

Abstract
Diazirines are important for photoaffinity labeling, and their photoisomerization is relatively well-known. This work shows how hyperpolarized NMR spectroscopy can be used to characterize an unstable diazo-compound formed via photoisomerization of a 15N2-labeled silyl-ether-substituted diazirine. This diazirine is prepared in a nuclear spin singlet state via catalytic transfer of spin order from para-hydrogen. The active hyperpolarization catalyst is characterized to provide insight into the mechanism. The photochemical isomerization of the diazirine into the diazo-analogue allows the NMR invisible nuclear singlet state of the parent compound to be probed. The identity of the diazo-species is confirmed by trapping with N-phenyl maleimide via a cycloaddition reaction to afford bicyclic pyrazolines that also show singlet state character. The presence of singlet states in the diazirine and the diazo-compound is validated by comparison of experimental nutation behavior with theoretical simulation. The magnetic state lifetime of the diazo-compound is determined as 12 ± 1 s in CD3OD solution at room temperature, whereas its chemical lifetime is measured as 100 ± 5 s by related hyperpolarized NMR studies. Indirect evidence for the generation of the photoproduct para-N2 is presented.
Introduction
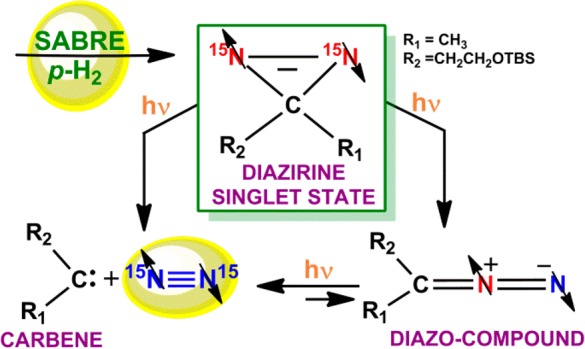
Diazirines exhibit significant photochemistry,1−3 and their photoisomerized diazo-products are versatile tools employed in chemical synthesis4 and chemical biology.5−7 In fact, their complex reactivity has interested the physical chemistry community for many years, with the importance of these compounds having increased due to their proven capabilities as good photoaffinity probes and molecular tags.8−14 Recently, the successful hyperpolarization of 15N2-diazirines via the SABRE-SHEATH (Signal Amplification By Reversible Exchange in Shield Enables Transfer to Heteronuclei) hyperpolarization technique has dramatically widened their relevance as it established that their 15N NMR signals can be enhanced to the point where they should be observable by in vitro or in vivo magnetic resonance imaging (MRI).15,16 In this Article, we exploit SABRE to prepare the diazirine in selected nuclear spin orientation and identify a diazirine complex as the active catalyst. We then use photoisomerization to produce the diazo-product, which is now available to NMR detection. We characterize this unstable intermediate and probe its kinetic evolution by NMR spectroscopy. Furthermore, we trap it by a 1,3 dipolar cycloaddition reaction, with singlet state retention, to provide further reaction rationalization. We finally discuss how this photochemical pathway can lead to the generation of para-N2.
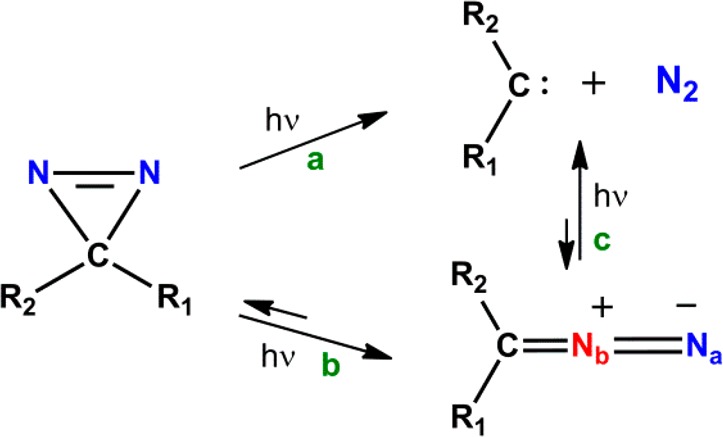
The two main photochemical pathways of diazirines that have been experimentally established by matrix photochemistry17−19 and laser flash photolysis20,21 (351 nm) correspond to the generation of a carbene with concomitant N2 extrusion (Scheme 1a), and photoisomerization to a linear diazo-isomer22 (Scheme 1b). The resulting diazo-species have been observed to decompose into a carbene and N2 gas as a result of secondary photolysis (Scheme 1c). Experimental studies of matrix photochemistry and theoretical calculations convincingly demonstrated that the formation of the diazo-isomer can take place through recombination of the carbene and N2.17 However, this photoprocess is again wavelength dependent with shorter or longer wavelengths favoring isomerization or N2 ejection depending on the reaction conditions or diazirine substitution pattern.23 While parent alkyldiazirines are stable compounds in solution, their diazo-analogues are very labile species, and as a consequence their trapping is often difficult, requiring the use of special methods.1
Scheme 1. Main Photochemical Pathways of Diazirines and Diazo-Compounds.
Diazirines are a special class of cyclic-azo compounds in which the electrons in the HOMO are located on the N atoms to just ∼55% with the remainder being delocalized within the molecule.24,25 Thus, diazirines are poor coordinating ligands, and very few of their metal complexes have been reported. For instance, the diazirine complex [(NH3)5Ru(N2C6H10)]2+ was prepared by reacting [(NH3)5Ru(H2O)]2+ with a stoichiometric amount of 3,3-pentamethylenediazirine under argon; coordination via a single nitrogen was established by X-ray crystallography, but no spectroscopic characterization in solution was achieved as rapid oxidation took place to yield the dinitrogen-Ru analogue and cyclohexanone.26 Nonacarbonyldiiron Fe2(CO)9 was also reported to react with 3,3-dimethyldiazirine in tetrahydrofuran to afford a relatively unstable complex in which two Fe(CO)4 groups were each σ-bound to a nitrogen atom of the diazirine (Scheme 2a). This complex was found to decompose slowly in solution affording complexes of the type shown in Scheme 2b where two molecules of diazirine have reacted with the loss of N2 and then coordinated to the Fe center.27 Cu(110) and Pd(110) surfaces have been employed to study the dissociative adsorption of diazirines at 124 K, and, while selective NN bond scission took place at Cu, both NN and CN bond scission were observed for Pd.28,29 In fact, most of the reactions between diazirines and a metal complex seem to afford a metal-carbene species via diazirine decomposition to the reactive carbene and N2.30−33
Scheme 2. Reported Dimethyldiazirine Coordination Modes Illustrated for Two Iron Centers.

THF plays a role in the solution-based decomposition to (b), which forms a structure with two dimethylketiminato groups.27
SABRE is a non hydrogenative PHIP (Para Hydrogen Induced Polarization) technique that builds on the earlier work of Bowers, Weitekamp, Eisenberg, and Bargon.34,35 It relies on the reversible binding to a suitable metal complex of both para-hydrogen (p-H2) and, in this case, a diazirine, to mediate hyperpolarization transfer into the free ligand.36 The SABRE-SHEATH variation uses a lower-field condition to build up 15N diazirine polarization efficiently. It has also been reported to create long-lasting signals that remain visible for 5.8 min after storage at 120 G in the case of 2-cyano-3-(d3-methyl-15N2-diazirine)-propanoic acid with >10 000-fold signal enhancements. Furthermore, a long-lived nuclear singlet spin order was created between the pair of chemically inequivalent 15N nuclei in this 15N2-labeled diazirine whose lifetime was 23 min at 3 G. However, when the same experiment was repeated at a 10 000 G (1 T) storage field, the lifetime reduced drastically to 12 s.15 Measurement of the hyperpolarized signal lifetimes of the diazirine as a function of substitution pattern gave useful insights into the magnetization transfer mechanism of SABRE-SHEATH, but the active SABRE species were not characterized experimentally, although possible products involving different coordination modes were proposed.16 Recently, 15N4-1,2,4,5-tetrazines were hyperpolarized via this approach and prepared in different spin orders. In addition, the bioorthogonal reaction of this species with cyclooctyne afforded hyperpolarized products. Indirect evidence for the generation of para-15N2 gas was also presented.37
Results
SABRE-SHEATH Mechanism
This study employed the SABRE catalyst Ir(d22-IMes)(COD)(Cl) 1 (where the IMes ligand is fully deuterated except for the pair of imidazolyl protons), and it involved examination of the silyl-ether-substituted diazirine 2 labeled with 15N2 by 1H NMR spectroscopy under SABRE and SABRE-SHEATH conditions (Scheme 3). To do this, solutions of 1 (5 mM) in the presence of excess pyridine-d5 (200 mM) and excess 2 (150 mM) were utilized. Pyridine plays a role as a coligand in this process and is essential to the formation of the active SABRE catalyst. These CD3OD (0.5 mL) solutions were activated via the addition of p-H2 (4 bar) to the NMR tube prior to monitoring.
Scheme 3. Structures of the Iridium Precatalyst 1 (Mesityl Groups Are Fully Deuterated) and the Silyl-Ether-Substituted Diazirine 2 Labeled with 15N2.
The SABRE-SHEATH process was then undertaken under conditions similar to those reported by Shen et al.16 Briefly, the solution was either manually shaken or bubbled for 20 s inside a μ-metal shield (at ∼0.5 μT measured by a milliGauss meter). These experimental conditions allowed hyperpolarization to be transferred from p-H2 to the 15N nuclei of 2 through SABRE (δ 465, Figure 1a,b). In addition to 15N sensitization, hyperpolarization is also transferred to the proton resonances of the CH3 and CH2 alkyl groups that are adjacent to the 15N center of 2 (Figure 1c). As these 1H resonances fail to show hyperpolarization when the sample is shaken outside the μ-metal shield, we conclude that this effect results from relayed transfer via the iridium hydrides formed by oxidative addition of p-H2 to the Ir precatalyst and the two 15N centers (see the Supporting Information). A peak for bound diazirine in the SABRE transfer catalyst is observed at δ 299 in the 15N{1H} spectrum (Figure 1b), and the following measurements secured its identity.
Figure 1.

Left: 15N{1H} NMR spectra from a reaction mixture of 1, 2, and pyr-d5 under p-H2 atmosphere in CD3OD. (a) Thermal reference NMR spectrum acquired with 1 scan and magnified (×8). (b) Corresponding hyperpolarized spectrum acquired identically after the sample was shaken with p-H2 inside a μ-metal shield. (c) Corresponding single scan 1H hyperpolarized NMR spectrum; the CH3 (blue ▲) and CH2 protons (purple ● and green star) of diazirine 2 receive hyperpolarization via transfer from 15N after the sample is shaken in a μ-metal shield.
Inspection of the hydride region of the 1H NMR spectra recorded in CD3OD at 298 K under both SABRE and SABRE-SHEATH conditions (p-H2 bubbled at 298 K) showed two broad peaks with chemical shifts indicating hydrides positioned trans to 15N (Figure 2a). In addition, resonances for the known [Ir(H)2(d22-IMes)(d5-pyr)3]Cl38 are seen at δ −22.7 alongside a minor signal for its hydride-deuteride analogue at δ −22.6. The broad nature of the new resonances suggested dynamic behavior, and hence experiments were repeated with NMR detection at low temperature. These broad resonances sharpened at 273 K resolving into four signals arising from structurally similar species in a 4:1 ratio. Resonances for the major compound were observed at δ −20.5 (antiphase doublet of doublets, JHH = 7.8 Hz, JNH = 31.8 Hz) and δ −16.2 (antiphase doublet, JHH = 7.8 Hz); the minor species displayed peaks at δ −20.7 (antiphase doublet of doublets, JHH = 9.3 Hz, JNH = 31.4 Hz) and δ −16.4 (antiphase doublet, JHH = 9.3 Hz) (Figure 2b). The doublets of doublets simplified to an antiphase doublet retaining only JHH coupling when 15N decoupling was applied (Figure 2c). All of the resonances exhibited p-H2-induced hyperpolarization. On the basis of these observations, we assigned these two compounds to two isomeric forms of the iridium dihydride complex 3 shown in Scheme 4.
Figure 2.
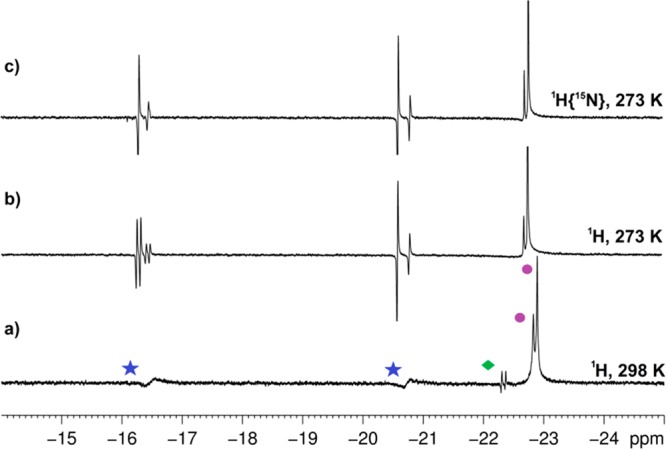
Hydride region of a series of 1H NMR spectra of a reaction mixture of 1, 2, and pyr-d5 in CD3OD under ∼4 bar of p-H2 pressure indicating the formation of 3 (blue ★). The purple ● correspond to [Ir(H)2(d22-IMes)(pyr-d5)3]Cl and its H/D analogue. The product indicated by the green ◆ is unidentified, and it is not detected at low temperature: (a) 1H NMR spectrum at 298 K; (b) 1H NMR spectrum at 273 K; and (c) 1H{15N} NMR spectrum at 273 K.
Scheme 4. (Top) Differential Ligand Binding in 3 Leading to 3a and 3b, Which Rapidly Interconvert at Room Temperature; and (Bottom) Key NMR Spins System Parameters for 3.

The two isomers of 3 are the active catalysts for SABRE as they possess one hydride that lies trans to a pyridine (resonances at lower δ) and the other hydride that lies trans to a 15N-labeled diazirine (resonances at higher δ that display 15N splitting). This experimental evidence agrees with previously reported DFT (Density Functional Theory) calculations that indicate η1 single-sided N-coordination.15 The two isomers 3a and 3b arise from binding of the Ir-center to one of the 15N centers of the diazirine, which renders them inequivalent. The two forms interconvert rapidly at room temperature such that broad hydride signals result (Scheme 4). The hopping of the Ir center from one 15N to the other is slowed at 273 K to the point where these two species become well distinguished. The formation of 3, and identical dynamic behavior, was observed when a similar reaction mixture was activated and analyzed on a larger scale via a flow reactor.39,40 Species related to 3 are also observed when a diazirine was employed in which the OTBS group of 2 is replaced by a carboxylic acid moiety (see the Supporting Information). Complex 3 is, to the best of our knowledge, the first example of a diazirine metal complex that has been characterized in solution by NMR spectroscopy where coordination to iridium proceeds through a single nitrogen atom. A theoretical analysis of the structure and J-coupling network associated with 3 is provided in the Supporting Information; although the reported theory has been described before,15 we include it to allow the reader to gain a better understanding of the experiments.
Photochemistry
A reaction mixture analogous to that described earlier was prepared and activated by bubbling p-H2 through the solution in the mixing chamber for 10 min. The hyperpolarization transfer process was then performed in a μ-metal shield (SABRE-SHEATH). Under these conditions, large signal enhancements were observed for the 15N response of 2 as already reported by Shen et al.16 For comparison purposes, the signal-to-noise ratio (SNR) in the corresponding 128 scan thermal reference spectrum was measured to be ≃7, while it became ≃230 in the single scan hyperpolarized spectrum. Zeeman polarization associated with 2 is therefore measured via this process.
The hyperpolarization of the 15N2-labeled-diazirine 2 will be the result of a mixture of direct polarization (SABRE-SHEATH) and singlet state transfer under ultralow magnetic field (mG). However, at relatively higher magnetic fields (1–1000 G), the formation of the singlet will be dominant with negligible contribution from direct magnetization.15 Furthermore, the singlet form can have a remarkably long magnetic state lifetime due to its symmetric character as exemplified by p-H2 whose reported lifetime is several months.37,41 However, it must be noted that because pure singlet states are nonmagnetic, they cannot be detected directly by NMR until their symmetry is broken.
We achieve symmetry breakage by irradiating the sample in a quartz mixing chamber, which is at the Earth’s magnetic field, with a broadband UV–vis lamp (either at λ > 290 nm or λ > 345 nm) for 20 s while p-H2 is bubbled through it. After this time, photolysis is stopped, and the solution is transferred into the NMR spectrometer (transfer time ≃ 2 s). A new hyperpolarized compound is then detected with an excellent SNR (≃165, 1 scan) by 15N{1H} NMR spectroscopy. Furthermore, a control experiment performed without irradiation confirmed this species as a photoproduct of 2. The new compound displayed two inequivalent nitrogen environments that are coupled to each other (δ 447 and δ 316, JNN = 11 Hz) (Figure 3a). Interestingly, when the experiment was repeated without 1H decoupling, the 15N peak at lower δ appeared as a doublet of sextets (Figure 3b). This fine structure suggested that one of the two inequivalent nitrogen centers interacts with the five nearby proton environments of CH2 and CH3 groups via a JNH coupling of 2.7 Hz. On the basis of these observations, we assign the new species to the predicted photoisomerization product alkyl diazo-4 of Scheme 5 where the signal at δ 316 can be assigned as the one corresponding to the N in the position α to carbon (Nb, Scheme 5). These chemical shift values are consistent with those reported in the literature for this type of compound.42,43 The generation of 2 in a nuclear singlet state is revealed through its photochemical conversion to 4 as the originally equivalent 15N nuclei now become inequivalent.
Figure 3.

(a) 15N{1H} NMR spectrum of a CD3OD hyperpolarized solution of 1, 2, and pyr-d5 after 20 s broadband photolysis displaying resonances for 4. (b) 15N NMR spectrum of the same solution.
Scheme 5. Structure of the Diazo-Compound 4 Formed by Photoisomerization of the 15N Labeled Diazirine, 2.
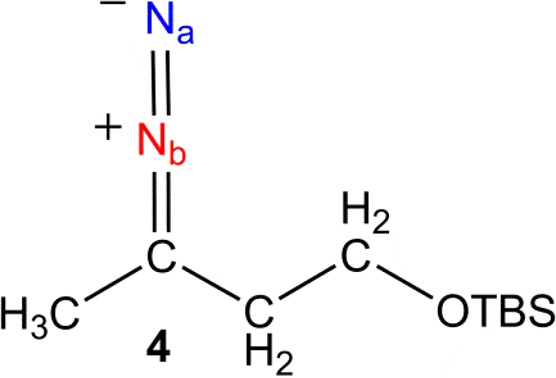
Figure 4 describes the evolution of the singlet state from low to high magnetic field. The singlet order will evolve under the depicted adiabatic pathways (Figure 4a) during this transfer to overpopulate the |αβ⟩ state. However, a 90° degree pulse will result in the signal shown in Figure 4b(i) where the four possible transitions are depicted in the color-coded energy level diagram. Thus, the best way to probe the singlet state is by applying a small flip angle pulse (avoiding the apparent coherence of the |αβ⟩ and |βα⟩ states) and only detecting the outer transitions, which are derived from the selective population of the |αβ⟩ state (Figure 4b(ii)).44−46 The resulting spectral pattern will therefore depend on the magnitude of the pulse flip angle. Figure 4d shows the SpinDynamica47,48 simulated NMR spectra of 4 according to the transitions of Figure 4a. Experimental findings show the predicted change in inner and outer peak intensities (Figure 4c). This behavior is striking evidence for these resonances being derived from a true singlet state.
Figure 4.

(a) Schematic depiction of a singlet order spin population for an inequivalent pair of coupled nuclear spins after hyperpolarization transfer in low field alongside the corresponding high-field populations after adiabatic transfer to high field. (b) Corresponding color-coded NMR spectral patterns predicted at high field after applying (i) a hard 90° flip angle pulse (purple, green, orange, and blue transitions in the above energy diagram) and (ii) a 10° flip angle pulse (purple and blue transitions). (c) 15N{1H} NMR spectra of a CD3OD reaction mixture containing the catalyst 1, diazirine 2, and pyr-d5. The solution was photolyzed before the acquisition of every spectrum that was hyperpolarized by 20 s of bubbling with p-H2. The pulse angle is increased from 10° to 170°, bottom to top. (d) SpinDynamica47,48 simulated 15N{1H} spectra for 4 assuming “pure” singlet order in the coupled 15N–15N system observed again by variable flip angle pulses of 10–170° in steps of 20° (bottom to top).
We have already established that 1H hyperpolarization transfer proceeds through nitrogen by reference to the parent diazirine. Analysis of the corresponding 1H NMR spectrum after irradiation showed the appearance of hyperpolarized peaks for the CH3 and CH2 groups, which are adjacent to 15N in the diazo-product 4. These resonances displayed a coupling of 2.4 Hz, which disappears when 15N is decoupled (see the Supporting Information). They are only visible under photolytic conditions indicating that transfer from the singlet state into these 1H resonances occurs.49−51
The detection of alkyl diazo-compounds has been achieved previously by time-resolved techniques and by matrix photochemistry.1,17 Very few of these compounds are isolable as a result of their extreme reactivity, and therefore they have not generally been characterized by NMR spectroscopy. Our success in using NMR spectroscopy to detect 4 is the result of combining the benefits of hyperpolarization and the long-lived singlet state character of 2. It should be noted that when the same experiment is performed employing the diazirine with the carboxylic acid group instead of the OTBS, the diazo-species was not detected, probably due to high reactivity and very short lifetime.
Diazoalkane 4 can also be detected when this reaction is performed in an NMR tube rather than a flow reactor under hyperpolarized conditions. The sealed nature of the system results in the detection of an extra resonance at δ 309 for ortho-15N2 alongside those for 4 (see the Supporting Information); we hypothesize that N2 gas is formed in a nonequilibrium state. The observation of N2 is consistent with the predicted photochemistry of 2, and concomitant formation of the corresponding carbene is required. Both CD3OD and pyr-d5 would be predicted to trap this intermediate under these reaction conditions.20,21,30,52 The presence of the methanol adduct was confirmed by LIFDI mass spectrometry analysis of the reaction mixture after photolysis (see the Supporting Information). We can therefore conclude that the formation of 4 runs alongside that of the carbene and N2, which also can be produced from 4.
Further control experiments were performed to validate these pathways. First, no hyperpolarization was observed in the absence of the iridium catalyst or pyridine, either with or without photolysis. In addition, diazo-4 could only be detected by NMR through its hyperpolarized response. A second set of measurements was then made on a CD3OD solution containing the catalyst 1, the diazirine 2, and pyr-d5 that was flushed with p-H2 for 20 s before 20 s of photolysis without further p-H2 bubbling (Figure 5a). At this point, the solution was transferred to the NMR spectrometer, and a 15N{1H} NMR spectrum recorded. Hyperpolarized resonances for 4 were now readily detected with both a 90° and a 30° pulse. More importantly, if the solution was sent back into the mixing chamber and rephotolyzed with no further p-H2 bubbling, resonances for 4 could still be detected at high field (30° pulse) even 90 s after initial singlet state preparation with 20% of the original signal intensity. Their appearance was again indicative of singlet state behavior (i.e., only outer peaks observed; see the Supporting Information). This confirms that the singlet state in diazirine 2 can be unlocked by photochemical conversion to 4.
Figure 5.
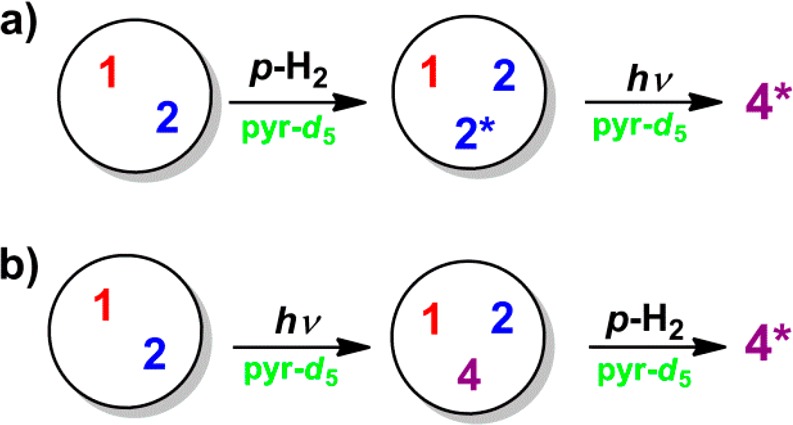
Schematic illustrating the steps in the control experiments elucidating the source and mechanism of hyperpolarization of 4 (the asterisks denote the hyperpolarized state of 2 and 4).
Furthermore, the same enhanced resonances of 4 were observed if the reaction mixture was photolyzed prior to bubbling with p-H2 (Figure 5b). Additionally, compound 4 could still be detected if the resulting solution was repolarized by flushing p-H2 through the previously irradiated solution. Hence, it can be concluded that diazo-4 can also be polarized by SABRE through its reversible coordination to iridium in a way similar to that of 2. This is not unexpected as diazo-compounds have been reported to act as good coordinating ligands.53 However, the diazo-compound 4 cannot be hyperpolarized in the absence of pyridine.15 We therefore propose a complex of the type [Ir(H)2(IMes)(py)2(4)]Cl to be the SABRE active catalyst. We noted, however, that the expected PHIP-enhanced hydride signals for the [Ir(H)2(IMes)(py)2(4)]Cl are not visible even at 253 K. Hence, very weak substrate binding or very fast ligand exchange is indicated.
Additional evidence for the formation of 4 was achieved by adding N-phenyl maleimide to the reaction mixture, which is well-known to act as a trapping agent for diazo-species.54,55 The resulting 1,3-dipolar cycloaddition reaction affords bicyclic pyrazoline (5a and 5b) in a 1:0.9 ratio of the diastereomers (Figure 6a). The 15N resonances for one diastereomer were observed at δ 509.2 and δ 462.2 (d, JNN = 16 Hz); the second isomer displayed peaks at δ 508.8 and δ 461.8 (d, JNN = 16 Hz) in this 15N{1H} NMR spectrum. However, as these products are now stable, they can subsequently be detected by normal NMR. The same bicyclic pyrazoline product ratio was determined under these non-hyperpolarized conditions, and no peaks for 4 were visible, confirming its instability and conversion into 5 (Figure 6b). We therefore conclude that the hyperpolarization levels exhibited by these secondary reaction products are indicative of their proportions. Thus, a common reaction pathway involving both retention of nuclear spin-correlation and diamagnetic electron-spin pair transfer during the cycloaddition reaction is indicated.56 These deductions are based on the up/down resonance phases seen for the 15N NMR peaks of 5a and 5b, which indicate they derive from an initial singlet state.
Figure 6.

(a) Schematic showing the products of a 1,3 dipolar cycloaddition reaction between the diazocompound 4 and N-phenyl maleimide; and (b) 15N{1H} NMR spectrum of the reaction mixture with trapping agent. The dots and the stars refer to the two inequivalent 15N responses of each diastereomer. The peaks are split because of the mutual 15N couplings.
Compounds 5a and 5b were again found to repolarize when p-H2 was flushed through the solution, thereby proving their ability also to coordinate to 16-electron [Ir(H)2(IMes)(py)2]Cl. Logically, these results demonstrate that the nuclear singlet state of 2 is retained during its conversion first to 4 and then to 5. We have therefore established the potential of such a hyperpolarization route to track a complex multistep organic reaction.57−60
Chemical and Nuclear Spin Order Lifetimes
The singlet state lifetime TS of the NMR invisible state of 2 can be probed indirectly via photochemistry once it has been successfully prepared by SABRE. To do this, a sample was subject to initial p-H2 polarization (20 s) prior to the cycling of low field photolysis and subsequent high field NMR detection of 4. Our estimate of the resulting lifetime of 2 is 17 ± 3 s (at 9.4 T), which lies close to that previously reported for related species under these reaction conditions.15,16 Unfortunately, we were able to collect only three points, which gives us the estimated value with large error (see the Supporting Information). Theis et al. have also demonstrated that the lifetimes of such states can be extended by storage at an appropriate low field.15
However, this situation is complicated by the fact that 4 should be unstable. We probed its lifetime TS directly by NMR. This involved a series of measurements where the sample was photolyzed for 20 s with concomitant p-H2 bubbling in the Earth’s magnetic field prior to storage at the same field for a set time before subsequent transfer into the high field for observation. After FID acquisition, the solution was left for a further 60 s in the NMR spectrometer to ensure full relaxation prior to transfer back to the mixing chamber. The resulting NMR spectra are shown in Figure 7 as the set time is increased from 0.25 to 45 s. The photolysis step converts a small and essentially equivalent amount of 2 into 4 each time. Thus, the only difference between these experiments is the amount of time that the solution waits in the Earth’s magnetic field before transfer to high field for monitoring. To confirm reproducibility, the same set of experiments was repeated in reverse, starting with the longest set time to the shortest (see the Supporting Information). The resulting signal decays were fit to a single exponential function with an average TS value for compound 4 at Earth’s magnetic field storage of 12 ± 1 s (Figure 8a,b).
Figure 7.

Series of 15N{1H} NMR spectra of a CD3OD solution of the photochemically generated diazo-compound 4 with encoding times of 0.25, 2, 5, 8, 12, 16, 25, 30, 35, and 45 s (from bottom to top).
Figure 8.

(a,b) Decay of signal intensity measurements for 4 after hyperpolarization and storage at low field in CD3OD solution prior to read-out. Points acquired from the shortest time to the longest. (c,d) Decay of signal intensity measurements for 4 in CD3OD solution with repolarization of the reaction mixture, which was photolyzed once prior to the first point. In all cases, ■ are the experimental points obtained by integrating the 15N{1H} NMR signal of compound 4, while red lines of best fit are added via an exponential decay function.
In a further series of measurements, the activated reaction mixture was first photolyzed for 20 s with p-H2 bubbling through it. After this time, the solution was transferred to the spectrometer and a 15N{1H} measurement completed. These spectra were acquired consecutively with a small flip angle pulse (30°) and a delay of 1.05 s between them. The resulting signal intensity decay was then fit to an exponential function (see the Supporting Information) to determine the TS of 4 at high field (9.4 T), which was 12 ± 3 s.
This process involving a single photolysis period was then repeated with repolarization. To do this, a sample was photolyzed for 20 s under p-H2 prior to making an initial 15N{1H} measurement. The solution was then transferred back to the mixing chamber and p-H2 added for 20 s before being sent back to the NMR spectrometer for FID acquisition. This sequence was repeated until no signal for 4 could be observed. In this way, we used the hyperpolarization of 4 to determine its chemical lifetime. These data were fitted to an exponential decay, and a chemical lifetime of 100 ± 5 s was estimated (Figure 8c,d). Our attempts to determine the chemical lifetime of 4 by UV–vis spectroscopy were unsuccessful, although we observed bleaching due to the consumption of 2.
Implications for the Formation of para-N2
We exploited the NMR signal integration of 5a and 5b formed in the cycloaddition process (Figure 6) to assess the ratio of N2 production versus the formation of the diazo-4. At 33% consumption of 2, where the products 5a and 5b account for 19%, the peak for ortho15N2 is 4% of the original signal for 2 (as derived from integration of the 15N spectrum under Boltzman conditions). The 10% of unassigned product could be rationalized by further N2 gas in the head space of the NMR tube (or by the NMR silent p-N2, see the Supporting Information). These results indicate an approximate 1:1 ratio for N2 liberation and photoisomerization processes assuming similar relaxation rates of 15N for the different species (see the Supporting Information). The observation of polarized N2 at δ 309 corroborates the potential formation of p-N2, the singlet nuclear spin isomer of N2 if the singlet state of 2 is preserved throughout the photochemical process. The detection of the hyperpolarized cycloaddition products (Figure 9, paths A and D) confirms retention of hyperpolarization. Similar reasoning has been used to justify p-N2 generation by others.37 Unfortunately, while diazo-4 was predicted to form by both photoisomerization (Figure 9, path A) and the recombination of N2 and the carbene17 (Figure 9, pathways B and C), we see no difference in product distribution when the photolysis step is performed under an N2 atmosphere. Therefore, we cannot attribute the detection of hyperpolarized 4 to the intermolecular recombination pathway (Figure 9, pathways B and C). We note that p-N2 can be formed by the direct route B of Figure 9 or as a secondary photoproduct via routes A and C. In either of these mechanisms, the p-N2 signal will however be NMR silent as it is perfectly symmetric and nonmagnetic in nature.
Figure 9.
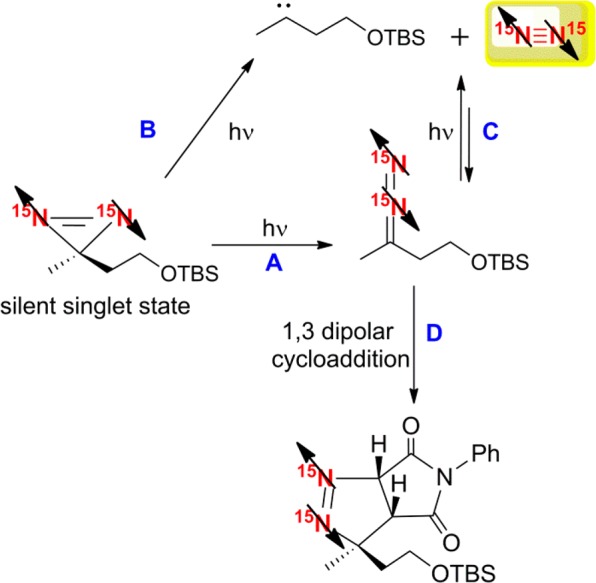
Reaction scheme summarizing the different pathways to the generation of p-N2.
Conclusions
In this Article, we have successfully used photochemistry to unlock the detection of a silent magnetic singlet state in 15N2-OTBS-diazirine 2 that was created via the SABRE mechanism (estimated TS at 9.4 T of 17 ± 3 s). Catalyst 3 of Scheme 4 with bound hydride, pyridine, and diazirine ligands was characterized by NMR spectroscopy and identified to play a key role in this step. The photoisomerization of 2 afforded unstable diazo-4 of Scheme 5, which was also characterized in solution by NMR spectroscopy. The TS of 4 was determined both at low field (≃500 mG) and at high field (9.4 T) to be 12 ± 1 s. This lifetime is relatively insensitive to the strength of the magnetic field. The nature of the singlet state of 4 has been validated by controlled radio frequency excitation and filtration experiments with the theoretical simulations detailed matching perfectly with the experimental findings. In contrast, the chemical lifetime of 4 is 100 ± 5 s and therefore substantially longer than its magnetic state lifetime.
We have further shown that 4 can be trapped by a suitable agent to afford complexes 5a and 5b. Remarkably, this process retains both hyperpolarization and singlet state in the products, indicating a 1,3-dipolar cycloaddition which must involve spin correlated electron transfer. Hence, we have established that hyperpolarization is a potential route to rationalize the outcome of organic pericyclic reactions. The different pathways for the photodecomposition of 2 are analyzed to account for generation of N2 and support the formation of p-N2. We are seeking to obtain more direct evidence for the formation of p-N2 and plan to exploit its hyperpolarization potential.
Acknowledgments
An anonymous donor, the Wellcome Trust (092506 and 098335), the University of York, and the EPSRC (EP/R51181X/1) are thanked for supporting this work. We thank Dr. Peter Rayner and Dr. Richard John for experimental help. Professor Robin N. Perutz, Dr. Pedro Aguiar, and Dr. William Unsworth are also acknowledged for stimulating discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10923. Raw NMR data can be downloaded at DOI 10.15124/8de06fe8-ea9d-49ff-bdd4-bb13cf605de5.
NMR characterization data, additional spectra, theoretical analysis, synthetic methods, and further technical descriptions (DOCX)
The authors declare no competing financial interest.
Supplementary Material
References
- Korneev S. M. Valence isomerization between diazo compounds and diazirines. Eur. J. Org. Chem. 2011, 2011, 6153. 10.1002/ejoc.201100224. [DOI] [Google Scholar]
- Griffin G. W. Generation of carbenes by photochemical cycloelimination. Angew. Chem., Int. Ed. Engl. 1971, 10, 537. 10.1002/anie.197105371. [DOI] [Google Scholar]
- Frey H. M.; Stevens I. D. R. Photolysis of dimethyldiazirine. J. Chem. Soc. 1963, 3514. 10.1039/jr9630003514. [DOI] [Google Scholar]
- Morandi B.; Carreira E. M. Iron-catalyzed cyclopropanation in 6 M KOH with in situ generation of diazomethane. Science 2012, 335, 1471. 10.1126/science.1218781. [DOI] [PubMed] [Google Scholar]
- Mix K. A.; Aronoff M. R.; Raines R. T. Diazo compounds: versatile tools for chemical biology. ACS Chem. Biol. 2016, 11, 3233. 10.1021/acschembio.6b00810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K. A.; Aronoff M. R.; McGrath N. A.; Raines R. T. Diazo groups endure metabolism and enable chemoselectivity in cellulo. J. Am. Chem. Soc. 2015, 137, 2412. 10.1021/ja5095815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath N. A.; Andersen K. A.; Davis A. K. F.; Lomax J. E.; Raines R. T. Diazo compounds for the bioreversible esterification of proteins. Chem. Sci. 2015, 6, 752. 10.1039/C4SC01768D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumranjit J.; Chung S. J. Recent advances in target characterization and identification by photoaffinity probes. Molecules 2013, 18, 10425. 10.3390/molecules180910425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky L.; Krom B. P.; Meijler M. M. Diazirine based photoaffinity labeling. Bioorg. Med. Chem. 2012, 20, 554. 10.1016/j.bmc.2011.06.066. [DOI] [PubMed] [Google Scholar]
- Hall M. A.; Xi J.; Lor C.; Dai S. P.; Pearce R.; Dailey W. P.; Eckenhoff R. G. m-Azipropofol (AziPm) a photoactive analogue of the intravenous general anesthetic propofol. J. Med. Chem. 2010, 53, 5667. 10.1021/jm1004072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebmann M.; Di Pasquale F.; Marx A. A new photoactive building block for investigation of DNA backbone interactions: photoaffinity labeling of human DNA polymerase beta. ChemBioChem 2006, 7, 7–1965. 10.1002/cbic.200600333. [DOI] [PubMed] [Google Scholar]
- Suchanek M.; Radzikowska A.; Thiele C. Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat. Methods 2005, 2, 261. 10.1038/nmeth752. [DOI] [PubMed] [Google Scholar]
- Kinoshita T.; Cano-Delgado A. C.; Seto H.; Hiranuma S.; Fujioka S.; Yoshida S.; Chory J. Binding of brassinosteroids to the extracellular domain of plant receptor kinase BRI1. Nature 2005, 433, 167. 10.1038/nature03227. [DOI] [PubMed] [Google Scholar]
- Chan E. W. S.; Chattopadhaya S.; Panicker R. C.; Huang X.; Yao S. Q. Developing photoactive affinity probes for proteomic profiling: hydroxamate-based probes for metalloproteases. J. Am. Chem. Soc. 2004, 126, 14435. 10.1021/ja047044i. [DOI] [PubMed] [Google Scholar]
- Theis T.; Ortiz G. X.; Logan A. W. J.; Claytor K. E.; Feng Y.; Huhn W. P.; Blum V.; Malcolmson S. J.; Chekmenev E. Y.; Wang Q.; Warren W. S. Direct and cost-efficient hyperpolarization of long-lived nuclear spin states on universal 15N2-diazirine molecular tags. Sci. Adv. 2016, 2, e1501438. 10.1126/sciadv.1501438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K.; Logan A. W. J.; Colell J. F. P.; Bae J.; Ortiz G. X.; Theis T.; Warren W. S.; Malcolmson S. J.; Wang Q. Diazirines as potential molecular imaging tags: probing the requirements for efficient and long-lived SABRE-induced hyperpolarization. Angew. Chem., Int. Ed. 2017, 56, 12112. 10.1002/anie.201704970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C. B.; Pimentel G. C. Matrix reaction of methylene with nitrogen to form diazomethane. J. Chem. Phys. 1964, 41, 3504. 10.1063/1.1725758. [DOI] [Google Scholar]
- Ammann J. R.; Subramanian R.; Sheridan R. S. Dicyclopropylcarbene - direct characterization of a singlet dialkylcarbene. J. Am. Chem. Soc. 1992, 114, 7592. 10.1021/ja00045a059. [DOI] [Google Scholar]
- Seburg R. A.; Mcmahon R. J. Photochemistry of matrix-isolated diazoethane and methyldiazirine - ethylidene trapping. J. Am. Chem. Soc. 1992, 114, 7183. 10.1021/ja00044a034. [DOI] [Google Scholar]
- Modarelli D. A.; Morgan S.; Platz M. S. Carbene formation, hydrogen migration, and fluorescence in the excited-states of dialkyldiazirines. J. Am. Chem. Soc. 1992, 114, 7034. 10.1021/ja00044a013. [DOI] [Google Scholar]
- Modarelli D. A.; Platz M. S. Interception of dimethylcarbene with pyridine - a laser flash-photolysis study. J. Am. Chem. Soc. 1991, 113, 8985. 10.1021/ja00023a079. [DOI] [Google Scholar]
- Amrich M. J.; Bell J. A. Photoisomerization of diazirine. J. Am. Chem. Soc. 1964, 86, 292. 10.1021/ja01056a044. [DOI] [Google Scholar]
- Yamamoto N.; Bernardi F.; Bottoni A.; Olivucci M.; Robb M. A.; Wilsey S. Mechanism of carbene formation from the excited-states of diazirine and diazomethane - an MC-SCF Study. J. Am. Chem. Soc. 1994, 116, 2064. 10.1021/ja00084a052. [DOI] [Google Scholar]
- Haselbach E.; Heilbron E.; Mannschr A.; Seitz W. Applications of photoelectron spectroscopy. Lone pair interaction in 3,3-dimethyldiazirine. Angew. Chem., Int. Ed. Engl. 1970, 9, 902. 10.1002/anie.197009021. [DOI] [Google Scholar]
- Kochanski E.; Lehn J. M. Electronic structure of cyclopropane, cyclopropene and diazirine an Ab Initio SCF-LCAO-MO study. Theor. Chim. Acta 1969, 14, 281. 10.1007/BF00527111. [DOI] [Google Scholar]
- Shur V. B.; Tikhonova I. A.; Aleksandrov G. G.; Struchkov Y. T.; Volpin M. E.; Schmitz E.; Jahnisch K. The diazirine complex [(NH3)5Ru(N2C6H10)]2+ - Synthesis, structure and conversion into the dinitrogen complex [(NH3)5RuN2]2+. Inorg. Chim. Acta 1980, 44, L275. 10.1016/S0020-1693(00)91034-X. [DOI] [Google Scholar]
- Albini A.; Kisch H. Transition-metal complexes of azo-compounds. Complexation and cleavage of N = N bond of diazirines by iron carbonyls. J. Organomet. Chem. 1975, 94, 75. 10.1016/S0022-328X(00)82532-2. [DOI] [Google Scholar]
- Roy J. R.; Monim S. S.; Mcbreen P. H. Totally selective NN bond-cleavage in the dissociative adsorption of diazirine on Cu(110). J. Am. Chem. Soc. 1994, 116, 11883. 10.1021/ja00105a031. [DOI] [Google Scholar]
- Mcbreen P. H.; Roy J. R.; Monim S. S. Non-synergistic pi-sigma chemisorption bonding - CH2N2 on Pd(110) and Cu(110). Chem. Phys. Lett. 1994, 223, 481. 10.1016/0009-2614(94)00482-X. [DOI] [Google Scholar]
- Barra M.; Fisher T. A.; Cernigliaro G. J.; Sinta R.; Scaiano J. C. On the photodecomposition mechanism of ortho-diazonaphthoquinones. J. Am. Chem. Soc. 1992, 114, 2630. 10.1021/ja00033a041. [DOI] [Google Scholar]
- Avent A. G.; Benyunes S. A.; Chaloner P. A.; Hitchcock P. B. Synthesis of mu-carbene complexes of rhodium from substituted diazirines. J. Chem. Soc., Chem. Commun. 1987, 1285. 10.1039/C39870001285. [DOI] [Google Scholar]
- Benyunes S. A.; Chaloner P. A. Synthesis of mu-carbene complexes of cobalt from substituted diazirines. J. Organomet. Chem. 1988, 341, C50. 10.1016/0022-328X(88)89122-8. [DOI] [Google Scholar]
- Avent A. G.; Benyunes S. A.; Chaloner P. A.; Gotts N. G.; Hitchcock P. B. Synthesis of mu-carbene and mu-carbyne complexes of rhodium and cobalt from substituted diazirines - crystal-structures of [Rh2(eta5-C5Me5)2(CO)2[μ-C(C6H4F4)(OMe)]] and [Rh2(η5-C5Me5)2(μ-CO)(μ-C6H4Me4)][BPh4]. J. Chem. Soc., Dalton Trans. 1991, 1417. 10.1039/DT9910001417. [DOI] [Google Scholar]
- Bowers C. R.; Weitekamp D. P. Para-hydrogen and synthesis allow dramatically enhanced nuclear alignment. J. Am. Chem. Soc. 1987, 109, 5541. 10.1021/ja00252a049. [DOI] [Google Scholar]
- Eisenschmid T. C.; Kirss R. U.; Deutsch P. P.; Hommeltoft S. I.; Eisenberg R.; Bargon J.; Lawler R. G.; Balch A. L. Para hydrogen induced polarization in hydrogenation reactions. J. Am. Chem. Soc. 1987, 109, 8089. 10.1021/ja00260a026. [DOI] [Google Scholar]
- Adams R. W.; Aguilar J. A.; Atkinson K. D.; Cowley M. J.; Elliott P. I. P.; Duckett S. B.; Green G. G. R.; Khazal I. G.; Lopez-Serrano J.; Williamson D. C. Reversible interactions with para-hydrogen enhance NMR sensitivity by polarization transfer. Science 2009, 323, 1708. 10.1126/science.1168877. [DOI] [PubMed] [Google Scholar]
- Bae J. N.; Zhou Z. J.; Theis T.; Warren W. S.; Wang Q. 15N4-1,2,4,5-tetrazines as potential molecular tags: integrating bioorthogonal chemistry with hyperpolarization and unearthing para-N2. Sci. Adv. 2018, 4, eaar2978. 10.1126/sciadv.aar2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley M. J.; Adams R. W.; Atkinson K. D.; Cockett M. C. R.; Duckett S. B.; Green G. G. R.; Lohman J. A. B.; Kerssebaum R.; Kilgour D.; Mewis R. E. Iridium N-heterocyclic carbene complexes as efficient catalysts for magnetization transfer from para-hydrogen. J. Am. Chem. Soc. 2011, 133, 6134. 10.1021/ja200299u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd L. S.; Adams R. W.; Bernstein M.; Coombes S.; Duckett S. B.; Green G. G. R.; Lewis R. J.; Mewis R. E.; Sleigh C. J. Utilization of SABRE-derived hyperpolarization to detect low-concentration analytes via 1D and 2D NMR methods. J. Am. Chem. Soc. 2012, 134, 12904. 10.1021/ja3051052. [DOI] [PubMed] [Google Scholar]
- Mewis R. E.; Atkinson K. D.; Cowley M. J.; Duckett S. B.; Green G. G. R.; Green R. A.; Highton L. A. R.; Kilgour D.; Lloyd L. S.; Lohman J. A. B.; Williamson D. C. Probing signal amplification by reversible exchange using an NMR flow system. Magn. Reson. Chem. 2014, 52, 358. 10.1002/mrc.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S. Conversion rate of para-hydrogen to ortho-hydrogen by oxygen: implications for PHIP gas storage and utilization. MAGMA 2014, 27, 195. 10.1007/s10334-013-0399-y. [DOI] [PubMed] [Google Scholar]
- Lown J. W.; Chauhan S. M. S.; Koganty R. R.; Sapse A. M. Alkyldinitrogen species implicated in the carcinogenic, mutagenic, and anticancer activities of N-nitroso compounds - characterization by 15N-NMR of 15N-enriched compounds and analysis of DNA-base situselectivity by abinitio calculations. J. Am. Chem. Soc. 1984, 106, 6401. 10.1021/ja00333a051. [DOI] [Google Scholar]
- Duthaler R. O.; Forster H. G.; Roberts J. D. 15N and 13C Nuclear magnetic-resonance spectra of diazo and diazonium compounds. J. Am. Chem. Soc. 1978, 100, 4974. 10.1021/ja00484a009. [DOI] [Google Scholar]
- Tayler M. C. D.; Marco-Rius I.; Kettunen M. I.; Brindle K. M.; Levitt M. H.; Pileio G. Direct enhancement of nuclear singlet order by dynamic nuclear polarization. J. Am. Chem. Soc. 2012, 134, 7668. 10.1021/ja302814e. [DOI] [PubMed] [Google Scholar]
- Natterer J.; Bargon J. Parahydrogen induced polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293. 10.1016/S0079-6565(97)00007-1. [DOI] [Google Scholar]
- Pravica M. G.; Weitekamp D. P. Net NMR alignment by adiabatic transport of para-hydrogen addition-products to high magnetic-field. Chem. Phys. Lett. 1988, 145, 255. 10.1016/0009-2614(88)80002-2. [DOI] [Google Scholar]
- Bengs C.; Levitt M. H. Spindynamica: symbolic and numerical magnetic resonance in a mathematica environment. Magn. Reson. Chem. 2018, 56, 374. 10.1002/mrc.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfram Research, I. Mathematica, Version 11.0; Wolfram Research, Inc.: Champaign, IL, 2018.
- Roy S. S.; Appleby K. M.; Fear E. J.; Duckett S. B. SABRE-Relay: a versatile route to hyperpolarization. J. Phys. Chem. Lett. 2018, 9, 1112. 10.1021/acs.jpclett.7b03026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S. S.; Norcott P.; Rayner P. J.; Green G. G. R.; Duckett S. B. A Hyperpolarizable 1H magnetic resonance probe for signal detection 15 minutes after spin polarization storage. Angew. Chem., Int. Ed. 2016, 55, 15642. 10.1002/anie.201609186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S. S.; Rayner P. J.; Norcott P.; Green G. G. R.; Duckett S. B. Long-lived states to sustain SABRE hyperpolarised magnetisation. Phys. Chem. Chem. Phys. 2016, 18, 24905. 10.1039/C6CP02844F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creary X.; Butchko M. A. beta-silylcarbenes from isolable diazosilanes. J. Org. Chem. 2002, 67, 112. 10.1021/jo0106546. [DOI] [PubMed] [Google Scholar]
- Alvarez M. A.; Garcia M. E.; Garcia-Vivo D.; Huergo E.; Ruiz M. A.; Vega M. F. Thermally Stable diazoalkane derivatives of the unsaturated ditungsten hydride [W2Cp2(H)(eta-PCy2)(CO)2]. Organometallics 2015, 34, 3833. 10.1021/acs.organomet.5b00466. [DOI] [Google Scholar]
- Stevens I. D. R.; Liu M. T. H.; Soundararajan N.; Paike N. The thermal-decomposition of diazirines - 3-(3-methyldiazirin-3-yl)propan-1-ol and 3-(3-Methyldiazirin-3-Yl)propanoic acid. J. Chem. Soc., Perkin Trans. 2 1990, 2, 661. 10.1039/p29900000661. [DOI] [Google Scholar]
- Molchanov A. P.; Diev V. V.; Kostikov R. R. Reactions of aliphatic diazo compounds: IV. Reaction of diphenyldiazomethane with substituted Imides of maleic and itaconic acids. Russ. J. Org. Chem. 2002, 38, 259. 10.1023/A:1015582104282. [DOI] [Google Scholar]
- Huisgen R. Kinetik Und Mechanismus 1.3-Dipolarer Cycloadditionen. Angew. Chem. 1963, 75, 742. 10.1002/ange.19630751603. [DOI] [Google Scholar]
- Battilocchio C.; Feist F.; Hafner A.; Simon M.; Tran D. N.; Allwood D. M.; Blakemore D. C.; Ley S. V. Iterative reactions of transient boronic acids enable sequential C-C bond formation. Nat. Chem. 2016, 8, 360. 10.1038/nchem.2439. [DOI] [PubMed] [Google Scholar]
- Poh J. S.; Tran D. N.; Battilocchio C.; Hawkins J. M.; Ley S. V. A Versatile room-temperature route to di- and trisubstituted allenes using flow-generated diazo compounds. Angew. Chem., Int. Ed. 2015, 54, 7920. 10.1002/anie.201501538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran D. N.; Battilocchio C.; Lou S. B.; Hawkins J. M.; Ley S. V. Flow chemistry as a discovery tool to access sp2-sp3 cross-coupling reactions via diazo compounds. Chem. Sci. 2015, 6, 1120. 10.1039/C4SC03072A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronoff M. R.; Gold B.; Raines R. T. 1,3-Dipolar cycloadditions of diazo compounds in the presence of azides. Org. Lett. 2016, 18, 1538. 10.1021/acs.orglett.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.