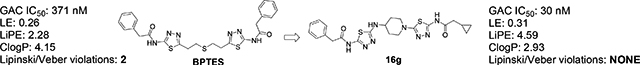
A novel set of GAC (Kidney Glutaminase Isoform C) inhibitors able to inhibit the enzymatic activity of GAC and the growth of the triple negative MDA-MB-231 breast cancer cells with low nanomolar potency is described. Compounds in this series have a reduced number of rotatable bonds, improved ClogPs, microsomal stability and ligand efficiency when compared to the leading GAC inhibitors BPTES and CB-839. Property improvements were achieved by the replacement of the flexible n-diethylthio or the n-butyl moiety present in the leading inhibitors by heteroatom substituted heterocycloalkanes.
Keywords: GAC, Novel glutaminase inhibitors, BPTES, CB-839
Graphical Abstract
1. Introduction
In 1955, Eagle, in his paper “Nutrition needs of mammalian cultured cells” reported that glutamine, a non-essential amino acid, is quite essential for the growth of tumor cells in culture media.1 Since that report, research in tumor cell metabolism has revealed that high glutamine utilization and dependence, a property termed “glutamine addiction”, is a key attribute for a number of tumor cell lines.2,3,4,5,6
The key step in glutamine processing in mitochondria is the hydrolysis of the glutamine amide group by the enzyme glutaminase. There are two main glutaminase isoforms, the kidney isoform (KGA or GLS1) and the liver isoform (LGA or GLS2). Starting with the 1969 paper of Knox et al., where it was shown that kidney glutaminase activity is proportional to the growth rate of tumors in rats, the evidence accumulated over the years point to the fact that KGA and its splice variant kidney glutaminase isoform C (GAC) particularly, is a target of interest for cancer therapy.7,8,9 GAC upregulation is present in multiple cancer cell lines, correlates with increased proliferative rates, and it is linked to the dysregulation of a number of pathways, including the dysregulation/amplification of the Myc oncogene.10,11,12,13
Inhibition of GAC through antisense, siRNA and/or synthetic molecules like BPTES, CB-839 and compound 968 leads to reduction of tumor cell proliferation in vitro and in vivo and suggests that glutaminase is a valid anticancer target.14,15,16,17,18 Among the selective small molecule GAC inhibitors, BPTES and CB-839 are the most similar, having as a key feature the presence of a lipophilic connecting chain (diethylthio in BPTES and n-butyl in CB-839) between two heterocyclic aromatic moieties (Figure 1). Being straight and lipophilic these chains contribute to the relatively high ClogPs and the high number of rotatable bonds (NRB) of these compounds (BPTES: ClogP=4.15, NRB=12; CB-839: ClogP=4.74, NRB=13). Herein we describe a novel and potent set of inhibitors with NRB values within the generally accepted drug-like range (≤10),19 improved ligand efficiency (LE),20 lipophilic efficiency (LiPE/LLE)21 and/or ClogPs when compared to the leading inhibitors.
Figure 1.

Glutaminase inhibitors
2. Results and Discussion
2.1. Design principles for new compounds
Catalytically active GAC units are tetrameric and recent evidence suggests that in cells GAC may in fact operate as an oligomer of tetramers.22
With respect to structural information there are three crystal structures of human GAC in complex with BPTES in the Protein Data Bank (PDB), namely structures 3UO9, 3VOZ and 3VP1.23,24 These structures show that BPTES binds in a stoichiometry of 2 molecules of inhibitor per GAC tetramer and at an allosteric pocket that is formed at the interface between GAC dimers (Figure 2).
Figure 2.

A & B: Binding of BPTES to glutaminase as appears in the 3UO9 x-ray structure. C: Heat map of B-factors for the BPTES atoms in the 3UO9 structure
Looking at the available BPTES/GAC crystal structures and particularly the bent conformation assumed by the thiadiazole-connecting diethylthio chain, it became apparent to us that this flexible connector could be replaced by small to medium size ring systems (Figure 3). Morphing this diethylthio chain connector into a cyclic structure would be highly beneficial as it would result in inhibitors with reduced number of rotatable bonds, a property inversely related to the probability of good absorption.19 An added benefit of this decrease in rotatable bonds would be a reduction in the entropic energy penalty for binding, that is inherently higher in molecules with a high number of rotatable bonds, and as such it could lead to greater potency inhibitors.25
Figure 3.

Design principles for new GAC inhibitors
As a means of maintaining the logP as low as possible, we envisioned the use of saturated ring systems that contained other-than-sulfur heteroatoms as surrogates for the conformation assumed by the BPTES flexible chain. Ease of synthesis considerations and our desire to have more than one heteroatom present on the small to medium size ring systems that would not clash with the walls of the binding pocket suggested to us that heteroatom substituents on prospective saturated ring systems should serve as connectors between the BPTES thiadiazoles and/or their isosters, and not as stand-alone substituents. In that regard, non-sulfur-containing ring systems such as 4-hyrdoxypiperidine, 4-aminopiperidine, 3-amino azetidine, etc. appeared as very suitable heteroatom containing rigid surrogates for the flexible connector chains of BPTES/CB-839.
B-factors in the 3UO9 x-ray structure suggest that one of the BPTES phenyls is particularly flexible/mobile (Figure 2c).23 This suggests that this phenyl moiety most likely does not contribute significantly to binding. As such, this phenyl group and possibly the whole phenylacetic acid moiety in that part of the molecule, could be replaced by smaller groups or perhaps completely eliminated from new compounds thus yielding compounds with even better properties. A recent paper on a series of BPTES analogs with flexible connector chains by the Tsukamoto group suggested that removal of one of the two phenylacetic acid moieties may indeed be viable.26
2.2. Chemistry
In order to assess the viability of replacing the flexible BPTES side chain with heteroatom containing saturated rings, and to also explore the possibility of replacing both of the phenyl moieties in constrained analogs with smaller groups, we pursued the synthesis of the “symmetrically” acylated compounds in Tables 1 and 2.
Table 1.
Properties and activity of “symmetrically” acylated bis-thiadiazoles with diamine containing saturated rings as surrogates for the flexible diethylthio moiety of BPTES
| Cmpd | Structure | MW | CLogPa | NRB | GAC IC50 (nM)b | MDA-MB-231 IC50 (nM)b | LEc | LiPEd |
|---|---|---|---|---|---|---|---|---|
| BPTES | 524.68 | 4.15 | 12 | 371 | 2,610 | 0.26 | 2.28 | |
| CB-839 | 571.57 | 4.74 | 13 | 180/25e | 33 | 0.23/0.24f | 1.99/2.86f | |
| 7a | 534.66 | 4.31 | 8 | >5,000 | NTg | NAg | NAg | |
| 7b | 520.63 | 4.25 | 8 | 3,070 | NTg | 0.21 | 1.26 | |
| 7c | 534.66 | 3.80 | 9 | 29 | 70 | 0.28 | 3.72 | |
| 7d | 520.63 | 3.74 | 9 | 70 | 1,200 | 0.27 | 3.41 | |
| 7e | 520.63 | 3.74 | 9 | 30 | 1,570 | 0.29 | 3.78 | |
| 7f | 534.66 | 3.74 | 10 | 50 | 320 | 0.28 | 3.56 | |
| 7g | 368.44 | 0.07 | 5 | 7,700 | >3,000 | 0.29 | 5.04 | |
| 7h | 368.44 | 0.07 | 5 | 720 | >3,000 | 0.36 | 6.07 |
Calculated using the ClogP module of the on-line/evaluation version of MavinSketch from ChemAxon
IC50 values are the result of a single experiment run in triplicate
LE= ΔG/N = −1.4log(IC50)/N
LiPE=pIC50-logP
Reported in ref 16
Calculated using the literature reported IC50
NT: Not tested; NA: Not assessed
Table 2.
Properties and activity of “symmetrically” acylated bis-thiadiazole with saturated hydroxysubstituted rings as surrogates for the flexible diethylthio moiety of BPTES
| Cmpd | Structure | MW | CLogPa | NRB | GAC IC50 (nM)b | MDA-MB-231 IC50(nM)b | LEc | LiPEd |
|---|---|---|---|---|---|---|---|---|
| 14a | 507.59 | 4.04 | 9 | 60 | >3,000 | 0.28 | 3.18 | |
| 14b | 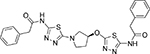 |
521.62 | 4.10 | 9 | 40 | 680 | 0.28 | 3.29 |
| 14c | 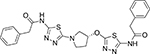 |
521.62 | 4.10 | 9 | 36 | 420 | 0.29 | 3.42 |
| 14d | 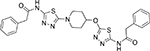 |
535.64 | 4.16 | 9 | 30 | 140 | 0.28 | 3.36 |
| 14e | 535.64 | 4.62 | 9 | 490 | 3,000 | 0.23 | 1.68 | |
| 14f | 535.64 | 4.22 | 10 | 30 | 230 | 0.28 | 3.30 | |
| 14g | 535.64 | 4.22 | 10 | 790 | >3000 | 0.23 | 1.88 | |
| 14h | 549.67 | 4.63 | 10 | 190 | >3000 | 0.25 | 2.09 | |
| 14i | 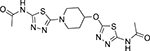 |
383.45 | 0.50 | 5 | 1,005 | >3,000 | 0.33 | 5.48 |
| 14j | 463.58 | 2.43 | 9 | 157 | 630 | 0.31 | 4.37 |
Calculated using the ClogP module of the on-line/evaluation version of MavinSketch from ChemAxon
IC50 values are the result of a single experiment run in triplicate
LE=ΔG/N= −1.4log(IC50)/N
LiPE=pIC50-logP
Compounds 7a-e (Table 1) were prepared via the reaction of 2-amino-5-bromothiadiazole (4) with 0.5 eq. of a cyclic diamines 5a-e and then acylation of the corresponding constrained bis thiadiazole intermediates 6a-e with phenylacetyl chloride (Scheme 1). Compounds 7g and 7h were prepared from bisthiadiazoles 6d and 6e after acylation with acetic anhydride (Scheme 1). Compound 7f was prepared from the known (±)-1-N-Boc-1,3-diaminopentane 827 by a reaction with 2-amino-5-bromothiadiazole to afford the N-Boc protected intermediate 9, followed by Boc deprotection, another reaction with 2-amino-5-bromothiadiazole and then acylation with phenylacetyl chloride (Scheme 1).
Scheme 1.

Synthesis of compounds 7a-h (Table 1). Reagents and conditions: a) NaHCO3 or Et3N or DIEA, EtOH, 80°C, sealed tube; b) 2 eq. of PhCH2COCl, Et3N, DMF, rt.; c) 6d or 6e, 2 eq. of (Ac)2O, Et3N, DMF, rt.; d) 30% TFA in CH2Cl2; e) NaHCO3, 4, EtOH 80 °C.
The preparation of compounds 14a-h (Table 2) involved first the building of the 2-aminothiadiazole moiety on the N-Boc-protected cyclic amino alcohols 10a-h to afford the corresponding intermediates 12a-h, then Boc deprotection, a reaction with 2-amino-5-bromothiadiazole and finally acylation of the resulting intermediates 13a-h with phenylacetyl chloride (Scheme 2).
Scheme 2.

Synthetic methods for preparation of compounds 14a-j and 16a-d. Reagents and conditions: a) NaH, CS2, MeI, THF, rt; b) NH2NH2 MeOH, rt; c) BrCN, Et3N, MeOH, rt; d) 4N HCl in dioxane, rt; e) 4, Et3N, EtOH or DMF, 80 °C, sealed tube.; (f) PhCH2COCl, Et3N, DMF, rt; g) 13d, (Ac)2O, Et3N, DMF, rt.; h) 13d, EDCI, cyclopropylacetic acid, DMF, rt.; i) 12a-d, PhCH2COCl, Et3N, DMF, rt.
Compounds 14i and 14j were prepared from their common bis thiadiazole intermediate 13d via acylation with either acetic acid anhydride or cyclopropyl acetic acid and EDCI respectively.
To test if removal of one of the phenyl/phenacetyl moieties from potent derivatives is tolerated we prepared the selected analogs shown in Table 3.
Table 3.
Properties and activity of constrained bis-thiadiazoles that lack one phenylacetic acid moiety
| Cmpd | Structure | MW | CLogPa | NRB | GAC IC50 (nM) b | MDA-MB-231 IC50(nM) b | LEc | LiPEd |
|---|---|---|---|---|---|---|---|---|
| 16a | 389.46 | 2.14 | 6 | 540 | >3,000 | 0.33 | 4.10 | |
| 16b | 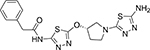 |
403.48 | 2.20 | 6 | 9,700 | >3,000 | 0.26 | 2.81 |
| 16c | 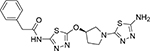 |
403.48 | 2.20 | 6 | 1,400 | >3,000 | 0.30 | 3.65 |
| 16d | 417.51 | 2.26 | 6 | 207 | 980 | 0.33 | 4.42 | |
| 16e | 388.47 | 1.77 | 6 | 227 | >3,000 | 0.35 | 4.87 | |
| 16f | 416.52 | 1.89 | 6 | NTe | NTe | NAe | NAe | |
| 16g | 498.62 | 2.93 | 9 | 30 | 90 | 0.31 | 4.59 |
Calculated using the ClogP module of the on-line/evaluation version of MavinSketch from ChemAxon
IC50 values are the result of a single experiment run in triplicate
LE=ΔG/N= −1.4log(IC50)/N
LiPE=pIC50-logP
NT: Not tested; NA: Not assessed
Derivatives 16a-d were prepared from the corresponding N-Boc intermediates 12a-d (Scheme 2) in three steps that involved first the reaction of 12a-d with phenylacetyl chloride to afford the corresponding N-phenylacetyl intermediates 15a-d, then Boc deprotection and reaction with 2-amino-5-bromothiadiazole. Compounds 16e and 16f were prepared from commercially available 2-amino-1-N-Boc-azetidine and 4-amino-1-N-Boc-piperidine, respectively, via reaction with 2-amino-5-bromothiadiazole, then acylation followed by Boc deprotection and again reaction with 2-amino-5-bromothiazole (Scheme 3). Compound 16g was prepared from 16f via acylation with cyclopropyl acetic acid and HATU.
Scheme 3.

Synthesis of compounds 16e-g (Table 3). Reagents and conditions: a) NaHCO3, EtOH, 80 °C; b) PhCH2COCl, Et3N, DMF, rt; c) 4N HCl in dioxane, rt.; d) 4, NaHCO3, EtOH, 80 °C, sealed tube.; e) phenylacetic acid, HATU, Et3N, DMF, rt.; f) cyclopropyl acetic acid, HATU, Et3N, DMF, rt.
Finally, with objective to expand our flexible-chain-rigidification strategy to analogs in which one or both of the BPTES thiadiazoles have bioisosterically been replaced by pyridazine, pyridines or pyrazine we prepared compounds 26a-f in Table 4.
Table 4.
Properties and activity of selected non bis-thiadiazole constrained derivatives
| Cmpd | Structure | MW | CLogPa | NRB | GAC IC50 (nM)b | MDA-MB-231 IC50(nM)b | LEc | LiPEd |
|---|---|---|---|---|---|---|---|---|
| 26a | 529.61 | 4.01 | 9 | 36 | 580 | 0.27 | 3.38 | |
| 26b | 523.59 | 3.86 | 9 | 98 | 2,200 | 0.25 | 3.14 | |
| 26c | 523.59 | 3.38 | 9 | >5,000 | NTe | NAe | NAe | |
| 26d | 521.61 | 4.63 | 9 | >5,000 | NTe | NAe | NAe | |
| 26e | 521.61 | 4.63 | 9 | >5,000 | NTe | NAe | NAe | |
| 26f | 521.61 | 4.63 | 9 | >5,000 | NTe | NAe | NAe |
Calculated using the ClogP module of the on-line/evaluation version of MavinSketch from ChemAxon
IC50 values are the result of a single experiment run in triplicate
LE= ΔG/N =−1.4log(IC50)/N
LiPE=pIC50-logP
NT: Not tested; NA: Not assessed
The synthesis of compounds 26a and 26c began with the reaction of 2,6-dichloropyridazine (19) or 2,5-dibromopyrazine (20) with 4-hydroxy-1-N-Boc-piperidine, under aromatic nucleophilic substitution conditions, to afford intermediates 21 and 22 which were then elaborated further to 26a and 26c as described in Scheme 4. The synthesis of 26b was effected via the initial reaction of 2,6-dichloropyridazine (19) with 4-hydroxypiperidine to afford intermediate 27 followed by the introduction of the second pyridazine moiety, amination and then acylation with phenylacetic acid (Scheme 4). A key step in the synthesis of compounds 26a-c was the Buchwald Pd-catalyzed amination of intermediates 21, 28 and 29 with benzophenone imine.28
Scheme 4.

Synthesis of 26a-c. Reagents and conditions: a) 4-hydroxy-1-N-Boc piperidine, NaH, THF or THF/DMSO 4:1, 50°C; b) 21, benzophenone imine, (±)-BINAP, Pd2(dba)3, Cs2CO3, toluene, 90 °C, sealed tube c) NH2OH.HCl, NaOAc, MeOH, rt.; d) phenylacetic acid, Et3N, HATU, DMF; e) 4N HCl dioxane, rt,; f) 2-amino-5-bromothiadiazole (4), Et3N, DMF, 80 °C; g) 19, Et3N, DMSO, 4-hydroxypiperidine, 50 °C; h) 19, NaH, THF, 50 oC i) 2–4 eq. benzophenone imine, (±)-BINAP, Pd2(dba)3, Cs2CO3, toluene, 90 °C, sealed tube, j) 22, 4N HCl, dioxane, then 20, Et3N, DMSO, 50 °C.
Finally, compounds 26d-f were prepared from intermediates 35, 38 and 39, respectively (Scheme 5). These intermediates were assessed via the reaction of 4-hydroxypiperidine with either 2-nitro-5-chloropyridine or 2-chloro-5-nitropyridine, under aromatic nucleophilic substitution conditions, and transformed further to compounds 24d-f upon introduction of a second nitropyridine moiety, followed by a nitro group reduction and then acylation with phenylacetic acid and HATU (Scheme 5).
Scheme 5.

Synthesis of compounds 26d-f. Reagents and conditions: a) 32, 4-hydroxypiperidine, K2CO3, DMSO, 90 °C; b) 34, 2-nitro-5-hydroxypyridine, Ph3P, DIAD, THF, rt. c) Fe, NH4Cl aq. EtOH, 80 °C d) phenylacetic acid, HATU, Et3N, DMF, rt.; e) 33, 4-hydroxypiperidine, Et3N, EtOH, 90 °C, sealed tube; f) 33, NaH, THF/DMSO (5:1), to afford 38; g) 33, NaH, 50 °C, THF to afford 39 h) H2, 10% Pd/C, EtOH/EtOAc (1:1).
2.3. Evaluation of novel compounds
All novel analogs were evaluated in a biochemical assay against recombinant GAC and against the MDA-MB-231 cancer cell line, using BPTES and CB-839 as comparators.
An overview of the data in Tables 1–4 suggests that the BPTES/CB-839 flexible chains can indeed be replaced by an appropriate heteroatom substituted ring system. Focusing on Tables 1 and 2 it is evident that small to medium size heteroatom substituted ring moieties, such as 4-piperidinyl, 3-pyrrolidinyl or 2-azetidinyl are good surrogates for the conformation assumed by the flexible BPTES connector chain. Compounds containing those rings are up to 6–10 fold more potent than BPTES in the GAC enzymatic assay (Tables 1 and 2). Larger rings or rings that cannot quite capture the flexible connector chain conformation seen in the 3UO9 x-ray structure, for instance the rings in compounds 7a, 7b (Table 1) and 14e (Table 2), lead to potency loss.
The GAC activity of compounds 7d,e,g,h and 14b,c,g,f which contain a pyrrolidine ring system (Tables 1 and 2), shows that there is a chirality preference for one of the two enantiomers.
Comparison of the GAC activity of potent bis-phenyl analogs and their direct bis-des-phenyl derivatives, for instance the a comparison between the activity of derivatives 7d, 7e and 14d with that of their bis-des-phenyl analogs 7g, 7h and 14i respectively (Tables 1 and 2), shows that removal of both phenyls from potent analogs leads to potency loss. However, as the data with compounds 14d, 14i and 14j in Table 2 indicate, this potency loss may be reversed to a great degree when the smaller cyclopropyl ring is introduced as replacement for the phenyl moiety.
Complete removal of only one of the phenylacetyl groups from potent compounds leads to derivatives with reduced potency in the GAC assay relative to that of their parent molecules (Tables 1–3). In this set, the piperidinyl and azetidinyl analogs suffered the least potency loss.
To assess if introduction of a cyclopropylacetic acid moiety may help in reversing the potency loss observed in analogs that lack a phenylacetic acid moiety, in effect test the premise that one phenyl ring may be replaced by a smaller group, and at the same time assess the viability of mixed phenylacetic acid cyclopropylacetic acid derivatives as lead optimization candidates, we prepared analog 16g (Table 3). Compound 7c (Table 1) was the best overall lead in our compound sets, as such, the most efficient way in answering this question was to synthesize 16g and see if its activity could match or approximate that of its parent 7c. What we found was the activity of 16g was practically identical to that of the parent compound 7c.
The GAC potency values for compounds 26a and 26b in Table 4 suggest that our chain-rigidification strategy may be expanded to derivatives in which one or both thiadiazole moieties have been replaced by a pyridazine ring.
The data in Table 4 also suggest that pyrazine or pyridine rings may not be ideal replacements for the thiadiazole/pyridazine ring systems.
Overall, potent leads in this series, defined as compounds with IC50≤100 nM in the GAC assay, have NRB ≤10 and better LE, LiPE and/or logPs when compared to the leading inhibitors BPTES and CB-839.
With respect to cell potency, compounds with 4-hydroxypiperidinyl and 4-aminopiperidinyl ring systems appear to consistently outperform derivatives with other ring systems which exhibit similar potency in the GAC enzyme assay (Tables 1–3). This trend does not appear to be ClogP driven and at this time is not quite understood.
Selected potent analogs 7c, 7f, 14c, 14d and 16g were tested in a human liver microsome (HLM) stability assay to ascertain if the absence of a labile group, such as sulfur, and/or their improved ClogP translates to better stability. Data from these experiments, shown in Figure 4, denote that these compounds have indeed improved microsomal stability over their comparators.
Figure 4.

HLM stability of compounds 7c, 7f, 14c, 14d, 16g, BPTES and CB-839
X-ray structures of GAC in complex with constrained derivatives
To confirm that our leads bind in the BPTES binding pocket and possibly understand the potency differences among the chiral derivatives with pyrrolidine rings we solved the crystal structure of GAC in complex with 7d, 7e, and 14d (shown in Figures 5 and 6).29 An additional x-ray structure of GAC in complex with 14b was also solved and has been included in the supplemental material.29 The crystal structures show that these leads bind in the allosteric pocket BPTES binds. The ring systems of these compounds occupy the same location the flexible connector chain of BPTES occupies and through their conformation allow for the projection of the thiadiazole groups to the same region the BPTES thiadiazoles lie in the 3UO9 x-ray structure (Figures 5, 6). Crystal structure overlays and the variable orientation of the phenylacetic acid moieties suggest that the BPTES binding pocket has a degree of plasticity which makes the explanation of the consistent potency difference observed for the R-and S-pyrrolidynyl analogs challenging. As can be seen from the overlay of the crystal structures of GAC with 7d and 7e (Figure 6) both of these enantiomeric compounds appear to be accommodated well in the BPTES allosteric pocket. Finally, our crystal structures, and the 3VOZ/3VP1 BPTES-GAC x-ray structures, suggest that the orientation of the thiadiazoles in the binding pocket is not critical for activity.
Figure 5.

Overlay of the BPTES (green, PDB: 3UO9) and 14d (brown) binding pockets
Figure 6.

Overlay of binding modes of BPTES (green, PDB: 3UO9), 7d (orange) and 7e (azure) with GAC
Conclusion
In conclusion, a novel set of GAC inhibitors/leads was synthesized with nanomolar range potency against GAC and against the MDA-MB-231 cancer cell line. These leads have less than 10 rotatable bonds, improved LE, LiPE and/or ClogPs when compared to leading GAC inhibitors BPTES and CB-839. In addition, in a microsomal stability assay, selected potent leads had greater stability than their comparators. Property improvements were the result of the replacement of the flexible lipophilic connector chains, seen in the leading GAC inhibitors, with heteroatom substituted cyclic ring systems that were chosen as surrogates for the conformation assumed by the BPTES flexible chain seen in the 3UO9 x-ray structure.
Experimental Section
1H NMR spectra were taken with a Bruker Avance 600 or Bruker Avance III 400 MHz Spectrometer. MS spectra under EI conditions were obtained with Shimadzu UFLC/Applied Biosystems 2000 MS mass spectrometer. MS spectra under APCI conditions were obtained using a Thar/Waters 3100 instrument. Infrared spectra were taken with a Bruker Alpha attenuated total reflectance (ATR) instrument. Column chromatography was performed using a Teledyne Isco Combiflash Rx instrument. Anhydrous solvents were obtained commercially and were used without further drying. Cell culture media and serum were obtained from Invitrogen (Carlsbad, CA). All other reagents were obtained from Fisher Scientific (Pittsburgh, PA) or Sigma Aldrich (St. Louis, MO). MDA-MB-231 cells were purchased from ATCC (Manassas, VA), and were cultured at 37°C, in 5% CO2, using RPMI-1640 media supplemented with 10% FBS. Recombinant GAC was expressed in E. coli and purified. Briefly, human GAC (residues 72–603) was cloned into the pET28a vector from Novagen, and was expressed as a His6-tagged fusion protein in E. coli. It was then purified by ion exchange and size exclusion chromatography
Biochemical assays:
The GAC inhibitors were solvated in DMSO. Assay vessels were charged with 1 μL of inhibitor in order to achieve the desired final concentration. To each vessel was added 95 μL of an aqueous solution containing 48 mM Trisacetate (pH 8.6), 21 mM glutamine, and 50 nM recombinant GAC. Fifteen μL of either water or 1 M potassium phosphate, pH 8.2, were added to the mixture to begin the reaction. The assay reagents were incubated for 10 minutes at room temperature, at which point 10 μL of ice-cold 2.4 M hydrochloric acid was added to quench the enzymatic reaction. A second reaction vessel contained 218 μL of an aqueous solution containing 114 mM Tris-HCl (pH 9.4), 0.35 mM ADP, 1.7 mM β-NAD, 238 mM hydrazine, and 1.3 units of glutamate dehydrogenase. A third reaction vessel contained an identical solution except that it lacked NAD+. Forty μL of the initial reaction mixture was added to each of the second and third vessels, which were then incubated at room temperature for one hour. The absorbance of both the second and third reactions was recorded at 340 nM. The third reaction was treated as a baseline, and its absorbance was subtracted from that of the second reaction prior to further data analysis. Dose curves and IC50 values were determined in Sigmaplot, using the built-in four parameter logistic function.
Cell assays:
MDA-MB-231 cells were cultured in RPMI-1640 media supplemented with 10% FBS. Prior to initiating the assay, cells that were 70–80% confluent were trypsinized. These cells were diluted, counted, and dispensed into 12-well culture plates at a density of 2 X 104 cells per well. Each well was then brought to 1 mL of media, total. The cells were allowed to adhere to the wells overnight, at which point they were recounted (day 0 of the assay). At this time, and every 48 hours thereafter, media was exchanged for media containing either the indicated amount of a given inhibitor, diluted from an appropriate DMSO stock, or an equivalent amount of DMSO without inhibitor (0.33% DMSO by volume). Cells were counted on the 6th day of culture. Cell counting was performed by aspirating media, rinsing the cells with room temperature PBS, and then incubating at 37°C for 5 minutes in 0.5 mL trypsin-EDTA solution. The culture plates were then agitated to fully dissociate cells from the plate surfaces, and 0.5 mL of RPMI-1640 complete media was added to quench trypsin activity. Cells were then counted on a hemocytometer, with 3 readings taken and averaged per sample. All experiments were performed in triplicate. Dose curves and IC50 values were determined in Sigmaplot, using the built-in four parameter logistic function.
Microsomal stability measurements:
The method for human liver microsome assays was adapted from Di et al. (Journal of Biomolecular Screening, 2003, 453–462) and/or Xu et al. Journal of the American Society for Mass Spectrometry, 2002, 155–165). Briefly, test compounds (diluted to final 1 μM (0.4% MeOH/ 0.1% DMSO)) were tested for metabolic stability by incubation against 1 mg protein/ml of pooled male human liver microsomes for 0 and 30 minutes at 37º C in the presence and absence of 1 mM NADPH. The reactions were terminated by addition of acetonitrile. Samples were centrifuged and the supernatant fractions analyzed by MS/MS. The instrument responses (i.e. peak areas, heights) at 30 min are referenced to the zero time-point samples to determine the percentage of compound remaining. Appropriate control compounds with known hepatic clearance were included in each assay like for instance metoprolol (control for moderate hepatic clearance), verapamil or testosterone (high hepatic clearance) warfarin (control for low hepatic clearance) etc.
Crystallization, data collection and structure determination:
Inhibitors 7d, 7e, 14b and 14d were dissolved in DMSO to make inhibitor stock solutions (30mM). The GAC-inhibitor complex was prepared by mixing 95μl of GAC solution (20mg/ml, in 150mM NaCl, 5mM Tris-HCl, pH7.5) with 5μl of inhibitor stock solution (mole ratio 1:4) and incubated on ice for one hour. Crystals were grown by the hanging drop vapor diffusion method at 20°C. Typically, 1.2μl of the complex solution was mixed with 1.2μl of the reservoir solution consisting of 10% PEG6000 (w/v), 1.0M LiCl and 0.1M Tris-HCl buffer, pH8.5. Crystals were observed after 24 hours, and reached a typical size of 100×100×200μm3, one week later. Crystals were pressurized at 350MPa for 30 minutes and then frozen to liquid nitrogen temperature using the high-pressure cryocooling method described by Huang et al. (Journal of Applied Crystalography, 2015, in press) before data collection. Diffraction data were collected at 100K at station A1 at MacCHESS. The diffraction data were reduced using the HKL package (Otwinowski & Minor, Methods in Enzymology, Volume 276: Macromolecular Crystallography, part A, p.307–326, 1997, C.W. Carter, Jr. & R. M. Sweet, Eds., Academic Press, New York). Crystal structures of the GAC-inhibitor complexes have been determined by molecular replacement with the program Phaser (McCoy et al. Journal of Applied Crystalography, 2007, 40:658–674) using the apo human GAC (PDB code 5D3O) as a search model. Four molecules of GAC were observed in an asymmetric unit. The model was examined and built in COOT (Emsley & Cowtan, Acta Crystallographica 2004, D60, 2126–2132) and subsequent refinement was carried out with Phenix_refine (Adams et al., Acta Crystallographica 2010, D66:213–221). Fitting of the inhibitor and refinements were performed in COOT and Phenix programs, respectively. Statistics of data collection and processing and refinement statistics are given in the supplementary material (Supplementary Table 1).
General procedure 1: Synthesis of compounds 6a-e
A mixture of 2-amino-5-bromothiadiazole (1 eq), desired diamine 5 (0.5 eq) and Et3N (4 eq) or NaHCO3 (6 eq) in EtOH in a sealed tube was heated at 75–80 °C until consumption of 2-amino-5-bromothiazole was complete. In case where Et3N was used as base the work up entailed solvent evaporation to small volume and then addition of water, filtration and trituration of the resulting solid with MeOH and drying to obtain the product. In case were NaHCO3 was used at the aprotic solvent the work-up entailed cooling to rt, filtration of the NaHCO3, evaporation of the filtrate and then purification of the crude product with silica column using a MeOH in CH2Cl2 gradient as elusion system.
General procedure 2: Preparation of compounds 7a-f, 14a-h
A mixture of a desired bis-thiadiazole 6 or 13 (1 eq.) and Et3N (3–4 eq) in DMF was treated with phenylacetyl chloride (2.1 eq when thiadiazoles 6 were the starting material or 3–4 eq when thiadiazoles 13 were the starting material) and the mixture was stirred at room temperature until consumption of the starting material was observed. Then followed concentration of the mixture to a small volume and addition of excess of water to precipitate the crude product. The crude product was purified by trituration and/or column chromatography with a MeOH/CH2Cl2 elution system.
General procedure 3: Preparation of 7g, 7h and 14i
A mixture of the bis-thiadiazole precursors 6 or 13 (1 eq), Et3N (3–4 eq) in DMF was treated with acetic anhydride (2 eq when 6 was starting material or 4 eq when 13 was starting material) and the mixture was allowed to stir until consumption of the limiting reagent material. The mixture was then concentrated to small volume and then treated with water to yield a precipitate that was filtered and then triturated with an organic solvent or washed with water and then dried to afford the product.
General procedure 4: Preparation of compounds 11a-h
A solution of desired N-Boc-protected cyclic alcohol 10 (1 eq) in THF was treated with NaH (60% suspension in mineral oil, 1.2 eq). The mixture was stirred at rt for approximately 20 min. Followed addition of CS2 (1.5 eq) and then approximately 5 min later addition of MeI (1.2 eq). The mixture was stirred until consumption of the limiting reagent was observed and then partitioned between CH2Cl2 or EtOAc and water. The water layer was extracted with CH2Cl2 or EtOAc (X2) and the combined organic layer was dried over Na2SO4, filtered and concentrated to a residue that was chromatographed on a silica gel column with an EtOAc in hexanes gradient to afford the product.
General procedure 5: Preparation of compounds 12a-h
A solution of desired xanthate 11 (1 eq) in MeOH was treated with 1.5 eq of H2NNH2 at room temperature. When the starting xanthate was consumed the mixture was evaporated under vacuum. The residue was re-dissolved in MeOH and the resulting solution was evaporated again. After repeating this dissolution and evaporation cycle one more time the residue was taken up again in MeOH and the solution was treated with Et3N (2 eq) and BrCN (1.2 eq). This mixture was stirred at room temperature until consumption of the starting material was complete. Followed solvent evaporation and partition of the resulting residue between EtOAc and water. The water layer was extracted with EtOAc (X2) and the combined organic layer was dried over Na2SO4 filtered and evaporated to the crude product. The crude product was purified via column chromatography and/or precipitation out of CH2Cl2 (or CHCl3) with excess of hexanes.
General procedure 6: Preparation of compounds 13a-h, 16a-f, 25
To a solution desired intermediates 12 or 15 or 18 or 24 in a small volume of dioxane was added 4N HCl in dioxane (2–3 eq). The mixture was stirred until consumption of the starting material was complete and then evaporated to dryness. The residue was triturated with hot CH2Cl2 fist and then hot hexanes and then dried to afford the corresponding intermediate HCl salt. This salt in EtOH or DMF was treated with Et3N (or NaHCO3 when compounds 18 were starting materials) (4–5 eq) and 2-amino-5-bromothiadiazole (1 eq) and the mixture was then heated at 75–80 °C in a sealed vessel until consumption of the staring material was complete. Followed evaporation of the solvent to a small volume and addition of excess of water to precipitate the crude product which was then collected by filtration, washed with water, dried and purified by trituration and/or column chromatography.
General procedure 7: Preparation of compounds 15a-d
A solution of desired thiadiazole 12 (1 eq) in CH2Cl2 was treated with Et3N (3–4 eq) and phenyl acetyl chloride (1.5 eq). The reaction was stirred until consumption of the starting material and then quenched with water and partitioned between CH2Cl2 and water. The water layer was extracted (X2) with CH2Cl2 and the combined organic layer was then dried over Na2SO4, filtered and concentrated to the crude product that was purified with silica gel column and an EtOAc in hexanes gradient as eluent.
General procedure 8: Preparation of derivatives 14j, 16g, 26a, 26d-f
A solution of the starting aromatic amine (13d or 16f or 25 or 36 or 40 or 41) (1 eq.) in DMF at room temperature was treated with the desired acetic acid derivative (1 or 2 eq depending the number of free amine moieties). The mixture was treated with EDCI or excess of DIEA or Et3N and then HATU stirred at room temperature until consumption of starting material was complete and then evaporated. The residue was treated with water to afford a suspension that was filtered to afford the crude product that was and then purified by trituration or column chromatography.
General procedure 9: Preparation of intermediates 21, 22, 28, 38 and 39
1-NBoc-4-hydroxy piperidine (1 eq) or desired heterocyclic 4-hydroxypyrolidine (27 or 34 or 37) (1 eq) in anhydrous THF or anhydrous THF/DMSO mixture was treated with NaH (60% dispersion in mineral oil, 1.5 eq) the mixture was stirred at room temperature for 5 min and then followed addition of the appropriate heterocycle (19 or 20 or 33) (1 eq). The mixture was stirred at 50 °C in a sealed vessel until consumption of the starting materials was complete then cooled and partitioned between EtOAc and NH4Cl saturated or water. Aqueous layer was extracted with EtOAc (X3) and combined organic layer was dried over Na2SO4 filtered and concentrated to the crude product that was purified with column chromatography.
General procedure 10: Preparation of benzophenone imines 23, 30, 31
A mixture of the appropriate halide starting material (21 or 28 or 29) (1 eq), benzophenone imine (2–4 eq), Cs2CO3 (6–8 eq) and (±)-BINAP (0.3 eq) in toluene in a sealed tube was degassed via N2 bubbling for 5 min. The mixture was then treated with Pd2(dba)3 (0.15 eq) sealed and heated at 90 °C (when starting halide had a Boc group on) or at 120 °C (when starting material was a dihalogen compound). The mixture was stirred until consumption of the starting material was observed, then cooled and filtered. The solids were washed with EtOAc portions thoroughly and the combined organic layer was evaporated to the crude product residue that was purified via column chromatography.
General procedure 11: Preparation of compounds 24, 26b, 26c
A mixture of starting benzophenone imine (23 or 30 or 31) in MeOH was treated with NaOAc (3–5 eq) and hydroxylamine hydrochloride (1.1 eq when 23 was used or 2.1 eq when 30or 31was used). The mixture was stirred at room temperature until consumption of the starting material was complete, evaporated to a solid residue which was chromatographed with a silica gel column to afford the corresponding purified deprotected aromatic amine intermediate. This intermediate was then dissolved in DMF and treated at room temperature with Et3N or DIEA (3–5 eq), phenylacetic acid (1.1 or 2.2 eq depending the number of amine groups to be acylated) and HATU (1.1–2.2 eq). The mixture was stirred at room temperature until consumption of the starting material was complete and then partitioned between EtOAc and water. The water layer was extracted with EtOAc (X3) and the combined organic layer was evaporated to the crude acylation product that was purified via column chromatography.
General procedure 12: Preparation of intermediates 27, 34, 37
Desired aromatic halide (19 or 32 or 33) (1eq), 4-hydroxypiperidine (1 eq) an aprotic base such as Et3N or K2CO3 (2–3 eq) in DMSO or EtOH was heated in a sealed vessel at until consumption of the starting material was observed. Work-up consisted of either partial mixture evaporation, addition of water and filtration of the resulting solid to afford the product (as in case of 37) or partition of the partially evaporated mixture between EtOAc and water, extraction of the aqueous layer with portions of EtOAc until no UV absorption in the organic extract and then evaporation of the combined organic layer to the crude product that was purified via column chromatography.
General procedure 13: Preparation of diamines 40 and 41
A slurry of the desired dinitro compound (1 eq) in 1:1 EtOH/EtOAc and 10% Pd/C (0.1–0.2 eq based on Pd content) was hydrogenated under 1 atm of H2 until consumption of the starting material was complete. Followed filtration and washing of the solids with MeOH portions until no UV absorption was detected in the solvent stream. Combined organic layer was then evaporated to afford the crude product that was chromatographed with column.
5-[4-(5-amino-1,3,4-thiadiazol-2-yl)-1,4-diazepan-1-yl]-1,3,4-thiadiazol-2-amine (6a)
Prepared according to general procedure 1 using Et3N as aprotic base. The product was isolated as a pinkish solid. (45% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.90 (apparent p, J=6.0 Hz, 2H), 3.44 (t, J=6.0 Hz, 4H), 3.60 (s, 4H), 6.34 (s, 4H). ATR IR (cm−1) 3272, 3111, 2945, 1624, 1571, 1500, 1437, 1383, 1357, 1318, 1295, 1250, 1213, 1191, 1085, 1047, 1026, 952, 922, 878, 742, 687, 653. MS (ESI) m/z for C9H14N8S2 calculated: 298.08, observed [M+H]: 299.0.
5-[4-(5-amino-1,3,4-thiadiazol-2-yl)piperazin-1-yl]-1,3,4-thiadiazol-2-amine (6b)
Prepared according to general procedure 1 using Et3N as aprotic base. The product was isolated as an off-white solid (83% yield). 1H NMR (600 MHz, DMSO d6) δ 3.34 (s, 8H), 6.55 (s, 4H). ATR IR (cm−1) 3261, 3125, 2961, 2848, 1626, 1546, 1494, 1447, 1375, 1323, 1273, 1234, 1150, 1065, 1048, 1023, 924, 762, 688. MS (ESI) m/z for C8H12N8S2 calculated: 284.06, observed [M+H]: 284.9
N2-(1-(5-amino-1, 3, 4-thiadiazol-2-yl) piperidin-4-yl)-1, 3, 4thiadiazole-2, 5-diamine (6c)
Prepared according to general procedure 1 using NaHCO3 was as aprotic base. The product was obtained as a tan solid after column chromatography with 0–35% MeOH in CH2Cl2 gradient (43% yield) 1H NMR (600 MHz, DMSO-d6) δ 1.43–1.529 (m,2H), 1.95–2.01 (m, 2H), 3.01–3.08 (apparent triplet, 2H), 3.53–3.63 (m, 2H), 6.32 (s, 2H), 6.47 (s, 2H), 6.86 (s, 1H). ATR IR (cm−1) 3257, 3137, 2926, 1611, 1560, 1490, 1445, 1375, 1359, 1309, 1263, 1220, 1121, 1082, 1029, 979, 900, 841, 803, 747, 669. MS (ESI) m/z for C9H14N8S2 calculated: 298.08, observed [M+H]: 299.0.
2-N-[(3S)-1-(5-amino-1,3,4-thiadiazol-2-yl)pyrrolidin-3-yl]-1,3,4-thiadiazole-2,5-diamine (6d)
Prepared according to general procedure 1 using NaHCO3 was as aprotic base. The product was obtained as a tan solid after column chromatography with a 0–30% MeOH in CH2Cl2 gradient (97% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.93–1.98 (m,1H), 2.19–2.24 (m,1H), 3.24 (dd, J= 10.2, 3.6Hz, 1H), 3.31–3.37 (m, 1H), 3.39–3.44 (m, 1H), 3.56 (dd, J=10.2, 6.0 Hz, 1H), 4.18–4.20 (m, 1H), 6.31 (s, 2H), 6.32 (s, 2H), 7.10 (d, J=6Hz, 1H). ATR IR (cm−1) 3252, 3136, 2920, 2860, 1606, 1563, 1492, 1470, 1334, 1299, 1235, 1188, 1117, 1028, 744. MS (ESI) m/z for C8H12N8S2 calculated: 284.06, observed [M+H]: 285.0.
2-N-[(3R)-1-(5-amino-1,3,4-thiadiazol-2-yl)pyrrolidin-3-yl]-1,3,4-thiadiazole-2,5-diamine (6e)
Prepared according to general procedure 1 using NaHCO3 was as aprotic base. The product was obtained as an off-white solid after column chromatography with 0–30% MeOH in CH2Cl2 gradient (48% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.95 (m, 1H), 2.21 (m, 1H), 3.23 (dd, J=10.2, 3.6 Hz, 1H), 3.43 (distorted dd, J= 16.8, 9.6 Hz, 1H), 3.56 (dd, J= 10.2, 6.0 Hz, 1H), 4.19 (m, 1H), 6.32 (s, 2H), 6.33 (s, 2H), 7.10 (d, J= 6Hz, 1H). ATR IR (cm−1) 3266, 3121, 2923, 2854, 1610, 1561, 1492, 1470, 1336, 1297, 1236, 1189, 1028, 743. MS (ESI) m/z for C8H12N8S2 calculated: 284.06, observed [M+H]: 285.0.
(±)-(anti)-3-[(5-Amino-[1,3,4]thiadiazol-2-ylamino)-cyclopentyl]-carbamic acid tert-butyl ester (9)
To a stirred solution of (±)-(8) (478 mg, 2.38 mmol) in EtOH (12 ml) was added NaHCO3 (300 mg, 3.57 mmol) and 2-amino-5-bromothiadiazole (515 mg, 2.86 mmol). The mixture was stirred in a sealed vessel at 80 °C until TLC showed consumption of the limiting reagent. The reaction mixture was then cooled and evaporated to a residue that was partitioned between EtOAc and water. The water layer was extracted with EtOAc (X2) and the combined organic layer was dried over Na2SO4 and evaporated. The resulting residue was purified via column chromatography with 0–30% methanol in methylene chloride gradient to yield the product, (±)-(anti)-3-[(5-amino-[1,3,4]thiadiazol-2-ylamino)-cyclopentyl]-carbamic acid tert-butyl ester (9) as an off-white/tan solid (170 mg, 24% yield). 1H NMR (600 MHz, MeOD) δ 1.41 (s, 9H), 1.47–1.54 (m, 2H), 1.85 (apparent t, J= 6.6 Hz, 2H), 2.03–2.09 (m, 1H), 2.11–2.19 (m, 1H), 3.95–4.06 (m, 2H). MS (ESI) m/z for C12H21N5O2S calculated: 299.14, observed [M+H]: 300.1.
(±)-(anti)-2-N-[3-[(5-Amino-1,3,4-thiadiazol-2-yl)amino]cyclopentyl]-1,3,4-thiadiazole-2,5-diamine (6f)
To a stirred solution of (±)-9 (170 mg, 0.57 mmol) in dichloromethane (7 mL) was treated with trifluoroacetic acid (3 mL) at 0 °C. The reaction mixture was then allowed to warm slowly to room temperature stirred for 1 h and then evaporated. The residue was dissolved in EtOH (3ml). Followed addition of NaHCO3 (167 mg, 1.98 mmol) and 2-amino-5-bromo-thiadiazole (122 mg, 0.68 mmol). The mixture was stirred in a sealed vessel at 80 °C until consumption of the limiting reagent, then cooled and evaporated. The residue was suspended in water then filtered, dried and purified via column chromatography with a 0–20% methanol in methylene chloride gradient to afford the product, (±)-(anti)-2-N-[3-[(5-amino-1,3,4-thiadiazol-2-yl)amino]cyclopentyl]-1,3,4-thiadiazole-2,5-diamine (6f), (91 mg, 54% yield). 1H NMR (600 MHz DMSO-d6) δ 1.41–1.48 (m, 2H), 1.83 (t, J=6.0 Hz, 2H), 2.02–2.08 (m, 2H), 3.91–3.96 (m, 2H), 6.21 (s, 4H), 6.81 (d, J=6.6 Hz, 2H).
2-Phenyl-N-{5-[4-(5-phenylacetylamino-[1,3,4]thiadiazol-2-yl)-[1,4]diazepan-1-yl]-[1,3,4]thiadiazol-2-yl}-acetamide (7a)
Prepared according to general procedure 2. The compound was obtained as an off white solid upon trituration of the crude product with hot hexanes and MeOH. 1H NMR (600 MHz, DMSO-d6) δ 1.92 (m, 2H), 3.54 (t, J= 6.0 Hz, 4H), 3.69 (s, 4H), 3.74 (s, 4H), 7.19–7.40 (m, 10H), 12.23 (s, 2H). ATR IR (cm−1) 3172, 3111, 3063, 2940, 2923, 2811, 2769, 2720, 1679, 1573, 1512, 1495, 1464, 1439, 1381, 1352, 1327, 1307, 1287, 1254, 1217, 1181, 1151, 1084, 1049, 1033, 972, 943, 921, 809, 752, 727, 693, 667. MS (ESI) m/z for C25H26N8O2S2 calculated: 534.16, observed [M+H]: 534.9.
2-Phenyl-N-(5-{4-[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]piperazin-1-yl}−1,3,4-thiadiazol-2-yl)acetamide (7b)
Prepared according to general procedure 2. The compound was obtained as an off-white solid upon trituration of the crude product with hot MeOH (43% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.52 (s, 8H), 3.74 (s, 4), 7.24 (m, 2H), 7.33 (m, 8H), 12.34 (s, 2H). ATR IR (cm−1) 3172, 2858, 2802, 2726, 1681, 1573, 1494, 1463, 1438, 1384, 1351, 1309, 1242, 1154, 1024, 924, 809, 732, 695. MS (ESI) m/z for C24H24N8O2S2 calculated: 520.15, observed [M+H]: 521.2.
2-Phenyl-N-(5-(4-((5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)amino)piperidin-1-yl)-1,3,4-thiadiazol-2-yl)acetamide (7c)
Prepared according to general procedure 2. The compound was obtained as a white solid upon column chromatography of the crude product with column using 0–10% methanol in methylene chloride gradient (55% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.46–1.55 (m, 2H), 2.02–2.06 (m, 2H), 3.16–3.22 (m, 2H), 3.70 (s, 2H), 3.73 (s, 2H), 3.71–3.78 (m, 3H), 7.23–7.39 (m, 11H), 12.18 (s, 1H), 12.29 (s, 1H). ATR IR (cm−1) 3389, 3243, 2852, 1682, 1662, 1572, 1493, 1356, 1315, 1299, 1128, 967, 833, 811, 757, 713, 693. MS (APCI), m/z for C25H26N8O2S2 calculated: 534.16, observed [M+H]: 535.2.
2-Phenyl-N-(5-{[(3S)-1-5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]pyrrolidin-3-yl]amino}−1,3,4-thiadiazol-2-yl)acetamide (7d)
Prepared according to general procedure 2. The compound was obtained as an off-white solid after column chromatography of the crude product with a 0–30% MeOH in CH2Cl2 gradient (41% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.99–2.06 (m, 1H), 2.24–2.32 (m, 1H), 3.35–3.39 (m, 1H), 3.42–3.54 (m, 2H), 3.71 (apparent singlet overlapping with a multiplet, 5H), 4.30–4.36 (m, 1H), 7.22–7.26 (m, 2H), 7.27–7.34 (m, 8H), 7.65 (broad s, 1H), 12.21 (s, 1H), 12.24 (s, 1H). ATR IR (cm−1) 3188, 2861, 1681, 1661, 1578, 1509, 1459, 1342, 1312, 1291, 1235, 1192, 1134, 1140, 1073, 1030, 970, 833, 756, 718, 693. MS (ESI) m/z for C24H24N8O2S2 calculated: 520.15, observed [M-H]: 518.8.
2-Phenyl-N-(5-{[(3R)-1-[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]pyrrolidin-3-yl]amino}−1,3,4-thiadiazol-2-yl)acetamide (7e)
Prepared according to general procedure 2. The compound was obtained as an off-white solid after column chromatography of the crude product with a 0–15% MeOH in CH2Cl2 gradient (37% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.00–2.06 (m, 1H), 2.25–2.32 (m, 1H), 3.36–3.39 (m, 1H), 3.43–3.53 (m, 2H), 3.69 (m, 1H), 3.71 (s, 2H), 3.72 (s, 2H), 4.32–4.37 (m, 1H), 7.23–7.34 (m, 10H) 7.65 (d, J=6.0 Hz, 1H), 12.21 (s, 1H), 12.25 (s, 1H). ATR IR (cm−1) 3339, 3186, 2868, 2735, 1682, 1662, 1577, 1512, 1457, 1358, 1316, 1296, 1226, 1185, 1160, 1132, 1147, 1073, 1029, 967, 842, 801, 755, 713, 693. MS (ESI) m/z for C24H24N8O2S2 calculated: 520.15, observed [M+H]: 520.9.
(±)-(anti)-2-Phenyl-N-{5-[3-(5-phenylacetylamino-[1,3,4]-thiadiazol-2-ylamino)-cyclopentylamino]-[1,3,4]thiadiazol-2-yl}-acetamide (7f)
Prepared according to general procedure 2. The compound was obtained as an off-white solid after column chromatography of the crude product with 0–10% MeOH in CH2Cl2 gradient (5% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.46–1.53 (m, 2H), 1.89 (t, J=6 Hz, 2H), 2.07–2.20 (m, 2H), 3.70 (s, 4H), 4.04–4.11 (m, 2H), 7.23–7.35 (m, 10H), 7.40 (d, J=6Hz, 2H) 12.16 (s, 2H). ATR IR (cm−1) 3214, 3062, 3015, 1655, 1574, 1531, 1494, 1414, 1350, 1296, 1191, 1074, 1029, 970, 844, 802, 756, 707, 693. MS (ESI) m/z for C25H26N8O2S2 calculated: 534.16, observed [M+H]: 535.1.
N-(5-{[(3S)-1-(5-acetamido-1,3,4-thiadiazol-2-yl)pyrrolidin-3-yl]amino}−1,3,4-thiadiazol-2-yl)acetamide (7g)
Prepared according to general procedure 3. The compound was isolated as an off-white/tan solid after washing of the crude product with water (25% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.07 (multiple overlapping with s, 1H), 2.08 (s, 3H), 2.10 (s, 3H), 2.26–2.33 (m, 1H), 3.39 (dd, J=10.2, 3.6 Hz, 1H), 3.44–3.55 (m, 2H), 3.71 (dd, J=10.2, 6.0 Hz, 1H), 4.32–4.38 (m, 1H), 7.63 (d, J=6.0 Hz, 1H), 11.94 (s, 1H), 11.97 (s, 1H). MS (ESI), m/z for C12H16N8O2S2 calculated: 368.08, observed [M+H]: 369.0.
N-(5-{[(3R)-1-(5-acetamido-1,3,4-thiadiazol-2-yl)pyrrolidin-3-yl]amino}−1,3,4-thiadiazol-2-yl)acetamide (7h)
Prepared according to general procedure 3. The compound was isolated as an off-white/tan solid after washing of the crude product with water (43% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.07 (multiple overlapping with s, 1H), 2.08 (s, 3H), 2.09 (s, 3H), 2.27–2.33 (m, 1H), 3.40 (dd, J=10.2, 3.6 Hz,1H), 3.45–3.55 (m, 2H), 3.71 (dd, J=10.2, 6.0 Hz, 1H,), 4.32–4.38 (m, 1H), 7.63 (d, J=6.0 Hz, 1H), 11.95 (s, 2H). ATR IR (cm−1) 3305, 3195, 3115, 2876, 2806, 2757, 1673, 1583, 1508, 1464, 1367, 1316, 1252, 1131, 1006, 962, 827, 696. MS (ESI) m/z for C12H16N8O2S2 calculated: 368.08, observed [M+H]: 368.7
tert-Butyl 3-(((methylthio)carbonothioyl)oxy)azetidine-1-carboxylate (11a)
Prepared according to general procedure 4. The product was obtained as a yellowish viscous oil after column chromatography with 0–20% EtOAC in hexanes gradient (89% yield). 1H NMR (600 MHz, CDCl3) δ 1.47 (s, 9H), 2.61 (s, 3H), 4.05 (m, 2H), 4.32 (m, 2H), 5.62 (m, 1H). ATR IR (cm−1) 2975, 2929, 2882, 1698, 1477, 1455, 1389, 1365, 1295, 1254, 1204, 1135, 1103, 1056, 1019, 965, 927, 858, 769.
tert-Butyl (3S)-3-{[(methylsulfanyl)methanethioyl]oxy} pyrrolidine-1-carboxylate (11b)
Prepared according to general procedure 4. The product was obtained as a colorless oil after column chromatography with 0–20% ethyl acetate in hexanes gradient (92% yield). 1H NMR (600 MHz, CDCl3) δ 1.47 (s, 9H), 2.11–2.20 (m, 1H), 2.21–2.25 (m, 1H), 2.56 (s, 3H), 3.38–3.51 (m, 1H), 3.52–3.73 (m, 3H), 5.97 (apparent s, 1H). MS (ESI) m/z for C11H19NO3S2 calculated: 277.08, observed [M+Na]: 300.1.
tert-Butyl (3R)-3-{[(methylsulfanyl)methanethioyl]oxy} pyrrolidine-1-carboxylate (11c)
Prepared according to general procedure 4. The product was obtained as a yellowish oil after column chromatography with 0–20% ethyl acetate in hexanes gradient (94% yield). 1H NMR (600 MHz CDCl3) δ = 1.46 (s, 9H) 2.11–2.21 (m, 1H), 2.22–2.28 (m, 1H), 2.56 (s, 3H), 3.40–3.51 (m, 1H), 3.52–3.73 (m, 3H), 5.96 (apparent s, 1H). MS (ESI) m/z C11H19NO3S2 calculated: 277.08, observed [M+Na]: 300.1.
tert-Butyl 4-(((methylthio)carbonothioyl)oxy)piperidine-1-carboxylate (11d)
Prepared according to general procedure 4. The product was obtained as a colorless viscous oil after column chromatography with 0–20% ethyl acetate in hexanes gradient (69% yield) 1HNMR (600 MHz, CDCl3) δ 1.49 (s, 9H), 1.84 (m, 2H), 1.99 (m, 2H), 2.58 (s, 3H), 3.37 (ddd, J= 11.4, 7.8, 3.6 Hz, 2H), 3.69 (broad m, 2H), 5.75 (apparent sep, J= 3.6 Hz). ATR IR (cm−1) 3002, 2973, 2925, 2864, 1687, 1476, 1452, 1418, 1364, 1311, 1273, 1237, 1209, 1162, 1129, 1049, 1008, 861, 767. MS (ESI) m/z for C12H21NO3S2 calculated: 291.1, observed [M+Na]: 314.1.
tert-Butyl 3-(((methylthio)carbonothioyl)oxy)piperidine-1-carboxylate (11e)
Prepared according to general procedure 4. The product was obtained as a colorless oil after column chromatography with 0–20% ethyl acetate in hexanes gradient (75% yield). 1H NMR (600 MHz, CDCl3) δ 1.44 (s, 9H), 1.56 (s, brd, 1H), 1.81 (s,brd,1H), 1.93 (s, brd, 2H), 2.53 (s, 3H), 3.18 (s, brd, 1H), 3.46 (s, brd, 1H), 3.71 (s, brd, 1H), 3.92 (s, brd, 1H), 5.53 (broad s, 1H). MS (ESI) m/z for C12H21NO3S2 calculated: 291.1, observed [M+Na]: 314.1.
tert-Butyl (3S)-3-({[(methylsulfanyl)methanethioyl]oxy} methyl)pyrrolidine-1-carboxylate (11f)
Prepared according to general procedure 4. The product was obtained as a light yellow/brown oil after column chromatography with 0–20% ethyl acetate in hexanes gradient (70% yield). 1H NMR (600 MHz CDCl3) δ 1.47 (s, 9H), 1.69–1.79 (m, 1H), 2.05 (m, 1H), 2.57 (d, 3H, J=8.4 Hz), 2.66–2.76 (m, 1H), 3.09–3.22 (m, 1H), 3.30–3.63 (m, 3H), 4.45–4.54 (m, 1H), 4.55–4.63 (m, 1H). ATR IR (cm−1) 2972, 2931, 2876, 1687, 1477, 1453, 1400, 1363, 1212, 1166, 1130, 1058, 965, 921, 882, 770.
tert-Butyl (3R)-3-({[(methylsulfanyl)methanethioyl]oxy} methyl)pyrrolidine-1-carboxylate (11g)
Prepared according to general procedure 4. The product was obtained as light yellow/brown oil after column chromatography with 0–20% ethyl acetate in hexanes gradient (75% yield). 1H NMR (600 MHz CD2Cl2) δ 1.44 (s, 9H), 1.68–1.76 (m,1H), 2.01–2.10 (m, 1), 2.56 (s, 3H), 2.66–2.76 (m, 1H), 3.09–3.15 (m, 1H), 3.28–3.45 (m, 1H), 3.40–3.46 (m, 1H), 3.51–3.56 (m, 1H), 4.49–4.55 (m, 1H), 4.57–4.62 (m, 1H). ATR IR (cm−1) 2972, 2931, 2876, 1687, 1477, 1453, 1400, 1363, 1212, 1166, 1130, 1058, 965, 882, 770. MS (ESI) m/z for C12H21NO3S2 calculated: 291.09, observed [M+Na]: 314.1
tert-Butyl-4-({[(methylsulfanyl)methanethioyl]oxy}methyl) piperidine-1-carboxylate (11h)
Prepared according to general procedure 4. The product was obtained as a colorless oil after column chromatography with a 0–50% EtOAc in hexanes gradient (82% yield).1H NMR (600 MHz, CDCl3) δ 1.22–1.29 (m, 2H), 1.46 (s, 9H), 1.74 (d, J=12.6Hz, 2H), 1.98–2.05 (m, 1H), 2.56 (s, 3H), 2.72 (s, brd, 2H), 4.14 (s, brd, 2H), 4.46 (d, J=6.6Hz, 2H). ATR IR (cm͞−1) 2973, 2927, 2852, 1685, 1448, 1418, 1364, 1288, 1274, 1213, 1165, 1139, 1060, 997, 970, 863, 768. MS (ESI) m/z calculated for C13H23NO3S2: 305.11, observed [M+Na]: 328.3.
tert-Butyl 3-((5-amino-1,3,4-thiadiazol-2-yl)oxy)azetidine-1-carboxylate (12a)
Prepared according to the general procedure 5. The compound was obtained as an off-white solid after a precipitation of the crude product out CH2Cl2 with excess of hexanes (81% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.39 (s, 9H), 3.86 (broad s, 2H), 4.21 (broad s, 2H), 5.25 (m, 1H), 6.85 (s, 2H). ATR-IR (cm−1) 3337, 3266, 3087, 2969, 2879, 1716, 1705, 1624, 1553, 1502, 1488, 1401, 1368, 1349, 1296, 1262, 1180, 1136, 1108, 1043, 858, 778, 691. MS (ESI) m/z for C10H16N4O3S calculated: 272.09, observed [M-H]: 270.8.
tert-Butyl (3S)-3-[(5-amino-1,3,4-thiadiazol-2yl)oxy] pyrrolidine-1-carboxylate (12b)
Prepared according to the general procedure 5. The compound was obtained after purification of the crude product via column chromatography with 0–5% MeOH in EtOAc gradient and a precipitation out of CHCl3 with excess of hexanes as an off-white solid (55% yield). 1H NMR (600 MHz, CDCl3) δ 1.46 (s, 9H), 2.12–2.16 (m, 1H), 2.25–2.37 (m, 1H), 3.44–3.77 (m, 4H), 4.63 (s, 2H), 5.49 (s, 1H). ATR-IR (cm−1) 3340, 3310, 3090, 2969, 2885, 1702, 1686, 1627, 1555, 1502, 1490, 1413, 1366, 1346, 1283, 1252, 1210, 1161, 1108, 1042, 968, 957, 930, 891, 875, 848, 765, 696. MS (ESI) m/z calculated for C11H18N4O3S: 286.11, observed [M-H]: 284.9.
tert-Butyl (R)-3-((5-amino-1,3,4-thiadiazol-2-yl)oxy) pyrrolidine-1-carboxylate (12c)
Prepared according to the general procedure 5. The compound was obtained as an off-white solid after precipitation of the crude product out of CHCl3 with excess of hexanes (48% yield). 1H NMR (600 MHz, CDCl3) δ = 1.46 (s, 9H), 2.09–2.19 (m, 1H), 2.26–2.35 (s, 1H), 3.40–3.79 (m, 4H), 4.63 (s, 2H), 5.49 (apparents, 1H). MS (ESI) m/z C11H18N4O3S calculated: 286.35, observed [M-H]: 284.9.
tert-Butyl 4-((5-amino-1,3,4-thiadiazol-2-yl)oxy)piperidine-1-carboxylate (12d)
Prepared according to the general procedure 5. The compound was isolated as a white powder after purification of the crude product via column chromatography and 0–100% EtOAc in CH2Cl2 to 0–5% MeOH in EtOAc gradient (40% yield). 1H NMR (600 MHz, CDCl3) δ 1.48 (s, 9H), 1.80 (m, 2H), 2.06 (m, 2H), 3.31 (ddd, J=12.0, 8.4, 3.6 Hz, 2H), 3.73 (broad m, 2H), 4.71 (s, 2H), 5.13 (apparent sep, J= 4.2 Hz, 1H). ATR IR (cm−1) 3383, 3255, 3086, 2973, 2935, 2872, 1657, 1558, 1496, 1427, 1365, 1280, 1243, 1228, 1165, 1125, 1074, 1052, 1024, 997, 901, 858, 843, 770, 749, 703, 675. MS (ESI) m/z for C12H20N4O3S calculated: 300.13, observed [M+H]: 301.0.
tert-Butyl 3-((5-amino-1,3,4-thiadiazol-2-yl)oxy)piperidine-1-carboxylate (12e)
Prepared according to the general procedure 5. The compound was isolated as a white powder after purification of the crude product via column and a 0–40% acetone in CH2Cl2 gradient (51% yield). 1H NMR (600MHz, DMSO-d6) δ 1.30 (s, brd, 9H), 1.42–1.44 (m, 2H), 1.65 (s, brd, 1H), 1.87(s, brd, 2H), 2.93 (s, brd, 1H), 3.69(s, brd, 1H), 4.09 (s, brd, 1H), 4.77(m, 1H), 6.74 (s, 2H). ATR IR (cm−1) 3326, 3275, 3129, 2953, 2863, 1682, 1623, 1542, 1488, 1422, 1366, 1345, 1277, 1236, 1167, 1150, 1130, 1097, 1073, 1003, 956, 925, 884, 854, 816, 766, 687. MS (ESI) m/z for C12H20N4O3S calculated: 300.13, observed [M+Na]: 323.2
tert-Butyl (3S)-3-{[(5-amino-1,3,4-thiadiazol-2-yl)oxy]methyl} pyrrolidine-1-carboxylate (12f)
Prepared according to the general procedure 5. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and a 0–8% MeOH in CH2Cl2 gradient (62% yield). 1H NMR (600 MHz CDCl3) δ 1.46 (s, 9H), 1.68–1.79 (m, 1H), 2.00–2.09 (m,1H), 2.64–2.73 (m, 1H), 3.10–3.25 (m, 1H), 3.30–3.61 (m,3H), 4.31–4.38 (m, 1H), 4.22 (dd, J= 10.2, 6.6 Hz, 1H), 4.66 (s, 2H). ATR IR (cm−1) 3328, 3275, 3119, 2974, 2874, 1681, 1625, 1549, 1497, 1459, 1412, 1392, 1364, 1331, 1252, 1167, 1136, 1087, 969, 888, 877, 770, 684. MS (ESI) m/z for C12H20N4O3S calculated: 300.13, observed [M-H]: 298.9.
tert-Butyl (3R)-3-{[(5-amino-1,3,4-thiadiazol-2-yl)oxy]methyl} pyrrolidine-1-carboxylate (12g)
Prepared according to the general procedure 5. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and a 0–5% MeOH in CH2Cl2 gradient as eluent (51% yield). 1H NMR (600 MHz CDCl3) δ 1.43 (s, 9H), 1.68–1.76 (m,1H), 1.98–2.07 (m,1H), 2.65–2.73 (m,1H), 3.10–3.20 (m,1H), 3.37–3.35 (m, 1H), 3.40–3.48 (brd m, 1H), 3.52 (dd, J=10.8, 7,8 Hz, 1H), 4.29–4.33 (m, 1H), 4.41 (dd, J=10.2, 6.6 Hz, 1H), 5.18 (very brd s, 2H). ATR IR (cm−1) 3324, 3280, 3105, 2970, 2878, 1684, 1623, 1555, 1495, 1459, 1408, 1364, 1329, 1256, 1170, 1136, 1113, 992, 962, 939, 885, 772, 687. MS (ESI) m/z for C12H20N4O3S calculated: 300.13, observed [M-H]: 298.9
4-(5-Amino-[1,3,4]thiadiazol-2-yloxymethyl)-piperidine-1-carboxylic acid tert-butyl ester (12h)
Prepared according to the general procedure 5. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and a 0–10% MeOH in CH2Cl2 gradient and then a precipitation out of CH2Cl2 with excess of hexanes (31% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.10(ddd, J= 24.6, 12.6, 4.2 Hz, 2H), 1.39 (s, 9H), 1.67 (d, J=12.6 Hz, 2H), 1.92–1.94 (m, 1H), 2.70 (broad s, 2H), 3.95 (broad s, 2H), 4.14 (d, J=6.6 Hz, 2H), 6.73 (s, 2H). ATR IR (cm−1) 3296, 3103, 2974, 2928, 2875, 1680, 1625, 1559, 1513, 1498, 1458, 1433, 1383, 1363, 1302,1262, 1233, 1175, 1132, 1092, 1068, 998, 979, 931, 902, 867, 827, 764, 686. MS (ESI) m/z calculated for C13H22N4O3S: 314.14, observed [M+H]: 315.2.
5-{3-[(5-amino-1,3,4-thiadiazol-2-yl)oxy]azetidin-1-yl}−1,3,4-thiadiazol-2-amine (13a)
Prepared according to general procedure 6. The compound was isolated as a tan solid after suspension of the crude product in hot MeOH and CH2Cl2 (63% yield). 1H NMR (600 MHz, DMSO-d6) δ 3.96 (dd, J=9.0, 3.6 Hz, 2H), 4.29 (dd, J=9.0, 6.0 Hz, 2H), 5.41 (m, 1H), 6.56 (s, 2H), 6.86 (s, 2H). ATR-IR (cm−1) 3256, 3085, 2954, 2858, 2786, 1631, 1567, 1497, 1455, 1366, 1330, 1237, 1174, 1113, 1060, 968, 760, 679. MS (ESI) m/z for C7H9N7OS2 calculated: 271.03, observed [M+H]: 272.0.
5-[(3S)-3-[(5-amino-1,3,4-thiadiazol-2-yl)oxy]pyrrolidin-1-yl]-1,3,4-thiadiazol-2-amine (13b)
Prepared according to general procedure 6. The compound was obtained as an off-white/tan solid after trituration of the crude product with boiling MeOH, boiling DCM, and boiling hexanes.1H NMR (600 MHz, DMSO-d6) δ 2.21–2.35 (m, 2H), 3.38–3.44 (m, 2H), 3.53 (d, J=12.0 Hz, 1H), 3.66 (dd, J=11.4, 4.8 Hz, 1H), 5.38–5.43 (m, 1H), 6.37 (s, 2H), 6.80 (s, 2H). ATR IR (cm−1) 3275, 3106, 2952, 2859, 2775, 1626, 1577, 1545, 1498, 1474, 1383, 1347, 1315, 1285, 1253, 1239, 1106, 1066, 1025, 890, 852, 754, 689. MS (ESI) m/z for C8H11N4OS calculated: 285.05, observed [M-H] 283.7.
5-[(3R)-3-[(5-amino-1,3,4-thiadiazol-2-yl)oxy]pyrrolidin-1-yl]-1,3,4-thiadiazol-2-amine (13c)
Prepared according to general procedure 6. The compound was obtained as an off-white/tan solid after trituration of the crude product with boiling MeOH, boiling DCM, and boiling hexanes (47% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.21–2.26 (m, 1H), 2.28–2.35 (m, 1H), 3.37–3.44 (m, 2H), 3.51–3.55 (m, 1H), 3.63–3.68 (m, 1H), 5.39 (apparent s, 1H), 6.37 (s, 2H), 6.80 (s, 2H). ATR IR (cm−1), 3270, 3103, 2950, 2852, 2771, 1626, 1577, 1543, 1503, 1469, 1380, 1346, 1312, 1286, 1250, 1238, 1107, 1064, 1025, 890, 850, 752, 688. MS (ESI) m/z for C8H11N7OS2 calculated: 285.05, observed [M-H]: 283.7.
5-(4-((5-amino-1,3,4-thiadiazol-2-yl)oxy)piperidin-1-yl)-1,3,4-thiadiazol-2-amine (13d)
Prepared according to general procedure 6. The compound was obtained as a brown solid after suspension/trituration of the crude product in CH2Cl2 (78% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.77 (m, 2H), 2.09 (m, 2H), 3.20 (ddd, J=12.6, 9.0, 3.6 Hz, 2H), 3.48 (m, 2H), 4.94 (apparent sep, J=4.2 Hz, 1H), 6.49 (s, 2H), 6.77 (s, 2H). ATR IR (cm−1) 3276, 3116, 2950, 2858, 1625, 1550, 1489, 1451, 1356, 1307, 1245, 1217, 1122, 1095, 1017, 951, 922, 878, 756, 677. MS (ESI) m/z for C9H13N7OS2 calculated: 299.06, observed [M-H]: 297.9.
5-{3-[(5-amino-1,3,4-thiadiazol-2-yl)oxy]piperidin-1-yl}−1,3,4-thiadiazol-2-amine (13e)
Prepared according to general procedure 6. The compound was obtained after purification of the crude product via column chromatography and using 0–25% MeOH in CH2Cl2 gradient as elution system (40% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.55–1.63 (m, 1H); 1.77–1.87 (m, 2H), 1.96–2.02 (m, 1H), 3.07–3.12 (m, 1H), 3.19–3.23 (m, 1H), 3.45 (dd, J=12.6, 6.0Hz, 1H), 3.63 (dd, 1H), 4.84–4.92 (m, 1H), 6.45 (s, 2H), 6.77 (s, 2H). MS (ESI) m/z for C9H13N7OS2 calculated: 299.06, observed [M-H]: 297.9
5-[(3S)-3-{[(5-Amino-1,3,4-thiadiazol-2-yl)oxy]methyl} pyrrolidin-1-yl]-1,3,4-thiadiazol-2-amine (13f)
Prepared according to general procedure 6. The compound was isolated as an off-white/tan solid after suspension/trituration of the crude product with boiling EtOH and then purification via column chromatography and 0–25% methanol in methylene chloride gradient (50% yield). 1H NMR (600 MHz DMSO-d6) δ 1.78–1.85 (m, 1H), 2.04–2.12 (m, 1H), 2.73–2.82 (m, 1H), 3.13 (dd, J=9.6, 6.6 Hz, 1H), 3.25–3.32 (m, 1H), 3.34–3.42 (m, 1H), 3.46 (dd, J=9.6, 7.8, 1H), 4.25–4.36 (m, 2H), 6.31 (s, 2H), 6.75 (s, 2H). ATR IR (cm−1) 3251, 3099, 2931, 2875, 1614, 1567, 1503, 1469, 1398, 1298, 1270, 1255, 1061, 1033, 1006, 989, 747, 682. MS (ESI) m/z for C9H13N7OS2 calculated: 299.06, observed [M+H]: 299.9.
5-[(3R)-3-{[(5-amino-1,3,4-thiadiazol-2-yl)oxy]methyl} pyrrolidin-1-yl]-1,3,4-thiadiazol-2-amine (13g)
Prepared according to general procedure 6. The compound was isolated as an off-white solid after suspension/trituration with boiling ethyl acetate, methanol and hexanes (16% yield). 1H NMR (600 MHz DMSO-d6) δ 1.74–1.85 (m, 1H), 2.06–2.14 (m, 1H), 2.73–2.82 (m, 1H), 3.14 (dd, J=9.6, 6.6 Hz, 1H), 3.27–3.33 (m, 1H), 3.36–3.42 (m, 1H), 3.45 (dd, J= 9.6, 7.8 Hz, 1H), 4.25–4.35 (m, 2H), 6.31 (s, 2H), 6.75 (s, 2H). MS (ESI) m/z for C9H13N7OS2 calculated: 299.06, observed [M+H]: 300.0.
5-(4-{[(5-amino-1,3,4-thiadiazol-2-yl)oxy]methyl}piperidin-1-yl)-1,3,4-thiadiazol-2-amine(13h)
Prepared according to general procedure 6. The compound was isolated as a tan solid after trituration of the crude product with boiling CH2Cl2 and hexanes (26% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.34 (ddd, J= 25.2, 12.6, 4.8 Hz, 2H) 1.73 (d, J=12.6 Hz, 2H), 1.93–2.02 (m, 1H), 2.88–2.95 (m, 2H), 3.63 (d, J=13.2 Hz, 2H), 4.17 (d, J=6.0 Hz, 2H), 6.43 (s, 2H), 6.74 (s, 2H). ATR IR (cm−1) 3260, 3112, 2946, 2843, 1616, 1551, 1492, 1457, 1384, 1328, 1291, 1262, 1215, 1049, 1000, 983, 959, 894, 834, 760, 685. MS (ESI) m/z for C10H15N7OS2 calculated: 313.08, observed [M-H]: 312.0.
2-Phenyl-N-[5-({1-[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]azetidin-3-yl}oxy)-1,3,4-thiadiazol-2-yl]acetamide (14a)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after suspension of the crude product in hot CHCl3 cooling and then addition of excess of hexanes (54% yield). 1H NMR (600 MHz, DMSO-d6) δ 3.74 (s, 2H), 3.77 (s, 2H), 4.14 (apparent broad d, J=9 Hz, 2H), 4.45 (apparent t, J=7.8 Hz, 2H), 5.57 (m, 1H), 7.25–7.42 (m, 10H), 12.40 (s, 1H), 12.67 (s, 1H). ATR-IR (cm−1) 3175, 3061, 3029, 2869, 1687, 1570, 1494, 1455, 1173, 1137, 1095, 1056, 1030, 969, 813, 758, 720, 694. MS (ESI) m/z for C23H21N7O3S2 calculated: 507.11, observed [M+H]: 508.1.
(S)-2-phenyl-N-(5-(3-((5-(2-phenylacetamido)-1, 3, 4thiadiazol-2-yl) oxy) pyrrolidin-1-yl)-1, 3, 4-thiadiazol-2-yl) acetamide (14b)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and a 0–20% MeOH in CH2Cl2 gradient followed by a trituration with boiling hexanes (21% yield). 1H NMR (400 MHz, DMSO-d6) δ 2.31–2.42 (m, 2H), 3.48–3.57 (m, 2H), 3.68 (apparent d, 1H), 3.72 (s, 2H), 3.76 (s, 2H), 3.78 (dd, J=12.0, 4.4 Hz, 1H), 5.55–5.65 (m, 1H), 7.20–7.40 (m, 10H), 12.27 (s, 1H), 12.59 (s, 1H). ATR IR (cm−1) 3179, 3060, 3031, 2915, 2851, 2808, 2750, 1684, 1573, 1530, 1495, 1454, 1360, 1298, 1256, 1198, 1147, 1080, 1028, 957, 857, 797, 758, 714, 694. MS (ESI) m/z for C24H23N7O3S2 calculated: 521.13, observed [M+H] 522.2.
2-phenyl-N-(5-{[(3R)-1-[5-(2-phenylacetamido)-1,3,4thiadiazol-2-yl]pyrrolidin-3-yl]oxy}−1,3,4-thiadiazol-2yl)acetamide (14c)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after trituration of the crude product with hot EtOH, then CH2Cl2 and then hexanes (26% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.31–2.40 (m, 2H), 3.48–3.56 (m, 2H), 3.68 (apparent d, 1H), 3.70 (s, 2H), 3.72 (s, 2H), 3.80 (dd, J=12.0, 4.8 Hz, 1H), 5.55–5.59 (m, 1H), 7.23–7.34 (m, 10H), 12.28 (s,1H) 12.60 (s,1H). ATR IR (cm−1), 3174, 3059, 3030, 2870, 2810, 2750, 1685, 1573, 1530, 1496, 1454, 1360, 1298, 1256, 1198, 1147, 1079, 1028, 958, 857, 798, 757, 714, 694. MS (ESI) m/z for C24H23N7O3S2 calculated: 521.13, observed [M+H]: 522.0.
2-Phenyl-N-{5-[1-(5-phenylacetylamino-[1,3,4]thiadiazol-2yl)-piperidin-4-yloxy]-[1,3,4]thiadiazol-2-yl}-acetamide (14d)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after suspension of the crude product in boiling CHCl3 then cooling and addition of excess of hexanes (73% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.84 (m, 2H), 2.13 (m, 2H), 3.37 (m, 2H), 3.65 (m, 2H), 3.76 (s, 2H), 3.78 (s, 2H), 5.13 (m, 1H), 7.25 (m, 2H), 7.31 (m, 8H), 12.31 (s, 1H), 12.58 (s, 1H). ATR IR (cm−1) 3176, 3060, 2955, 2856, 2809, 1686, 1569, 1503, 1455, 1351, 1351, 1315, 1268, 1218, 1141, 1087, 1016, 969, 947, 880, 830, 813, 758, 717, 695. MS (ESI) m/z for C25H25N7O3S2 calculated: 535.15, observed [M+H]: 535.9.
2-Phenyl-N-(5-(3-((5-(2-phenylacetamido)-1,3,4-thiadiazol-2yl)oxy)piperidin-1-yl)-1,3,4-thiadiazol-2-yl)acetamide (14e)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and a 0–10% MeOH in CH2Cl2 gradient (57% yield). 1H NMR (600 MHz, DMSO-d6) 1.59–1.66 (m, 1H), 1.81–1.88 (m, 1H), 1.90–1.96 (m, 1H), 1.99–2.06 (m, 1H), 3.36–3.41 (m, 1H), 3.46–3.52 (m, 1H). 3.67–3.81 (m, 2H), 3.71 (s, 2H), 3.75 (s, 2H), 5.03–5.09 (m, 1H), 7.23–7.35 (m, 10H), 12.30 (s, 1H), 12.60 (s, 1H). ATR IR (cm−1) 3177, 3063, 3029, 2850, 2752, 1684, 1574, 1503, 1454, 1355, 1315, 1252, 1199, 1142, 1073, 1008, 968, 927, 857, 807, 758, 716, 693. MS (ESI) m/z for C25H25N7O3S2 calculated: 535.15, observed [M+H]: 536.1.
2-Phenyl-N-{5-[(3S)-3-({[5-(2-phenylacetamido)-1,3,4thiadiazol-2-yl]oxy}methyl)pyrrolidin-1-yl]-1,3,4-thiadiazol-2-yl}acetamide (14f)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after trituration of the crude product with boiling CH2Cl2, then EtOAc and then hexanes (39% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.81–1.92 (m, 1H), 2.11–2.19 (m, 1H), 2.83–2.92 (m, 1H), 3.26 (dd, J=9.6, 6.6 Hz, 1H), 3.38–3.43 (m, 1H), 3.47–3.52 (m, 1H), 3.56 (dd, J= 9.6, 7.8 Hz, 1H), 3.72 (s, 2H), 3.76 (s, 2H), 4.41–4.51 (m, 2H), 7.23–7.36 (m, 10H) 12.24 (s, 1H), 12.57 (s, 1H). ATR IR (cm−1) 3176, 3063, 3030, 2866, 1686, 1575, 1505, 1454, 1394, 1358, 1294, 1268, 1162, 1074, 1031, 968, 812, 753, 720, 694. MS (ESI) m/z for C23H25N7OS2 calculated: 535.15, observed [M+H]: 536.0.
2-Phenyl-N-{5-[(3R)-3-({[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]oxy}methyl)pyrrolidin-1-yl]-1,3,4-thiadiazol-2-yl}acetamide (14g)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid after trituration of the crude product with hot ethyl acetate, methanol, methylene chloride and then hexanes (37% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.82–1.92 (m, 1H), 2.11–2.19 (m, 1H), 2.83–2.91 (m, 1H), 3.26 (dd, J=9.6, 6.6 Hz, 1H), 3.38–3.43 (m, 1H), 3.45–3.51 (m, 1H), 3.56 (dd, J=9.6, 7.8 Hz, 1H), 3.72 (s, 2H), 3.76 (s, 2H), 4.41–4.49 (m, 2H), 7.23–7.35 (m, 10H), 12.24 (brd s, 1H), 12.57 (brd s, 1H). ATR IR (cm−1) 3178, 3065, 3032, 2918, 2850, 1685, 1573, 1505, 1452, 1394, 1354, 1290, 1315, 1253, 1159, 1074, 1030, 966, 811, 750, 719, 692. MS (ESI) m/z for C25H25N7O2S2 calculated: 535.64, observed [M+H] 536.1.
2-Phenyl-N-{5-[1-(5-phenylacetylamino-[1,3,4]thiadiazol-2-yl)-piperidin-4-ylmethoxy]-[1,3,4]thiadiazol-2-yl}-acetamide (14h)
Prepared as described in general procedure 2. The compound was obtained as an off-white solid upon purification of the crude product via column chromatography and a 0–15% MeOH in CH2Cl2 gradient (52% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.34–1.39 (m, 2H), 1.74–1.80 (m, 2H), 2.07–2.12 (m, 1H), 3.04 (t, J=12 Hz, 2H), 3.72 (s, 2H), 3.75 (s, 2H), 3.79–3.83 (m, 2H), 4.27–4.30 (m, 2H), 7.24–7.34 (m, 10H), 12.27 (s, 1H), 12.55 (s, 1H). ATR IR (cm−1) 3389, 3264, 3156, 2940, 2849, 1693, 1563, 1494, 1255, 969, 693. MS (ESI) m/z calculated for C26H27N7O3S2: 549.16, observed [M+H]: 550.1.
N-{5-[1-(5-Acetylamino-[1,3,4]thiadiazol-2-yl)-piperidin-4-yloxy]-[1,3,4]thiadiazol-2-yl}-acetamide (14i)
Prepared as described in general procedure 3.The product was obtained as a tan solid after suspension of the crude product in hot CHCl3 then cooling and addition of excess of hexanes (70% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.85 (m, 2H), 2.11 (s, 3H), 2.13 (s, 3H), 2.15 (m, 2H), 3.37 (m, 2H), 3.67 (m, 2H), 5.14 (m, 1H), 12.04 (s, 1H), 12.30 (s, 1H). ATR-IR (cm−1) 3181, 3099, 2876, 2806, 1686, 1574, 1503, 1448, 1368, 1308, 1267, 1249, 1113, 1096, 1009, 968, 947, 927, 881, 833, 816, 794, 696, 673. MS (ESI) m/z for C13H17N7O3S2 calculated: 383.08, observed (M-H): 381.9.
2-Cyclopropyl-N-(5-(4-((5-(2-cyclopropylacetamido)-1,3,4thiadiazol-2-yl)oxy)piperidin-1-yl)-1,3,4-thiadiazol-2-yl)acetamide (14j).
Prepared as described in general procedure 8 from 13d via treatment with 4 eq of cyclopropyl acetic acid and EDCI (4 eq) as coupling agent. The compound was obtained as an offwhite/beige solid after trituration of the crude product with MeOH, hexanes and then hot CHCl3 (51% yield). 1H NMR (600 MHz, DMSO-d6) δ 0.18 (broad d, J=4.2 Hz, 4H), 0.47 (broad d, J=3.6 Hz, 4H), 1.02 (m, 2H), 1.86 (m, 2H), 2.15 (m, 2H), 2.29 (d, J=7.2 Hz, 2H), 2.31 (d, J=6.6 Hz, 2H), 3.39 (m 2H), 3.68 (m, 2H), 5.15 (m, 1H), 11.98 (s, 1H), 12.25 (s, 1H). ATR IR (cm−1) 3171, 3080, 2998, 2929, 2905, 2854, 2759, 1685, 1678, 1561, 1498, 1458, 1450, 1386, 1375, 1324, 1263, 1250, 1208, 1187, 1115, 1018, 951, 926, 901, 875, 830, 805, 776, 728, 691. MS (ESI) m/z for C19H25N7O3S2 calculated: 463.15, observed [M+H]: 464.0.
tert-Butyl 3-((5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)oxy)azetidine-1-carboxylate (15a)
Prepared according to the general procedure 7. The compound was obtained as a white solid after purification of the crude product via column chromatography and 0–100% EtOAc in hexanes gradient. (45% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.38 (s, 9H), 3.77 (s, 2H), 3.91 (broad s, 2H), 4.25 (broad s, 2H), 5.35–5.37 (m, 1H), 7.25–7.34 (m, 5H), 12.65 (s, 1H). ATR IR (cm−1) 3162, 3092, 2971, 2926, 2888, 2815, 1700, 1688, 1565, 1504, 1480, 1453, 1392, 1349, 1308, 1273, 1176, 1137, 1089, 1022, 1005, 972, 948, 872, 857, 839, 813, 766, 723, 693. MS (ESI) m/z for C18H22N4O4S calculated: 390.14, observed [M+Na]: 413.0.
tert-Butyl (3S)-3-{[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]oxy}pyrrolidine-1-carboxylate (15b)
Prepared according to the general procedure 7. The compound was obtained as a white solid after purification of the crude product via column chromatography with a 0–100% EtOAc in hexanes gradient. (76% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.37–1.40 (two overlapping singlets, 9H), 2.12–2.21 (m, 2H), 3.20–3.30 (m, 1H), 3.41–3.45 (m, 1H), 3.49–3.61 (m, 2H), 3.76 (s, 2H), 5.43 (broad s,1H), 7.24–7.27 (m, 1H), 7.29–7.34 (m, 4H), 12.59 (s, 1H). ATR IR (cm−1) 3168, 3093, 3060, 2974, 2928, 2887, 2814, 2762, 2717, 1689, 1570, 1502, 1458, 1410, 1362, 1328, 1306, 1262, 1174, 1148, 1120, 960, 885, 850, 771, 749, 725, 694. MS (ESI) m/z calculated for C19H24N4O4S: 404.15, observed [M-H]: 402.9.
tert-Butyl (3R)-3-{[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]oxy}pyrrolidine-1-carboxylate (15c)
Prepared according to the general procedure 7. The compound was obtained as an off-white solid after purification of the crude product via column chromatography with a 0–100% EtOAc in hexanes gradient (71% yield). 1H NMR (600 MHz, CDCl3) δ 1.46 (two overlapping singlets, 9H), 2.10–2.19 (brd m, 1H), 2.28–2.45 (brd m, 1H) 3.46–3.82 (cluster of m, 4H), 3.98 (s, 2H), 5.52 (s, 1H) 7.31 (distorted triplet, 1H), 7.36 (t, J=7.2, 2H), 7.43 (distorted d, J=7.8, 2H), 12.05 (brd d, 1H). MS (ESI) m/z for C19H24N4O4S calculated: 404.15, observed [M-H]: 403.0.
4-(5-Phenylacetylamino-[1,3,4]thiadiazol-2-yloxy)-piperidine-1-carboxylic acid tert-butyl ester (15d)
Prepared according to the general procedure 7. The compound was obtained as a white solid after purification of the crude product via column chromatography with a 0–70% EtOAc in hexanes gradient (73% yield). 1H NMR (600 MHz, CDCl3) δ 1.44 (s, 9H), 1.83 (broad m, 2H), 2.01 (broad m, 2H), 3.27 (ddd, J=13.8, 10.2, 5.4 Hz, 2H), 3.67 (broad m, 2H), 3.90 (s, 2H), 5.14 (m, 1H), 7.30 (m, 3H), 7.40 (d, J=7.2 Hz, 2H). 11.86 (brd s, 1H) ATR IR (cm−1) 2967, 2950, 2914, 2882, 2799, 2729, 1684, 1567, 1503, 1477, 1453, 1425, 1358, 1328, 1308, 1284, 1262, 1229, 1151, 1130, 1060, 1017, 895, 866, 830, 818, 769, 724, 693. MS (ESI) m/z for C20H26N4O4S calculated: 418.17, observed [M+H]: 419.1.
N-(5-((1-(5-amino-1, 3, 4-thiadiazol-2-yl) azetidin-3-yl) oxy)-1, 3, 4-thiadiazol-2-yl)-2-phenylacetamide (16a)
Prepared according to the general procedure 6. The compound was obtained as a tan solid after purification of the crude product via column chromatography with a 0–20% MeOH in CH2Cl2 gradient (25% yield). 1H NMR (600 MHz, DMSO-d6) δ 3.77 (s, 2H), 4.01 (dd, J=9.6, 3.6 Hz, 2H), 4.34 (dd, J=9.0, 6.6 Hz, 2H), 5.52–5.54 (m, 1H), 6.56 (s, 2H), 7.25–7.35 (m, 5H), 12.66 (s, 1H). ATR IR cm−1 3408, 3277, 3137, 3089, 2919, 2868, 1686, 1567, 1493, 1457, 1350, 1308, 1264, 1155, 1103, 1047, 975, 951, 881, 828, 748, 725, 692. MS (ESI) m/z for C15H15N7O2S2 calculated: 389.07, observed [M+H]: 389.8.
N-(5-{[(3S)-1-(5-amino-1,3,4-thiadiazol-2-yl)pyrrolidin-3-yl]oxy}−1,3,4-thiadiazol-2-yl)-2-phenylacetamide (16b)
Prepared according to the general procedure 6. The compound was obtained as a tan solid after purification of the crude product via column chromatography with a 0–10% MeOH in CH2Cl2 gradient (32% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.25–2.33 (m, 1H), 2.35–2.39 (m, 1H), 3.43 (dd, J=9.0, 4.8Hz, 2H), 3.58 (d, J=12.0 Hz, 1H), 3.71 (dd, J=12.0, 4.8 Hz, 1H), 3.76 (s, 2H), 5.53–5.56 (m, 1H), 6.38 (s, 2H), 7.25–7.36 (m, 5H), 12.60 (s, 1H). ATR IR (cm−1) 3400, 3263, 3157, 2926, 2847, 2653 1664, 1562, 1494, 1470, 1365, 1351, 1331, 1313, 1284, 1252, 1217, 1190, 1157, 1096, 1078, 1055,1031,948, 916, 850, 790, 752, 726, 713, 692. MS (ESI) m/z calculated for C16H17N7O2S2: 403.09, observed [M+H]: 404.1.
N-(5-{[(3R)-1-(5-amino-1,3,4-thiadiazol-2-yl)pyrrolidin-3-yl]oxy}−1,3,4-thiadiazol-2-yl)-2-phenylacetamide (16c)
Prepared according to the general procedure 6. The compound was obtained as an off-white solid after purification of the crude product via column chromatography with a 0–10% MeOH in CH2Cl2 gradient (40% yield). 1H NMR (600 MHz, DMSO-d6) δ 2.25–2.31 (m, 1H), 2.32–2.39 (m, 1H), 3.43 (dd, J= 9.0, 4.8 Hz, 2H), 3.58 (d, J=12.0 Hz, 1H), 3.71 (dd, J=12.0, 4.8 Hz, 1H), 3.76 (s, 2H), 5.52–5.57 (m, 1H), 6.37 (s, 2H), 7.22–7.35 (m, 5H), 12.60 (s, 1H). ATR IR (cm−1) 3397, 3271, 3160, 2918, 2848, 2650, 1664, 1564, 1504, 1470, 1366, 1351, 1331, 1313, 1284, 1252, 1218, 1190, 1159, 1096, 1078, 1052, 1031, 949, 917, 850, 790, 752, 727, 714, 692. MS (ESI) m/z for C16H17N7O2S2 calculated: 403.09, observed [M-H]: 404.1.
N-{5-[1-(5-Amino-[1,3,4]thiadiazol-2-yl)-piperidin-4-yloxy]-[1,3,4]thiadiazol-2-yl}−2-phenyl-acetamide (16d)
Prepared according to the general procedure 6. The compound was obtained as a white solid after trituration of the crude product with MeOH, and then boiling hexanes (44% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.81 (m, 2H), 2.12 (m, 2H), 3.22 (m, 2H), 3.51 (m, 2H), 3.76 (s, 2H), 5.10 (m, 1H), 6.49 (s, 2H), 7.247.36 (m, 5H), 12.55 (s, 1H). ATR IR (cm−1) 3421, 3266, 3141, 2959, 2834, 2747, 1679, 1591, 1558, 1508, 1497, 1463, 1367, 1357, 1299, 1253, 1226, 1194, 1116, 1016, 954, 909, 845, 797, 781, 756, 708, 694, 687. MS (ESI) m/z for C17H19N7O2S2 calculated: 417.10, observed [M-H]: 416.0.
tert-butyl 3-((5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)amino)azetidine-1-carboxylate (18a)
A mixture of 1-N-Boc-3-aminoazetidine (520 mg, 3.02 mmol), 2-amino-5-bromothiadiazole (650 mg, 3.61 mmol) and NaHCO3 (380 mg, 4.53 mmol) in EtOH (15 mL) in a sealed vessel was heated at 80 °C until consumption of the limiting reagent was indicated by TLC. The mixture was then cooled and partitioned between EtOAC and water. The water layer was extracted twice more with EtOAc and the combined organic layer was evaporated to a residue that was chromatographed on a silica gel column with 100% EtOAC to afford a solid that without further characterization was dissolved in DMF (10 mL). This mixture was treated with Et3N (0.84 mL, 6.04 mmol) and phenylacetyl chloride (560 mg, 3.63 mmol) and heated in a sealed vessel at 80 °C for 48h. The reaction mixture was then cooled and partitioned between CH2Cl2 and water. The aqueous layer was extracted twice more with CH2Cl2 and the combined organic layer evaporated. The residue was chromatographed on a silica gel column with 0–100% EtOAc in hexanes to afford the intermediate tert-butyl 3-((5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl)amino)azetidine-1-carboxylate (18a) as a grey solid (154 mg, 0.39 mmol, 13% yield). 1H NMR (600 MHz, CDCl3) δ 1.42 (s, 9H), 3.83 (dd, J=9.0, 4.8 Hz, 2H), 3.92 (s, 2H), 4.27 (dd, J=9.0, 7.8 Hz, 2H), 4.45 (bm, 1H), 5.45 (bs, 1H), 7.26–7.36 (m, 3H), 7.42 (apparent d, J=7.8 Hz, 2H), 12.23 (broad s, 1H).
tert-Butyl 4-{[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]amino}piperidine-1-carboxylate (18b)
A solution of 4-amino-1-N-Boc-piperidine (200 mg, 1.00 mmol,) in EtOH (11 mL) was treated with NaHCO3 (336 mg, 4.00 mmol,) and 2-amino-5-bromothiadiazole (180 mg, 1.00 mmol) and the resulting mixture was stirred in a sealed vessel at 80 °C until consumption of the starting materials was indicated by TLC. The mixture was then cooled and evaporated to dryness. The residue was treated with water to yield a suspension that was then filtered. The yellow solid obtained from this operation was washed with water, dried and chromatographed with column and 0–15% MeOH in CH2Cl2 gradient to afford the intermediate thiadiazole addition product as a pale-yellow solid (135mg, 45% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.21–1.29 (m, 2H), 1.39 (s, 9H), 1.87–1.92 (m, 2H), 2.81–2.98 (broad s, 2H), 3.51–3.58 (m, 1H), 3.76–3.83 (m, 2H), 6.24 (s, 2H), 6.75 (d, J=7.2 Hz, 1H). This addition product (118 mg, 0.39 mmol) was dissolved in anhydrous DMF (1.5 ml) and then treated with phenylacetic acid (59 mg, 0.43 mmol,), DIEA (0.21 mL, 1.18 mmol) and HATU (225 mg, 0.59 mmol). This mixture was stirred at room temperature until consumption of the limiting reagent was complete and evaporated to a residue to which water was added. The suspension formed was filtered to afford a reddish-yellow solid that was dried and chromatographed with column and 0–100% EtOAc in hexanes gradient as eluent to give the product, tert-butyl 4-{[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl] amino}piperidine-1-carboxylate (18b), as a pale-white solid (101 mg, 62% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.23–1.29 (m, 2H), 1.39 (s, 9H), 1.90–1.95 (m, 2H), 2.89 (broad s, 2H), 3.63–3.69 (m, 1H), 3.70 (s, 2H), 3.76–3.84 (m, 2H), 7.23–7.34 (m, 6H), 12.16 (s, 1H). ATR IR (cm−1) 3192, 3109, 3007, 2974, 2929, 2849, 1692, 1647, 1581, 1558, 1508, 1452, 1414, 1361, 1319, 1297, 1272, 1239, 1176, 1136, 1090, 1028, 1005, 971, 943, 874, 844, 802, 754, 712. MS (ESI) m/z for C20H27N5O3S calculated: 417.18, observed [M+H]: 418.9.
N-(5-{[1-(5-amino-1,3,4-thiadiazol-2-yl)azetidin-3-yl]amino}−1,3,4-thiadiazol-2-yl)-2-phenylacetamide (16e)
Prepared according to the general procedure 6. The compound was obtained after purification of the crude product via column chromatography with 0–10% MeOH in CH2Cl2 and trituration/suspension in hot CH2Cl2 as an off-white solid. 1H NMR (600 MHz, DMSO-d6) δ 3.71 (s, 2H), 3.75 (m, 2H), 4.20 (apparent t, J=7.8 Hz, 2H), 4.57 (m, 1H), 6.52 (s, 2H), 7.22–7.40 (m, 5H), 7.99 (d, J=6.6 Hz, 1H), 12.26 (s, 1H). ATR IR (cm−1) 3413, 3357, 3261, 3078, 3062, 2920, 2851, 1666, 1622, 1576, 1351, 1492, 1462, 1311, 1293, 1258, 1180, 1134, 1028, 966, 917, 868, 827, 758, 721, 694. MS (ESI) m/z for C15H16N8OS2 calculated: 388.09, observed [M+H]:388.9.
N-(5-{[1-(5-Amino-1,3,4-thiadiazol-2-yl)piperidin-4yl]amino}−1,3,4-thiadiazol-2-yl)-2-phenylacetamide (16f)
Prepared according to the general procedure 6. The compound was obtained as an orange solid after purification of the crude product via column chromatography and a 0–15% MeOH in CH2Cl2 gradient (57% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.45–1.53 (m, 2H), 1.98–2.03 (m, 2H), 3.02–3.09 (m, 2H), 3.54–3.60 (m, 2H), 3.69–3.75 (singlet overlapping with m, 3H), 6.46 (s, 2H), 7.23–7.37 (m, 6H), 12.17 (s, 1H). ATR IR cm−1 3306, 3240, 3140, 3050, 3027, 2934, 2914, 2843, 2739, 2693, 1649, 1567, 1527, 1489, 1467, 1441, 1386, 1317, 1244, 1218, 1195, 1124, 1098, 1046, 1018, 971, 922, 884, 843, 814, 789, 756, 721, 693. MS (ESI) m/z for C17H20N8OS2 calculated: 416.12, observed [M+H]: 417.8.
2-Cyclopropyl-N-[5-(4-{[5-(2-phenylacetamido)-1,3,4-thiadiazol-2-yl]amino}piperidin-1-yl)-1,3,4-thiadiazol-2-yl]acetamide (16g)
Prepared according to general procedure 8 from 16f by treatment with 1 eq of cyclopropyl acetic acid, DIEA (3 eq) and HATU (1.5 eq). The compound was obtained as an off-white solid after purification of the crude product via column chromatography with a 0–15% MeOH in CH2Cl2 gradient (30%). 1H NMR (600 MHz, DMSO-d6) δ 0.15–0.19 (m, 2H), 0.44–0.48 (m, 2H), 0.99–1.05 (m, 1H), 1.48–1.55 (m, 2H), 2.02–2.08 (m, 2H), 2.28 (d, J=6.6Hz, 2H), 3.15–3.23 (m, 2H), 3.70 (s, 2H), 3.73–3.79 (m, 3H), 7.23–7.37 (m, 6H), 11.95 (s, 1H), 12.18 (s, 1H). ATR IR cm−1 3330, 2950, 2925, 2837, 2769, 1671, 1561, 1507, 1460, 1380, 1325, 1288, 1255, 1190, 1126, 1104, 967, 927, 831, 752, 695. MS (ESI) m/z for C22H26N8O2S2 calculated: 498.16, observed [M+H]: 499.6.
4-(6-Chloro-pyridazin-3-yloxy)-piperidine-1-carboxylic acid tert-butyl ester (21)
Prepared according to general procedure 9 from 1-NBoc-4-hydroxypiperidine and 3, 6-dichloropyridazine using THF/DMSO (4/1) as solvent. The compound was obtained after purification of the crude product with silica gel column and 0–100% EtOAc in hexanes as a white solid (73 % yield). 1H NMR (600 MHz DMSO-d6) δ 1.41 (s, 9H), 1.58–1.63 (m, 2H), 1.982.02 (m, 2H), 3.15–3.26 (m, 2H), 3.67–3.71 (m, 2H), 5.31–5.34 (m, 1H), 7.32 (d, J=9.6 Hz, 1H), 7.80 (d, J=9.6 Hz, 1H). ATR IR (cm−1) 3051, 3004, 2967, 2927, 2876, 1672, 1581, 1480, 1421, 1363, 1293, 1274, 1231, 1165, 1127, 1075, 1022, 938, 914, 858, 764, 715, 673. MS (ESI) m/z calculated for C14H20ClN3O3: 313.12, observed [M+Na]: 336.0.
4-[6-(Benzhydrylidene-amino)-pyridazin-3-yloxy]-piperidine-1-carboxylic acid tert-butyl ester (23)
Prepared according to general procedure 10 using 21 as starting material. The compound was obtained as a yellow solid after chromatography of the crude product with column and 0–100% EtOAc in hexanes (74% yield). 1H NMR (600 MHz DMSO-d6) δ 1.39 (s, 9H), 1.49–1.52 (m, 2H), 1.92–1.96 (m, 2H), 3.10–3.12 (m, 2H), 3.68–3.70 (m, 2H), 5.19–5.20 (m, 1H), 7.02 (d, J=9.0 Hz, 1H), 7.10 (d, J=9.0 Hz, 1H), 7.14–7.18 (m, 2H), 7.34–7.38 (m, 3H), 7.51 (t, J=7.2 Hz, 2H), 7.59 (t, J=7.2 Hz, 1H) 7.70 (d, J=7.8Hz, 2H). ATR IR (cm−1) 3058, 2976, 2961, 2935, 2850, 1686, 1624, 1597, 1578, 1476, 1425, 1363, 1331, 1293, 1269, 1237, 1163, 1132, 1091, 1027, 957, 842, 818, 773, 697, 658. MS (ESI) m/z calculated for C27H30N4O3: 458.23, observed [M+H]: 459.3
4-(6-Phenylacetylamino-pyridazin-3-yloxy)-piperidine-1-carboxylic acid tert-butyl ester (24)
Prepared according to general procedure 11 from 23. The purified aromatic amine intermediate from the first step was obtained after column chromatography with a 0–10% MeOH in CH2Cl2 gradient. The final product (compound 24) was obtained as a white solid after column chromatography of the crude acylation product from the second step with a 0–5% MeOH in CH2Cl2 gradient (68% yield). 1H NMR (600 MHz DMSO-d6) δ 1.41 (s, 9H) 1.55–1.62 (m, 2H) 1.98–2.03 (m, 2H) 3.21 (broad s, 2H) 3.64–3.73 (m, 2H) 3.74 (s, 2H), 5.27–5.33 (m, 1H), 7.19 (d, J=9.6Hz, 1H), 7.23–7.45 (m, 5H), 8.20 (d, J=9.6Hz, 1H), 11.12 (s, 1H). ATR IR (cm−1) 3191, 3130, 3061, 3006, 2977, 2962, 2931, 2861, 1687, 1609, 1568, 1522, 1477, 1423, 1379, 1364, 1344, 1302, 1268, 1235, 1165, 1134, 1103, 1074, 1022, 970, 936, 864, 844, 814, 768, 713, 695. MS (ESI) m/z calculated for C22H28N4O4: 412.21, observed [M+H]: 413.3.
N-{6-[1-(5-Amino-[1,3,4]thiadiazol-2-yl)-piperidin-4-yloxy]-pyridazin-3-yl}−2-phenyl-acetamide (25)
Prepared according to general procedure 6. The compound was obtained as a tan solid from the crude product via column chromatography with a 0–20% MeOH in CH2Cl2 gradient (85% yield). 1H NMR (600 MHz DMSO-d6) δ 1.70–1.82 (m, 2H) 2.05–2.13 (m, 2H), 3.20–3.30 (m,2H) 3.52–3.56 (m, 2H), 3.74 (s, 2H), 5.32–5.37 (m, 1H), 6.48 (s,2H), 7.21–7.38 (m, 6H), 8.21 (d,J=10.2Hz, 1H) 11.13 (s, 1H). ATR IR (cm−1) 3452, 3306, 3231, 3166, 3131, 3063, 3018, 2992, 2959, 2896, 2822, 2992, 1695, 1593, 1566, 1521, 1485, 1464, 1435, 1380, 1339, 1306, 1266, 1255, 1223, 1193, 1142, 1119, 1101, 1032, 963, 942, 895, 857, 813, 770, 756, 722, 697, 668. MS (ESI) m/z calculated for C19H21N7O2S: 411.15, observed [M+H]: 412.3.
2-Phenyl-N-{6-[1-(5-phenylacetylamino-[1,3,4]thiadiazol-2-yl)-piperidin-4-yloxy]-pyridazin-3-yl}-acetamide (26a)
Prepared according to general procedure 8 from 25 by treatment with phenylacetic acid (1.3 eq), Et3N (4 eq) and HATU (1.2 eq.) The compound was obtained as an off-white solid after suspension/trituration of the crude product with water (42 % yield). 1H NMR (600 MHz DMSO-d6) δ 1.75–1.83 (m, 2H), 2.09–2.13 (m, 2H), 3.35–3.42 (m, 2H), 3.63–3.68 (m, 2H), 3.72 (s, 2H), 3.73 (s, 2H), 5.34–5.37 (m, 1H), 7.19–7.25 (m, 3H), 7.287.35 (m, 8H), 8.19 (d, J=9.6 Hz, 1H), 11.12 (s, 1H), 12.29 (s, 1H). ATR IR (cm−1) 3364, 3179, 3088, 3063, 3029, 2976, 2955, 2930, 2838, 2798, 2838, 2798, 2746, 1688, 1580, 1499, 1437, 1385, 1350, 1320, 1296, 1257, 1181, 1129, 1100, 1033, 962, 861, 759, 712, 693. MS (ESI) m/z calculated for C27H27N7O3S: 529.19, observed [M+H]: 530.1.
1-(6-Chloro-pyridazin-3-yl)-piperidin-4-ol (27)
Prepared according to general procedure 12 from 3,6-dichloropyridazine and 4-hydroxypiperidine using Et3N as aprotic base and DMSO as solvent. The compound was obtained as a white solid after purification of the crude product via silica gel column chromatography with a 0–100% EtOAc in hexanes gradient (89% yield). 1H NMR (600 MHz, CD3OD) δ 1.54 (m, 2H), 1.94 (m, 2H), 3.28 (m, 2H), 3.89 (apparent sep, J= 4.2 Hz, 1H), 4.11 (apparent dt, J=13.2, 4.8 Hz, 2H), 7.32 (d, J=9.6 Hz, 1H), 7.4 (d, J=9.6 Hz, 1H). ATR IR (cm−1) 3199, 3077, 3008, 2991, 2941, 2847, 1583, 1530, 1429, 1362, 1312, 1298, 1257, 1237, 1224, 1184, 1167, 1148, 1107, 1061, 1032, 1012, 981, 921, 835, 766, 745, 670. MS (ESI) m/z for C9H12ClN3O calculated: 213.07, observed [M+H]: 214.0.
3-Chloro-6-{4-[(6-chloropyridazin-3-yl)oxy]piperidin-1-yl}pyridazine (28)
Prepared as per the general procedure 9 from 3,6-dichloropyridazine and 27 using THF as the reaction solvent. The compound was obtained as a white solid after purification of the crude product via column chromatography and a 0–100% EtOAc in hexanes gradient (82% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.76 (m, 2H), 2.14 (m, 2H), 3.50 (ddd, J=13.2, 9.0, 3.6 Hz, 2H), 4.04 (m, 2H), 5.45 (apparent sep, J=4.2 Hz, 1 H), 7.34 (d, J=9.6 Hz, 1H), 7.46 (d, J=10.2 Hz, 1H), 7.54 (d, J=9.6 Hz, 1H), 7.82 (d, J=9.0 Hz, 1H). ATR IR (cm−1) 3051, 2973, 2958, 2938, 2920, 2858, 1583, 1529, 1440, 1415, 1369, 1330, 1301, 1246, 1236, 1188, 1167, 1150, 1121, 1082, 1047, 1030, 972, 946, 919, 848, 827, 764, 700, 675. MS (ESI) m/z for C13H13Cl2N5O calculated: 325.05, observed [M+H]: 325.9.
N-(diphenylmethylidene)-6-[4-({6-[(diphenylmethylidene) amino]pyridazin-3-yl}oxy)piperidin-1-yl]pyridazin-3-amine (30)
Prepared from 28 according to general procedure 10. The compound was obtained as an off-white solid after purification of the crude product via column chromatography with a 0–100% EtOAc in CH2Cl2 gradient and a precipitation of the chromatographed product out of CH2Cl2 with excess of hexanes (30% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.62 (m, 2H), 2.04 (m, 2H), 3.25 (apparent t, J=9.6H, 2H), 3.95 (m, 2H), 5.26 (broad m, 1H), 6.88 (d, J=9.6 Hz, 1H), 7.04 (d, J=9.0 Hz, 1H), 7.12 (d, J=9.0 Hz, 1H), 7.16 (m, 4H), 7.21 (d, J=9.6 Hz, 1H), 7.36 (m, 6H), 7.52 (m, 4H), 7.60 (m, 2H), 7.71(dd, J=13.2, 7.2 Hz, 4H). ATR IR (cm−1) 3062, 2987, 2963, 2940, 2917, 1625, 1594, 1571, 1534, 1475, 1444, 1421, 1308, 1297, 1250, 1231, 1176, 1155, 1122, 1097, 1074, 1018, 995, 958, 906, 836, 780, 763, 734, 695, 650. MS (ESI) m/z for C39H33N7O calculated: 615.27, observed [M+H]: 616.2.
2-Phenyl-N-{6-[1-(6-phenylacetylamino-pyridazin-3-yl)-piperidin-4-yloxy]-pyridazin-3-yl}-acetamide (26b)
Prepared from 30 according to general procedure 11. The purified aromatic diamine intermediate from the first step was obtained after column chromatography with 0–15% MeOH in CH2Cl2 gradient. The final product (compound 26b) was obtained as a white solid after purification of the crude acylation product from the second step via column chromatography with a 0–100% EtOAc in CH2Cl2 gradient. 1H NMR (600 MHz, DMSO-d6) δ 1.72 (m, 2H), 2.11 (m, 2H), 3.40 (apparent t, J=10.2 Hz, 2H), 3.71 (s, 2H), 3.75 (s, 2H), 3.97 (m, 2H), 5.39 (m, 1H), 7.21 (d, J=9.6 Hz, 1H) 7.24 (m, 2H), 7.34 (m, 8H), 7.39 (d, J=9.6 Hz, 1H), 8.01 (d, J=9.6 Hz, 1H), 8.20 (d, J=9.6 Hz, 1H), 10.92 (s, 1H), 11.13 (s, 1H). ATR IR (cm−1) 3348, 3221, 3192, 3102, 3059, 3028, 2949, 2919, 2839, 1685, 1509, 1421, 1356, 1313, 1295, 1258, 1234, 1227, 1137, 1119, 1095, 1020, 997, 951, 907, 864, 834, 813, 773, 731, 713, 692, 654. MS (ESI) m/z for C29H29N7O3 calculated: 523.23, observed [M+H]: 524.1.
tert-butyl 4-[(5-bromopyrazin-2-yl)oxy]piperidine-1-carboxylate (22)
Prepared according to general procedure 9 from 1-NBoc-4-hydroxypiperidine and 2,5-dibromopyrazine using THF as the reaction solvent. The compound was obtained as a white solid after purification of the crude product via column chromatography with a 0–30% EtOAc in hexanes gradient. (74% yield). 1H NMR (600 MHz, CDCl3) δ 1.73 (broad m, 2H), 1.96 (broad m, 2H), 3.28 (m, 2H), 3.76 (broad m, 2H), 5.13 (m, 1H), 7.98 (d, J=0.6 Hz, 1H), 8.15 (d, J=0.6 Hz, 1H). ATR IR (cm−1) 3001, 2976, 2933, 2858, 1687, 1560, 1525, 1476, 1452, 1437, 1413, 1363, 1330, 1300, 1265, 1234, 1189, 1160, 1129, 1106, 1086, 1029, 1008, 970, 943, 920, 893, 858, 816, 771, 759, 694, 666. MS (ESI) m/z calculated for C14H20BrN3O3: 357.07, observed [M+H]: 357.8.
2-Bromo-5-{4-[(5-bromopyrazin-2-yl)oxy]piperidin-1-yl}pyrazine (29)
A solution of tert-butyl 4-[(5-bromopyrazin-2-yl)oxy]piperidine-1-carboxylate (22) (540 mg, 1.51 mmol) in dioxane (3 mL) was treated with 4N HCl in dioxane (1 mL) and the mixture was stirred at rt for 6 hr. The solvent was then evaporated to a white residue which was suspended in CH2Cl2 (2 mL) and excess (10 mL) of hexanes. Followed filtration of the solids, wash again (X2) with hexanes and drying to afford the intermediate dihydrochloride salt. This salt without delay was added to a 1:1 mixture of THF/DMSO (12 mL). Followed addition of Et3N (0.84 mL, 6.02 mmol), 2,5-dibromopyrazine and then stirring at 50 °C for 24 h. The solvents were then evaporated and the residue was partitioned between EtOAc and water. The water layer was extracted with EtOAc once more and the combined organic layer was evaporated. The residue was chromatographed with a column and 0–30% EtOAc in hexanes gradient as elution system to afford the product 2-bromo-5-{4-[(5-bromopyrazin 2 yl)- oxy]piperidin 1 yl}pyrazine as an off-white solid (110 mg, 18% yield) 1H NMR (600 MHz, CDCl3) δ 1.88 (m, 2H), 2.10 (m, 2H), 3.51 (ddd, J=12.6, 7.8, 3.6 Hz, 2H), 3.91 (m, 2H), 5.25 (m, J= 10.8, 7.2, 3.6 Hz, 1H), 7.92 (s, 1H), 8.01 (s, 1H), 8.14 (s, 1H), 8.17 (s, 1H). ATR IR (cm−1) 3064, 2985, 2945, 2841, 1554, 1519, 1505, 1473, 1450, 1437, 1392, 1334, 1313, 1278, 1245, 1234, 1187, 1160, 1122, 1101, 1056, 1028, 1009, 996, 970, 933, 890, 866, 772, 755, 694, 667. MS (ESI) m/z for C13H13Br2N5O calculated: 414.95, observed [M+H]: 415.9
N-(Diphenylmethylidene)-5-[4-({5-[(diphenylmethylidene)amino]pyrazine-2-yl}oxy)piperidin-1-yl]pyrazine-2-amine (31)
Prepared from 29 according to the general procedure 10. The compound was obtained as a yellow solid after purification of the crude product via column chromatography with a 0–100% EtOAc in CH2Cl2 gradient (34% yield).1HNMR (600 MHz, CDCl3-d6) δ 1.79 (dddd, J=16.8, 12.6, 8.4, 4.2 Hz, 2H), 2.02 (m, 2H), 3.33 (ddd, J= 12.6, 8.4, 3.0 Hz, 2H), 3.82 (m, 2H), 5.09 (ddd, J=11.4, 7.8, 3.6 Hz, 1H), 7.16 (distorted t, 4H), 7.31 (m, 5H), 7.42 (m, 5H), 7.48 (m, 3H), 7.55 (d, J=0.6 Hz, 1H), 7.78 (t, J=7.8 Hz, 4H), 7.87 (s, 1H), 7.91 (d, J=0.6 Hz, 1H). ATR IR (cm−1) 3055, 2957, 2921, 2849, 1619, 1604, 1594, 1568, 1480, 1456, 1442, 1391, 1341, 1316, 1297, 1266, 1252, 1236, 1156, 1116, 1073, 1059, 1031, 1015, 975, 955, 916, 877, 843, 815, 781, 769, 738, 720, 690. MS (ESI) m/z for C39H33N7O calculated: 615.27, observed [M]: 615.3
2-Phenyl-N-[5-({1-[5-(2-phenylacetamido)pyrazine-2-yl]piperidin-4-yl}oxy)pyrazin-2-yl]acetamide (26c)
Prepared from 31 according to the general procedure 11. The purified aromatic diamine intermediate, from the first step, was obtained after column chromatography with 0–20% MeOH in CH2Cl2 gradient. The final product (compound 26c) was obtained as a white solid after purification of the crude acylation product, from the second step, via column chromatography with a 0–90% EtOAc in CH2Cl2 gradient. (38%yield). 1H NMR (400 MHz, DMSO-d6) δ 1.67 (m, 2H), 2.04 (m, 2H), 3.34 (m, 2H), 3.68 (s, 2H), 3.72 (s, 2H), 3.94 (m, 2H), 5.18 (m, 1H), 7.25 (m, 2H), 7.32 (m, 8H), 8.08 (d, J=1.2 Hz, 1H), 8.13 (d, J=1.2 Hz, 1H), 8.76 (s, 1H), 8.83 (d, J=1.2 Hz, 1H), 10.53 (s, 1H), 10.77 (s, 1H). ATR IR (cm−1) 3301, 3087, 3062, 3023, 2925, 2850, 2831, 1670, 1588, 1550, 1510, 1454, 1408, 1376, 1354, 1268, 1251, 1224, 1164, 1122, 1064, 1041, 1020, 965, 928, 886, 846, 767, 710, 692, 662. MS (ESI) m/z for C29H29N7O3 calculated: 523.23, observed [M+H]: 523.8
1-(6-Nitropyridin-3-yl)piperidin-4-ol (34)
Prepared according to general procedure 12 from 2-nitro-5-chloropyridine and 4 hydroxypiperidine using K2CO3 as the aprotic base, DMSO as solvent and 90 °C as the reaction temperature. The compound was obtained as a yellow solid upon purification of the crude product via column chromatography with a 30–100% EtOAc in hexanes gradient (65% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.42 (m, 2H), 1.81 (m, 2H), 3.24 (ddd, J= 13.2, 9.6, 3.0 Hz, 2H), 3.75 (m, 1H), 3.83 (m, 2H), 4.78 (d, J=4.2 Hz, 1H), 7.45 (dd, J=9.0, 3.0 Hz, 1H), 8.11 (d, J=9.0 Hz, 1H), 8.23 (d, J=3.0 Hz, 1H). ATR IR (cm−1) 3393, 3321, 3107, 3088, 2947, 2930, 2855, 1569, 1507, 1490, 1420, 1382, 1324, 1270, 1260, 1219, 1173, 1101, 1072, 1016, 1003, 962, 901, 824, 745, 692, 664. MS (ESI) m/z for C10H13N3O3 calculated: 223.10, observed [M+H]: 224.1.
2-Nitro-5-{4-[(6-nitropyridin-3-yl)oxy]piperidin-1-yl}pyridine (35)
A solution of 1-(6-nitropyridin-3-yl)piperidin-4-ol (34) (110 mg, 0.49 mmol), 2-nitro-5-hydroxypyridine (69 mg, 0.49 mmol) and Ph3P (136 mg, 0.52 mmol) in THF (7 mL) was treated with diisopropyl azodicarboxylate, (0.10 mL, 0.52 mmol) and stirred at room temperature for 4 h. The mixture was then partitioned between CH2Cl2 and water. The organic layer was evaporated and the residue was chromatographed with a silica gel column and 0–20% acetone in CH2Cl2 gradient to afford the product. 1H NMR (600 MHz, DMSO-d6) δ 1.78 (dddd, J=16.2, 12.0, 7.8, 3.6 Hz, 2H), 2.14 (m, 2H), 3.48 (ddd, J= 12.6, 9.0, 3.6 Hz, 2H), 3.89 (m, 2H), 5.01 (ddd, J= 12.0, 7.8, 4.2 Hz, 1H), 7.55 (dd, J= 9.0, 3.0 Hz, 1H), 7.85 (dd, J= 9.0, 3.0 Hz, 1H), 8.17 (d, J=9.0 Hz, 1H), 8.32 (d, J= 3.0 Hz, 1H), 8.35 (d, J= 9.0 Hz, 1H), 8.38 (d, J= 3.0 Hz, 1H) MS (ESI) m/z for C15H15N5O5 calculated: 345.11, observed [M+H]: 346.0.
5-{4-[(6-Aminopyridin-3-yl)oxy]piperidin-1-yl}pyridin-2-amine (36)
2-nitro-5-{4-[(6-nitropyridin-3-yl)oxy]piperidin-1-yl}pyridine (35) (210 mg, 0.61 mmol), was dissolved in EtOH (10 mL). To this solution was added Fe (680 mg, 12.17 mmol), aq. saturated NH4Cl (4 mL) and the mixture was heated for 15 min at 80 °C then cooled and partitioned between EtOAc and water. The water layer was extracted with EtOAC until no UV absorption could be detected in the organic extract. The combined organic layer was then dried over Na2SO4, filtered, concentrated and the residue was chromatographed with a silica gel column and 0–10% MeOH in CH2Cl2 gradient to afford the product an off-white/tan solid. (45 mg, 32% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.68 (m, 2H), 1.97 (m, 2H), 2.77 (ddd, J= 12.0, 9.6, 3.0 Hz, 2H), 3.22 (m, 2H), 4.19 (apparent sep, J= 3.6 Hz, 1H), 5.39 (s, 2H), 5.51 (s, 2H), 6.39 (d, J= 8.4 Hz, 1H), 6.41 (d, J=8.4 Hz, 1H), 7.16 (dd, J=8.4, 3.0 Hz, 1H), 7.18 (dd, J=9.0, 3.0 Hz, 1H), 7.63 (d, J=3 Hz, 1H), 7.67 (d, J= 3Hz, 1H). ATR IR (cm−1) 3414, 3398, 3307, 3167, 3038, 2944, 2817, 1639, 1566, 1489, 1463, 1400, 1372, 1312, 1259, 1232, 1221, 1183, 1162, 1138, 1114, 1062, 1039, 1012, 979, 912, 822, 787, 743, 669. MS (ESI) m/z for C15H19N5O calculated: 285.16, observed [M+H]: 286.0.
2-Phenyl-N-[5-(4-{[6-(2-phenylacetamido)pyridin-3-yl]oxy}piperidin-1-yl)pyridin-2-yl]acetamide (26d)
Prepared according to general procedure 8 from 36 using phenylacetic acid (4 eq), Et3N (6 eq) and HATU (3 eq.). The compound was obtained as an off-white solid after purification of the crude product by trituration with MeOH and then EtOAc (67% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.73 (m, 2H), 2.04 (m, 2H), 3.05 (m, 2H), 3.48 (m, 2H), 3.66 (s, 2H), 3.68 (s, 2H), 4.56 (m, 1H), 7.24 (m, 2H), 7.33 (m, 8H), 7.41 (dd, J=9.0, 3.0 Hz, 1H), 7.48 (dd, J=9.0, 3.0 Hz, 1H), 7.90 (d, J=9.6 Hz, 1H), 7.99 (d, J=9.0 Hz, 1H), 8.04 (d, J=3 Hz, 1H), 8.07 (d, J=3 Hz, 1H), 10.46 (s, 1H), 10.59 (s, 1H). ATR IR (cm−1) 3392, 3238, 3084, 3059, 3026, 2927, 2825, 1666, 1583, 1508, 1493, 1464, 1390, 1344, 1297, 1278, 1217, 1139, 1030, 962, 910, 830, 764, 720, 693. MS (ESI) m/z for C31H31N5O3 calculated: 521.24, observed [M+H]: 522.0.
1-(5-Nitropyridin-2-yl)piperidin-4-ol (37)
Prepared according to general procedure 12 from 2-chloro-5-nitropyridine and 4-hydroxypiperidine using Et3N as the aprotic base, EtOH as solvent and 70 °C as the reaction temperature. The product was obtained as a yellow solid after water suspension/trituration (80% yield). 1H NMR (600 MHz, CDCl3) δ 1.54 (d, J= 4.2 Hz, 1H) 1.64 (m, 2H), 1.99 (m, 2H), 3.50 (ddd, J= 12.6, 8.4, 3.0 Hz, 2H), 4.06 (m, 1H), 4.16 (m, 2H), 6.60 (d, J= 11.4 Hz, 1H), 8.19 (dd, J=9.6, 3.0 Hz, 1H), 9.03 (d, J=2.4, 1H). ATR IR (cm−1) 3463, 3104, 3084, 2942, 2916, 2873, 1589, 1571, 1514, 1478, 1433, 1338, 1328, 1285, 1243, 1213, 1160, 1129, 1115, 1067, 1016, 995, 976, 950, 933, 919, 819, 761, 744, 722. MS (ESI) m/z for C10H13N3O3 calculated: 223.10, observed [M+H]:224.1.
2-Nitro-5-{4-[(5-nitropyridin-2-yl)oxy]piperidin-1-yl}pyridine (38)
Prepared from 37 and 2-chloro-5-nitropyridine according to general procedure 9 using a mixture of anhydrous THF and DMSO (5:1) as solvent. The compound was obtained as a yellow solid after purification of the crude product via column chromatography with 0–80% EtOAc in CH2Cl2 gradient (73% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.81 (m, 2H), 2.14 (m, 2H), 3.50 (ddd, J= 12.6, 9.0, 3.0 Hz, 2H), 3.90 (m, 2H), 5.44 (apparent sep, J= 4.2 Hz 1H), 7.05 (d, J=9.0 Hz, 1H), 7.54 (dd, J=9.6, 3.0 Hz, 1H), 8.17 (d, J=7.2 Hz, 1H), 8.31 (d, J=2.4 Hz, 1H), 8.50 (dd, J=9.0, 2.4 Hz, 1H), 9.11 (d, J=3.0 Hz, 1H). ATR IR (cm−1) 3058, 2963, 2921, 2854, 1684, 1600, 1569, 1502, 1469, 1418, 1399, 1343, 1311, 1283, 1270, 1222, 1182, 1170, 1108, 1090, 1021, 1000, 971, 948, 911, 829, 760, 746, 720, 693, 681, 658. MS (ESI) m/z for C15H15N5O5 calculated: 345.11, observed [M+H]: 345.9.
5-Nitro-2-{4-[(5-nitropyridin-2-yl)oxy]piperidin-1-yl}pyridine (39)
Prepared from 37 and 2-chloro-5-nitropyridine according to general procedure 9 using THF as solvent. The compound was obtained as a yellow solid after purification of the crude product via column and 0–100% EtOAc in hexanes gradient as eluent (47% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.74 (m, 2H), 2.11 (m, 2H), 3.67 (m, 2H), 4.16 (m, 2H), 5.45 (apparent sep, J= 4.2 Hz, 1H), 7.01 (d, J=9.6 Hz, 1H), 7.03 (d, J=9.0 Hz, 1H), 8.21 (dd, J=9.6, 3.0 Hz, 1H), 8.48 (dd, J=9.6, 3.0 Hz, 1H), 8.96 (d, J=2.4 Hz, 1H), 9.09 (d, J=3.0 Hz, 1H). ATR IR (cm−1) 3097, 3080, 3050, 2967, 2923, 2869, 1594, 1574, 1508, 1471, 1460, 1429, 1399, 1342, 1313, 1291, 1268, 1226, 1165, 1139, 1113, 1095, 1024, 993, 954, 925, 867, 833, 805, 759, 742, 720, 681, 659. MS (ESI) m/z for C15H15N5O5 calculated: 345.11, observed [M+H]: 346.0.
5-{4-[(5-Aminopyridin-2-yl)oxy]piperidin-1-yl}pyridine-2-amine (40)
Prepared from 38 according to general procedure 13. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and 0–10% MeOH in CH2Cl2 gradient as eluent (69% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.68 (m, 2H), 2.02 (m, 2H), 2.80 (ddd, J=12.4, 9.6, 2.8 Hz, 2H), 3.23 (m,2H), 4.72 (broad s, 2H), 4.87 (apparent sep, J= 4.0 Hz, 1H), 5.37 (broad s, 2H), 6.40 (dd, J=8.8, 0.4 Hz), 6.52 (dd, J=8.8, 0.8 Hz, 1H), 7.00 (dd, J=8.8, 3.2 Hz, 1H), 7.19 (dd, J=8.8, 2.8 Hz, 1H), 7.50 (dd, J=2.8, 0.4 Hz, 1H), 7.64 (d, J=2.8 Hz, 1H). MS (ESI) m/z for C15H19N5O calculated: 285.16, observed [M+H]: 286.1.
6-{4-[(5-aminopyridin-2-yl)oxy]piperidin-1-yl}pyridine-3-amine (41)
Prepared from 39 according to general procedure 13. The compound was obtained as an off-white solid after purification of the crude product via column chromatography and 0–5% MeOH in CH2Cl2 gradient as eluent (77% yield) 1H NMR (600 MHz, DMSO-d6) δ 1.57 (m, 2H), 1.96 (m, 2H), 2.98 (ddd, J=13.2, 10.2, 3 Hz, 2H), 3.74 (m, 2H), 4.55 (broad s, 2H), 4.72 (broad s, 2H), 4.92 (apparent sep, J= 4.2 Hz, 1H), 6.50 (d, J=9 Hz, 1H), 6.65 (d, J=9.0 Hz, 1H), 6.90 (dd, J=9.0, 3.0 Hz, 1H), 6.98 (dd, J=8.4, 2.4 Hz, 1H), 7.48 (d, J=2.4 Hz, 1H), 7.59 (d, J=3 Hz, 1H). MS (ESI) m/z for C15H19N5O calculated: 285.16, observed [M+H]: 286.1.
2-Phenyl-N-[6-({1-[6-(2-phenylacetamido)pyridin-3-yl]piperidin-4-yl}oxy)pyridin-3-yl]acetamide (26e)
Prepared from 40 according to the general procedure 8 using phenylacetic acid (2.5 eq), Et3N (6 eq) and HATU (2 eq.) The compound was obtained as a white solid after purification of the crude product via column chromatography and 0–100% EtOAc in hexanes (65% yield). 1H NMR (600 MHz, DMSO-d6) δ 1.74 (m, 2H), 2.06 (m, 2H), 3.03 (apparent t, J= 10.2 Hz, 2H), 3.50 (m, 2H), 3.63 (s, 2H), 3.67 (s, 2H), 5.09 (m, 1H), 6.77 (d, J=9.0 Hz, 1H), 7.25 (m, 2H), 7.32 (m, 8H), 7.41 (broad d, J=9.0 Hz, 1H), 7.90 (m, 2H), 8.04 (s, 1H), 8.34 (s, 1H), 10.20 (s, 1H), 10.46 (s, 1H). ATR IR (cm−1) 3268, 3030, 2934, 2816, 2794, 1659, 1611, 1578, 1522, 1482, 1452, 1410, 1391, 1360, 1340, 1295, 1279, 1248, 1231, 1185, 1144, 1119, 1049, 1034, 1019, 980, 972, 916, 826, 748, 726, 704, 692, 657. MS (ESI) m/z for C31H31N5O3 calculated: 521.24, observed [M+H]:521.9.
2-Phenyl-N-[6-(4-{[5-(2-phenylacetamido)pyridin-2-yl]oxy}piperidin-1-yl)pyridin-3-yl]acetamide (26f)
Prepared from 41 according to general procedure 8 using phenylacetic acid (2.5 eq), Et3N (6 eq) and HATU (2 eq.). The compound was obtained as an off-white solid after purification of the crude product by trituration with MeOH and then with EtOAc. 1H NMR (600 MHz, DMSO-d6) δ 1.61 (m, 2H), 2.01 (m, 2H), 3.23 (m, 2H), 3.60 (s, 2H), 3.63 (s, 2H), 3.93 (m, 2H), 5.15 (m, 1H), 6.76 (d, J=9.0 Hz, 1H), 6.85 (d, J=9.0 Hz, 1H), 7.26 (m, 2H), 7.33 (m, 8H), 7.77 (dd, J=9.0, 2.4 Hz, 1H), 7.89 (dd, J=9.0, 3.0 Hz, 1H), 8.28 (d, J=3.0 Hz, 1H), 8.34 (d, J=2.4 Hz, 1H), 10.01 (s, 1H), 10.20 (s, 1H). ATR IR (cm−1) 3214, 3167, 3062, 3028, 2971, 2942, 2844, 1663, 1636, 1600, 1549, 1529, 1484, 1453, 1402, 1387, 1367, 1355, 1303, 1270, 1241, 1225, 1199, 1152, 1116, 1058, 1038, 1015, 983, 967, 930, 908, 844, 817, 767, 735, 710, 692. MS (ESI) m/z for C31H31N5O3 calculated: 521.24, observed [M+H]: 522.0.
Supplementary Material
Acknowledgments
Funding support was provided by the University of Pittsburgh Medical Center Competitive Medical Research Fund (CMRF). CHESS is supported by the NSF & NIH/NIGMS via NSF award DMR-1332208 and the MacCHESS resource is supported by NIGMS award GM-103485.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Eagle H “Nutrition needs of mammalian cells in tissue culture” Science, 1955, 122, 501–04. [DOI] [PubMed] [Google Scholar]
- 2.Reviews in references 3–6 (below) show how our understanding of glutamine importance in tumor metabolism has evolved over the years.
- 3.Kovacevic Z; McGivan JD “Mitochondrial metabolism of glutamine and glutamate and its physiological significance” Phys. Rev 1983, 63, 547–605. [DOI] [PubMed] [Google Scholar]
- 4.Matsuo T “bioenergetics of tumor cells: Glutamine metabolism in tumor cell mitochondria” Int. J. Bioch 1987, 19, 303–07. [DOI] [PubMed] [Google Scholar]
- 5.Medina MA “Glutamine and cancer” J. Nutr 2001, 131 (9 Supp) 2539S–42S. [DOI] [PubMed] [Google Scholar]
- 6.DeBerardinis RJ; Cheng T “Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer” Oncogene, 2010, 29, 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knox WE, Horowitz ML, Friedell GH. “The proportionality of glutaminase content to growth rate and morphology of rat neoplasms” Cancer Res. 1969, 29, 669–80. [PubMed] [Google Scholar]
- 8.Gomez-Perez C; Campos-Sandoval JA; Alonso FJ; Segura JA; Manzanares E; Ruis-Sanchez P; Gonzales ME; Marquez J; Mates HM “Co-expression of glutaminase K and L isoenzymes in human tumor cells” Biochem. J 2005, 386, 535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassago A; Ferreira APS; Ferreira IM; Fornezari C; Gomes ERM; Greene KS; Pereira HM; Garratt RC; Dias SMG; Ambrosio ALB “Mitochondrial localization and structure-based phosphate activation mechanism of glutaminase C with implication for cancer metabolism” PNAS 2012, 109, 1092–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szeliga M; Obara-Michlewska M “Glutamine in neoplastic cells: Focus on the expression and roles of glutaminases” Neurochem. Intern 2009, 55, 71–75. [DOI] [PubMed] [Google Scholar]
- 11.Quiet S; Chu C; Li W; Wang C; Regina to M; Sang N “ErbB2 activation up-regulates glutaminase 1 expression via NF-kobo pathway” Cancer Res. 2012, 72 (8 Suppl) Abstract. 5143 [Google Scholar]
- 12.Wise DR; DeBerardinis RJ; Mancuso A; Sayed N; Zhang X-Y;Pfeiffer HK;Nissim I;Daikhin E;Yudkoff M;McMahon SB;Thompson CB “Myc regulates a transcriptional program that stimulated mitochondrial glutaminolysis and leads to glutamine addiction” Proc. Natl. Acad. Sci. USA 2008, 105, 18782–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao P; Tchernyshyov I; Chang T-C;Lee Y-S;Kita K;Ochi T;Zeller K;De Marzo AM;Van Eyk JE;Mendell JT, Dang CV “c-Myc suppression of miR-23 enhances mitochondrial glutaminase and glutamine metabolism” Nature, 2009, 458, 762–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le A; Lane AN; Hamaker M; Bose S; Gouw A; Barbi J; Tsukamoto T; Rojas CJ; Slucher BS; Zhang H; Zimmerman LJ; Liebler DC; Slebos RJC; Lorkiewicz PK; Higashi RM; Fan TWM; Dang CV “Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells” Cell Metab. 2012, 15, 110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J-B; Erickson JW; Fuji R; Ramachandran S; Gao P; Dinavahi R; Wilson KF; Ambrosio ALB; Dias SMG; Dang CV; Cerione RA “Targeting mitochondrial glutaminase activity inhibits oncogenic transformation” Cancer Cell, 2010, 18, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross MI; Demo SD; Dennison JB; Chen L; RoganChernov T; Goyal B; Janes JR; Laidig GJ; Lewis ER; Li L; MacKinnon AL; Parlati F; Rodriguez MLM;Shwonek PJ;Sjogren EB;Stanton TF;Wang T;Yang J;Zhao F;Bennett MK “Antitumor activity of glutaminase inhibitor CB-839 in triple negative breast cancer” Mol. Cancer Ther. 2014, 13, 890–901 [DOI] [PubMed] [Google Scholar]
- 17.Lobo C; Ruiz-Bellido MA; Aledo JC; Marquez J; Nunez de Castro I; Alonso FJ “inhibition of glutaminase expression by antisense mRNA decreases growth and tumorigenicity of tumour cells” Biochem. J 2000, 348, 257–61. [PMC free article] [PubMed] [Google Scholar]
- 18.Seltzer MJ; Bennett BD; Joshi AD; Gao P; Thomas AG; Ferraris DV; Tsukamoto T; Rojas CJ; Slusher BS; Rabinowitz JD; Dang CV; Riggins GJ “Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1” Cancer Res. 2010, 70, 8981–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD “Molecular properties that influence the oral bioavailability of drug candidates” J. Med. Chem 2002, 45, 2615–2623 [DOI] [PubMed] [Google Scholar]
- 20.Hopkins AL; Groom CR; Alex A Drug Discovery Today 2004, 9, 430–31 [DOI] [PubMed] [Google Scholar]
- 21.Leeson PD; Springthorp B Nat. Rev. Drug Disc 2007, 6, 881–90. [DOI] [PubMed] [Google Scholar]
- 22.Ferreira APS; Cassago A; Goncalves K, Dias MM; Adamoski D; Ascencao CFR; Honorato RV; de Oliveira JF; Ferreira IM; Fornezari C; Bettini J; Oliveira PSL; Leme AFP; Portugal RV; Ambrosio ALB; Dias SMG “Active glutaminase C selfassembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor” J. Biol. Chemistry 2013, 288, 28009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLaBarre B; Gross S; Fang C; Gao Y; Jha A; Jiang F; Song J J; Wei W; Hurov JB Full-Length Human Glutaminase in Complex with an Allosteric Inhibitor. Biochemistry 2011, 50 (50), 10764–10770. [DOI] [PubMed] [Google Scholar]
- 24.Thangavelu K; Pan CQ; Karlberg T; Balaji G; Uttamchandani M; Suresh V; Schuler H; Low BC; Sivaraman J Structural Basis for the Allosteric Inhibitory Mechanism of Human Kidney-Type Glutaminase (KGA) and Its Regulation by Raf-Mek-Erk Signaling in Cancer Cell Metabolism. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (20), 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CA; Chen W; Gilson MK “Ligand configurational entropy and protein binding” PNAS, 2007, 104, 1534–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shukla K; Ferraris DV; Thomas AG; Stathis M; Duval B; Delahanty G; Alt J; Rais R; Rojas C; Gao P; Xiang Y; Dang CV; Slusher BS; Tsukamoto T “Design and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) analogs as glutaminase inhibitors” J. Med. Chem 2012, 55, 10551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian Y; Knape-Conde K; Erickson SD; Falcioni F; Gillespie P; Hakimi I; Mennona F; Ren Y; Salari H; So SS; Tilley JW “Potent MCH-1 receptor antagonists from cis-1,4-diaminocyclohexane-derived indane analogs” Biorg. Med. Chem. Letter 2013, 23, 4216–20. [DOI] [PubMed] [Google Scholar]
- 28.Wolfe JP; Ahman J; Sadighi JP; Singer RA; Buchwald SL “An ammonia equivalent for Pd-catalyzed amination of aryl halides and triflates” Tetrahedron Lett. 1997, 36, 6367–70. [Google Scholar]
- 29.Crystal structure coordinates have been deposited in the PDB for the crystals of GAC in complex with compounds 7d (PDB: 5FI2), 7e (PDB: 5FI6), 14b (PDB: 5FI7) and 14d (PDB: 5I94).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.