

Abstract

Knowledge about the functions of individual proteins on a system-wide level is crucial to fully understand molecular mechanisms underlying cellular processes. A considerable part of the proteome across all organisms is still poorly characterized. Mass spectrometry is an efficient technology for the global study of proteins. One of the most prominent methods for accurate proteome-wide comparative quantification is stable isotope labeling by amino acids in cell culture (SILAC). However, application of SILAC to prototrophic organisms such as Saccharomyces cerevisiae, also known as baker’s yeast, is compromised since they are able to synthesize all amino acids on their own. Here, we describe an advanced strategy, termed 2nSILAC, that allows for in vivo labeling of prototrophic baker’s yeast using heavy arginine and lysine under fermentable and respiratory growth conditions, making it a suitable tool for the global study of protein functions. This generic 2nSILAC strategy allows for directly using and systematically screening yeast mutant strain collections available to the scientific community. We exemplarily demonstrate its high potential by analyzing the effects of mitochondrial gene deletions in mitochondrial fractions using quantitative mass spectrometry revealing the role of Coi1 for the assembly of cytochrome c oxidase (respiratory chain complex IV).
Knowledge about protein functions is mandatory to fully understand cellular processes. Although numerous genomes have been sequenced decades ago, the functions of many proteins are still unknown or only poorly characterized. The genome of the budding yeast Saccharomyces cerevisiae, an important and widely used eukaryotic model organism to study biological processes, contains approximately 6700 genes, of which more than 10% are still uncharacterized.1,2
A powerful strategy to elucidate the function of poorly characterized proteins and the cellular process in which they are involved is the proteome-wide analysis of cells deficient for a gene encoding such protein by quantitative mass spectrometry (MS).3 For most accurate quantitative MS analysis at a proteome-wide scale, stable isotope labeling by amino acids in cell culture (SILAC) is used,4,5 which is based on the metabolic incorporation of isotope-coded “heavy” amino acids into the proteome during cell growth. This allows for mixing of differentially labeled cells directly after harvesting, which minimizes differences caused by experimental variations during sample handling. This is of particular advantage for quantitative proteomic studies of subcellular structures including organelles that are purified following multistep protocols. To exploit its full potential, SILAC is best performed with isotopically labeled lysine and arginine.6 It is frequently observed, though, that heavy arginine is converted to heavy proline,7−11 which compromises quantification of SILAC peptide pairs.12 Effective strategies have been developed to minimize arginine-to-proline conversion, including culturing of cells in the presence of unlabeled proline.8,13
Since yeast is prototrophic and, thus, able to synthesize all amino acids endogenously, SILAC studies are usually performed using strains in which genes coding for enzymes of the arginine and lysine biosynthesis pathways have been deleted to ensure complete labeling of the proteome with the heavy versions of these amino acids.14,15 However, this strategy precludes the use of yeast mutant strain libraries that are readily accessible for the scientific community,16−20 but lack the auxotrophies required for SILAC.
Previous studies showed that prototrophic yeast can be labeled with heavy amino acids when grown on glucose.21−23 Walther and co-workers introduced this concept as “native SILAC” (nSILAC) using heavy lysine for labeling of the yeast strain W303 grown on glucose22 and further applied this concept to study protein turnover in yeast.24
In this work, we developed “2nSILAC”, a strategy that allows for complete metabolic labeling of prototrophic yeast with both isotope-coded lysine and arginine. We show that this strategy works efficiently for yeast grown on fermentable and respiratory carbon sources making it well-suited for the study of mitochondria, organelles with essential functions in eukaryotic cells, which usually requires respiratory growth conditions. We demonstrate the high potential of 2nSILAC for proteome-wide studies by using it for loss-of-function studies of selected mitochondrial proteins in S. cerevisiae under respiratory conditions.
Experimental Section
Yeast Strains, Cultivation, and Metabolic Labeling
S. cerevisiae strains used in this study were BY4741 (and derivatives thereof) and W303, both prototrophic for arginine and lysine. Yeast were cultured in synthetic complete (SC) medium containing 0.17% (w/v) yeast nitrogen base without amino acids, 0.5% (w/v) ammonium sulfate, CSM-Arg-Lys amino acid dropout mix (Sunrise Science Products), l-arginine and l-lysine (50 mg/L each), and 3% (w/v) glycerol/0.02% (w/v) glucose, or 2% (w/v) galactose as carbon source. For growth on 2% (w/v) glucose, the medium was supplemented with l-histidine, l-leucine, l-methionine, l-tryptophan, adenine, and uracil (20 mg/L each) instead of the CSM amino acid dropout mix. l-Proline (200 mg/L) was added as indicated. Metabolic labeling was performed using stable isotope-coded “heavy” arginine (13C6/15N4; Arg10) and lysine (13C6/15N2; Lys8) or “medium-heavy” arginine (13C6/14N2; Arg6) and lysine (2H4; Lys4) instead of the “light” counterparts.
Preparation of Samples for Mass Spectrometry
Aliquots of whole cell lysates and mitochondria-enriched fractions were proteolytically digested using LysC, trypsin, or a combination of both proteases. Tryptic peptides obtained from whole cell extracts of unlabeled cells were chemically labeled using stable isotope dimethyl-labeling. Proteolytic peptides of whole cell extracts derived from mixtures of differentially light, medium-heavy, and heavy SILAC-labeled cells were fractionated (8 fractions) by high pH reversed-phase chromatography. For more details, see Supporting Information.
Mass Spectrometry and Data Analysis
Peptide mixtures were analyzed on an LTQ Orbitrap XL or a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). For protein identification and relative quantification, mass spectrometric raw data were processed using the software MaxQuant/Andromeda.25,26 Database searches were performed against all entries of the Saccharomyces Genome Database (http://www.yeastgenome.org/; downloaded September 2011). Detailed information and specific parameters for MS and data analysis are given in Supporting Information.
Results and Discussion
Effects of Exogenous Arginine and Lysine on the Amino Acid Metabolism of Prototrophic Yeast
To analyze the effects of exogenously added arginine and lysine on the yeast proteome, we selected the S. cerevisiae strains BY4741, on which many commonly used yeast collections are based,16−20 and W303, which was previously used for nSILAC.22 We grew cells in the presence and absence of unlabeled arginine and lysine using glucose as carbon source and quantitatively compared their proteomes by LC-MS following peptide stable isotope dimethyl labeling (n = 3). Enzymes of the α-aminoadipate pathway of lysine biosynthesis (i.e., Lys1, Lys2, Lys4, Lys9, Lys12, Lys20, and Aco2) were considerably decreased in both BY4741 and W303 cells (Figure 1a,b, Tables S-1 and S-2). These changes in protein abundance result from feedback inhibition of the lysine biosynthesis, in which the expression of the respective genes is repressed by lysine.22,27
Figure 1.

Regulation of arginine and lysine biosynthetic enzymes in prototrophic yeast strains. (a, b) BY4741 and W303 cells were grown in in the presence (+) or absence (−) of external arginine (Arg) and lysine (Lys). For quantitative proteome analysis, stable isotope peptide dimethyl labeling was performed followed by LC-MS (n = 3). Proteins of the arginine (circles) and lysine (triangles) metabolic pathways are labeled. Larger symbols (blue, red, or gray) indicate proteins significantly down-/up-regulated (i.e., both t-test and Significance B p-value <0.05). Dashed horizontal lines mark the t-test p-value of 0.05. (c) Arginine biosynthesis pathway in S. cerevisiae. Colors indicate relative protein abundances of enzymes of the arginine biosynthesis and degradation as determined in (a). n.i., not identified. (d) Transcriptional regulation of the arginine biosynthesis pathway.66 ARC, arginine control elements.
In BY4741 cells, exogenous arginine led to a significant downregulation of numerous enzymes of the arginine biosynthesis pathway (Arg1, Arg3, Arg5/6, Arg8, and Cpa1; Figure 1a, Table S-1). The biosynthesis of arginine starts with the acetylation of glutamate to form N-acetylglutamate, which is ultimately converted to ornithine via the acetylated derivatives cycle28,29 (Figure 1c). These first five steps of the arginine biosynthesis occur in the mitochondrial matrix29 and are catalyzed by the acetylglutamate synthase (Arg2), the acetylglutamate kinase/N-acetyl-γ-glutamyl-phosphate reductase (Arg5/6), the acetylornithine aminotransferase (Arg8), and the mitochondrial ornithine acetyltransferase (Arg7). Arg7 is a bifunctional enzyme that may also catalyze the acetylation of glutamate.30 Ornithine is exported into the cytosol via the ornithine transporter Ort1 in the inner mitochondrial membrane30 and converted to arginine in three steps that require the enzymes ornithine carbamoyltransferase (Arg3), arginosuccinate synthetase (Arg1), and argininosuccinate lyase (Arg4). Alternatively, arginine can be produced from cytosolic glutamine, which is converted to citrulline via carbamoyl phosphate by the arginine-specific carbamoyl phosphate synthetase Cpa1 and Arg3. Except for Arg2, all enzymes required for arginine biosynthesis were identified in our study of BY4741 cells (Figure 1a, Table S-1). We hypothesize that Arg2 eluded detection because its abundance is very low (∼100 molecules per cell), while Arg7, for example, is approximately 180-fold higher expressed (∼18000 molecules per cell) in cells grown on glucose.1
S. cerevisiae is able to import exogenous arginine, preferentially via the arginine permease Can1,31,32 and, to a minor extent, through its paralog Alp133,34 and the general amino acid permease Gap134,35 located in the plasma membrane (Figure 1c). In case of excess availability, free cytosolic arginine is either transported into the vacuole via the permease Vba2 (Figure 1c),32,36 or converted to proline.37,38 The arginase Car1 and the ornithine aminotransferase Car2, which catalyze the initial steps of the arginine-to-proline degradation,37,38 were considerably upregulated in BY4741 cells (Figure 1a,c, Table S-1), indicating that the conversion is higher in cells grown with exogenous arginine.
Arginine biosynthesis and degradation are tightly balanced in S. cerevisiae, allowing the organism to adapt to varying nutritional conditions. Free arginine is known to repress genes involved in arginine biosynthesis,39 whereas CAR1 and CAR2 are positively regulated by arginine.40 This fits our observation that levels of proteins involved in arginine biosynthesis are reduced and Car1 and Car2 are increased (Figures 1a, c, Table S-1). The arginine-dependent regulatory mechanism in S. cerevisiae relies on the ArgR protein complex, a transcription factor, which in a complex with Mcm1 directly senses the concentration of arginine and affects the abundances of distinct arginine metabolic enzymes41 (Figure 1d). The reduced levels of arginine biosynthetic enzymes observed in BY4741 cells grown with arginine (Figure 1a,c) suggest that the cells preferably use the arginine supplied with the medium, which, after import, leads to shutdown of arginine biosynthesis by repression of arginine-sensitive genes of this pathway (Figure 1d). W303 cells differ from BY4741 cells by a loss-of-function mutation in the CAN1 gene.42 As a consequence, import of exogenous arginine is impaired, which is in line with our finding of virtually unaltered levels of arginine biosynthetic enzymes in cells grown with or without exogenous arginine (Figure 1b, Table S-2).
In summary, our data suggest that the lysine and arginine prototrophic strain BY4741 can be used for metabolic labeling using both heavy arginine and lysine, in the following referred to as “2nSILAC”, while W303 cells are only amenable to labeling with heavy lysine.
Quantitative Labeling of Prototrophic BY4741 Cells with Heavy Arginine and Lysine
We monitored the incorporation of stable isotope-coded arginine (Arg10) and lysine (Lys8) into the proteome of BY4741 grown on different carbon sources. In addition to glucose (2%; fermentable conditions), we used 3% glycerol/0.02% glucose (nonfermentable conditions) and 2% galactose (alternative carbon source), which are commonly used to study mitochondria in S. cerevisiae. Cells were harvested at different times during growth (i.e., at OD600 of 0.5, 1.0, 1.5, and ∼4.0) to assess Arg10/Lys8 incorporation by LC-MS (n = 3). For precultured BY4741 cells, we determined an incorporation of 65–90% with lysine generally exhibiting a higher incorporation than arginine (Figure 2a–c, t0; Table S-3). In BY4741 cells harvested at early to mid log phase (OD600 0.5–1.5) incorporation of Arg10/Lys8 increased to >98% for all carbon sources tested. At a higher OD600 of ∼4, incorporation of Arg10/Lys8 was decreased in cells grown in glucose and galactose, while it remained high (>98%) in BY4741 cells grown on glycerol. In agreement with their genotype, W303 cells efficiently incorporated Lys8 (>98%) but not Arg10 (Figure 2d; Table S-3).
Figure 2.

Metabolic incorporation of heavy arginine (Arg10) and lysine (Lys8) into proteins of BY4741 and W303 cells during growth on different carbon sources as indicated (a–d). Overnight cultures were diluted to an OD600 of 0.025, cells were taken immediately after dilution (t0) and at different ODs (0.5, 1.0, 1.5, ∼4) and analyzed by LC-MS. Incorporation of Arg10 and Lys8 per condition and OD were calculated based on 8333–21267 peptide features (for more details, see Supporting Information and Table S-3). Error bars, standard deviation (n = 3); gal, galactose; glc, glucose; gly, glycerol.
Taken together, these data confirm that the strain BY4741 preferentially uses exogenous arginine and lysine, making it well applicable for 2nSILAC of yeast under fermentable and nonfermentable growth conditions. However, since distinct growth conditions or genetic modifications of BY4741 may affect labeling efficiencies for heavy SILAC amino acids, complete incorporation of SILAC amino acids should routinely be checked.
Suppression of Arginine-to-Proline Conversion
To examine the extent of arginine-to-proline conversion in BY4741, cells were grown in the presence or absence of unlabeled proline.13 Quantitative proteome analysis did not reveal an effect of exogenous proline on the abundance of enzymes involved in proline biosynthesis (Pro1, Pro2, Pro3)43,44 or the conversion of arginine to proline (Car1, Car2, Pro3)37,38 (Figure S-1a,b and Table S-4).
Without exogenous proline, we generally observed a higher number of Pro6-containing peptides of up to 16% (Tables 1 and S-3; see Supporting Information for details about the determination of arginine-to-proline conversion). Adding proline largely prevented heavy arginine-to-proline conversion, and at an OD600 of 0.5–1.5, less than 5% of all identified peptides contained Pro6. At a higher OD600 of 4.0, however, conversion increased to up to 8.9% of Pro6-containing peptides under galactose condition (Tables 1 and S-3), which needs to be taken into account when 2nSILAC is used for addressing biological questions that require culturing of yeast to a higher OD, for example in aging experiments. Similar results were obtained when peptide intensities were used for the calculation of arginine-to-proline conversion (Table S-5 and Figure S-2). Thus, to minimize the metabolic conversion of heavy arginine to heavy proline and thereby ensure a most accurate relative protein quantification, it is essential to add unlabeled proline to the growth medium. Furthermore, our data indicate that it is advisable to routinely check the extent of arginine-to-proline conversion. In addition, performing label-switch experiments has been shown to counterbalance quantification inaccuracies based on arginine-to-proline conversion.67
Table 1. Extent of Heavy Arginine-to-Proline Conversion During Native SILAC of Cells Grown Using the Indicated Carbon Sourcesa.
| Pro6-containing peptides (% ± SD) |
|||||
|---|---|---|---|---|---|
| C-source | Pro | OD 0.5 | OD 1.0 | OD 1.5 | OD 4.0 |
| glc | – | 6.3 ± 2.27 | 8.7 ± 3.72 | 12.9 ± 3.86 | 14.6 ± 3.79 |
| + | 2.7 ± 0.25 | 3.5 ± 0.12 | 3.9 ± 0.17 | 6.6 ± 0.15 | |
| gal | – | 8.1 ± 0.15 | 8.6 ± 0.56 | 10.9 ± 0.83 | 10.8 ± 0.78 |
| + | 0.2 ± 0.02 | 2.8 ± 0.6 | 5.0 ± 0.53 | 8.9 ± 0.53 | |
| gly/glc | – | 1.2 ± 0.46 | 2.9 ± 0.95 | 6.9 ± 1.47 | 16.8 ± 2.64 |
| + | 0.5 ± 0.21 | 0.9 ± 0.28 | 1.6 ± 0.17 | 3.0 ± 1.30 | |
Cells were grown as described in Figure 2. The numbers indicate the relative number of peptides containing Pro6 in % in relation to the total number of peptides identified in the respective dataset. SD, standard deviation (n = 3); OD, optical density at 600 nm; glc, glucose; gal, galactose; gly, glycerol.
2nSILAC for Global Proteome Quantification in S. cerevisiae
To assess 2nSILAC for global proteome quantification in S. cerevisiae, we grew cells under respiratory conditions and labeled them with either light, medium-heavy, or heavy lysine only (Lys0/4/8; nSILAC) or both arginine and lysine (Arg0/6/10, Lys0/4/8; 2nSILAC) in a triple labeling experiment. We mixed cells in defined amounts (6:1.5:1 [L/M/H]; n = 4) followed by protein digestion and LC-MS analysis. For nSILAC, we used the protease LysC, for 2nSILAC trypsin or a combination of both proteases. A total of 2191 proteins were identified, of which 1879 (85.8%) were detected in all three approaches (Figure 3a, Table S-6). Most proteins (93, 4.2%) were exclusively identified using LysC/trypsin. 2nSILAC resulted in significantly higher numbers of peptides (16783 ± 737 for LysC/trypsin and 15265 ± 567 for trypsin) compared to “lysine only” labeling with LysC digest (11789 ± 844; Figure 3b, Table S-6), while numbers of proteins identified and quantified were similar between all three approaches (1864 ± 70 to 1901 ± 44 proteins identified; 1801 ± 70 to 1806 ± 32 proteins quantified; Table S-6). The average sequence coverage of quantified proteins was consistently higher following LysC/trypsin tandem digestion, considering both the overall set of proteins (“total”) and proteins grouped according to their subcellular localization (Figure 3c, Table S-6), which is in line with the observation that this approach provided the highest number of peptides (Figure 3b).
Figure 3.
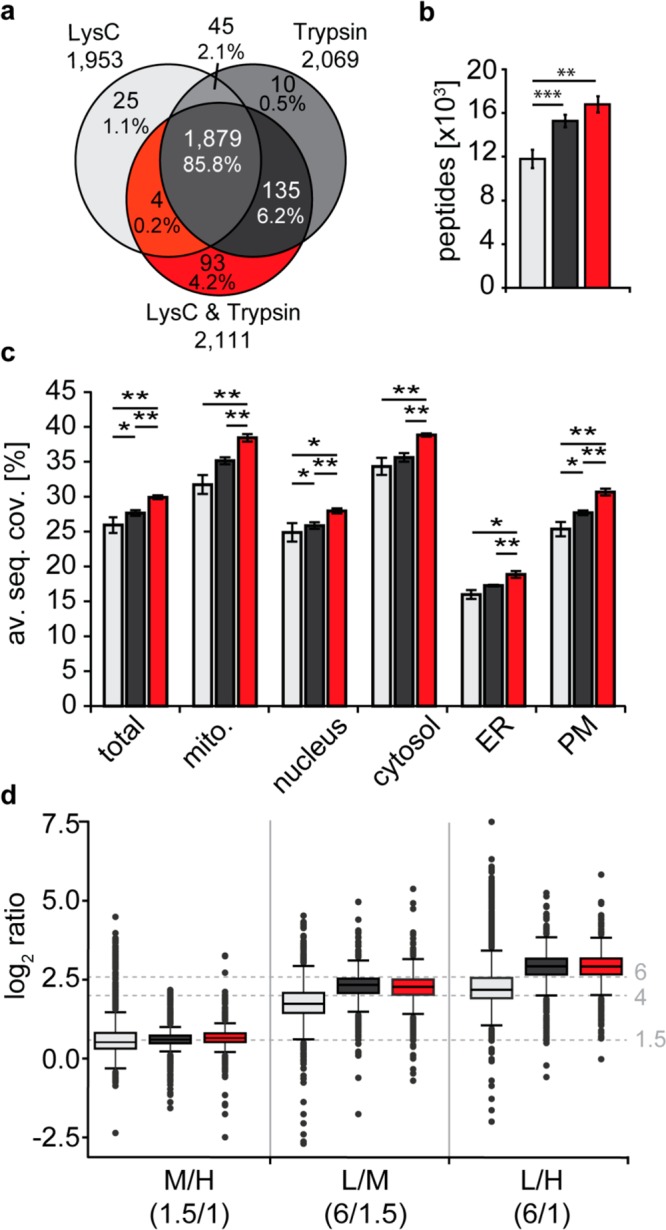
Evaluation of 2nSILAC for quantitative proteome analysis. BY4741 cells labeled with stable isotope-coded lysine (Lys4, Lys8) only or lysine and arginine (Arg6, Arg10) and unlabeled cells (Arg0, Lys0) were mixed 6:1.5:1 (L/M/H; n = 4). Proteins were digested with LysC (nSILAC using lysine only) or trypsin or a combination of both proteases in case of 2nSILAC. (a, b) Overlap of proteins (a) and average number of peptides (b) identified by LC-MS. (c) Average sequence coverage (av. seq. cov.) of proteins of different subcellular localization. ER, endoplasmic reticulum; mito., mitochondria; PM, plasma membrane. (d) Accuracy of relative protein quantification. Dashed horizontal lines mark the experimental mixing ratios. (b, c) *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001; error bars, std. dev.
We next examined the accuracy of quantification (Figure 3d; Table S-6). At a low mixing ratio of 1.5:1, median ratios (1.44–1.56) were close to the expected ratio for all three approaches. At higher mixing ratios, however, the median ratios determined for the LysC/trypsin tandem digest in 2nSILAC experiment (4.76 and 7.39) showed a lower deviation from the expected ratios compared to values obtained with the other approaches (LysC, 3.21 and 4.44; trypsin, 4.96 and 7.48). The data further show that in 2nSILAC experiments, the precision of the data was consistently higher than in nSILAC experiments as reflected by a smaller interquartile range (Figure 3d). Taken together, 2nSILAC is well applicable to global proteome quantification of BY4741 cells and best combined with LysC/trypsin digestion to enhance sequence coverage and precision of quantification.
2nSILAC Applied to the Study of Gene Deletion Effects on the Mitochondrial Proteome
As proof of principle, we employed the 2nSILAC strategy to analyze global effects of gene deletions on the mitochondrial proteome. BY4741 wild-type cells and cells with deletion of a distinct mitochondrial gene were grown under nonfermentative conditions. Following 2nSILAC labeling of wild-type and deletion strains (n = 3 each; incorporation >98% for both heavy arginine and lysine, Figure S-3 and Table S-7), cells were mixed and mitochondria-enriched fractions were prepared by differential centrifugation using a small-scale protocol which we specifically developed to allow for fast processing of multiple strains in parallel from low-volume cultures (see Supporting Information for experimental details). Proteins were digested with LysC/trypsin for LC-MS analysis and statistical outlier analyses were performed to determine proteins significantly altered in abundance in the gene deletion cells (see Supporting Information). Quantitative MS data were corroborated by immunoblotting using antibodies specifically recognizing mitochondrial proteins.
Deletion of SDH5, which codes for an assembly factor of the succinate dehydrogenase complex45 (complex II of the mitochondrial respiratory chain), resulted in reduced levels of all four core components of complex II (Sdh1–Sdh4) while proteins of complex III (Rip1), complex IV (Cox2), and the F1F0-ATP synthase (Atp2, Atp17) were not affected (Figure 4a,b; Table S-8). Sdh5 is a highly conserved mitochondrial protein that is required for the flavinylation of Sdh1,45 which in turn is necessary for assembly and stability of complex II46 as underscored by our 2nSILAC-MS and immunoblot data. In line with these data, results of a Gene Ontology (GO) term enrichment analysis indicates that the components of the succinate dehydrogenase complex were significantly decreased in abundance in sdh5Δ cells (Figure S-4a, Table S-9). Interestingly, the mitochondrial protein Fmp16 was strongly upregulated (>4-fold) upon SDH5 deletion (Figure 4a; Table S-8). Its exact molecular function is still unknown but it has been reported to be of higher abundance in cells undergoing stress.47,48 Our data indicate a potential role for Fmp16 in cellular stress response to dysfunctional mitochondria lacking complex II of the respiratory chain.
Figure 4.

2nSILAC for the study of mitochondrial protein functions. (a, c, f) The mitochondrial proteome of sdh5Δ (a), phb1Δ (c), and coi1Δ (f) cells was quantitatively compared to wild-type (WT) cells using the 2nSILAC strategy. The dashed horizontal lines indicate a t-test p-value of 0.05 (n = 3). Larger circles indicate proteins significantly altered in abundance (i.e., both t-test and Significance B p-value < 0.05). Mitochondrial proteins are highlighted in blue except for proteins associated with complex II (purple) in (a) or with complex III (orange) and IV (red) in (f); subunits of the prohibitin complex are marked green (c). (b, d, e) Mitochondria-enriched fractions of WT and deletion cells were analyzed by SDS-PAGE and immunoblotting using antisera against selected mitochondrial proteins. (g) Detergent-lysed mitochondria-enriched fractions of WT and coi1Δ cells were analyzed by blue native PAGE and immunoblotting using antisera against Rip1 (complex III) and Cox1 (complex IV) decorating the respiratory chain complexes and supercomplexes (III2IV and III2IV2). Mito, mitochondria
Phb1 and Phb2 form the ring-shaped 1.2-MDa prohibitin complex in the inner mitochondrial membrane,49 which has been described to play a role in mitochondrial membrane organization, lipid homeostasis and mitophagy.50,51 2nSILAC analysis of phb1Δ cells revealed that the level of Phb2 was drastically reduced when PHB1 was deleted (Figure 4c and Table S-10). According to the immunoblot analysis shown in Figure 4d, Phb2 was virtually absent in phb1Δ cells, which is in agreement with previous data showing an interdependence of Phb1 and Phb2: loss of Phb1 leads to destabilization of Phb2 and vice versa.52 Mic10, a component of the mitochondrial contact site and cristae organizing system, which has been reported to interact with prohibitin,53 was not affected by PHB1 deletion similar to the mitochondrially encoded subunits of the respiratory chain complexes54 (Figure 4c,d and Table S-10). Moreover, our quantitative analysis of the mitochondrial proteome shows that the cytochrome c oxidase (COX, CIV) regulatory subunit Cox13 was decreased upon PHB1 deletion, revealing a specific functional connection between the prohibitins and the respiratory chain apart from mitochondrial protein synthesis (Figure 4c,d).55 Interestingly, GO term enrichment analysis of proteins with increased abundance in crude mitochondrial fractions of phb1Δ cells shows an overrepresentation of proteins involved in cytoplasmic translation (Figure S-4b and Table S-9), which functionally links the prohibitins to the translation apparatus in the cytosol.
Coi1/Mco13 is a ∼13 kDa inner membrane protein with a C-terminal intermembrane space domain with an increased expression (up to 200%) upon respiratory growth.1,56Coi1Δ cells show a severe respiratory growth defect, a reduced mitochondrial membrane potential (Δψ), and the major interaction partners of Coi1 are subunits of the respiratory chain complexes III and IV.56 Singhal et al. (2017) analyzed coi1Δ mitochondria and detected reduced steady state protein amounts of Cox2 (complex IV), Qcr6, and Cor1 (complex III), further inner and outer membrane proteins and proposed a role of Coi1 as respiratory chain supercomplex assembly factor. However, the analysis of deletion mutants of other respiratory chain supercomplex assembly factors like Rcf1, Rcf2, Rcf3, and Aim24 did neither display such severe growth defects, nor such a broad reduction of mitochondrial protein steady state levels compared to coi1Δ.57−61 Therefore, we analyzed the role of Coi1 and grew wild-type and coi1Δ cells on respiratory medium, isolated mitochondria, and performed Western blot analysis. We also observed a severe reduction of Cox2 levels as Singhal et al. (2017) and additionally a significant reduction of the cytochrome c oxidase (complex IV) subunits Cox12 and Cox13 (Figure 4e). In contrast, we did neither observe a reduction for the cytochrome c reductase (complex III) subunits Cor1, Cyt1, Rip1, Qcr6, and Qcr8, nor for other inner or outer mitochondrial membrane proteins. To unambiguously quantify mitochondrial protein levels, we performed a 2nSILAC analysis with wild-type and coi1Δ cells. We observed a significantly decreased abundance of four subunits of the cytochrome c oxidase (Cox2, Cox12, Cox13, and Cox26) in coi1Δ cells (Figure 4f), while the remaining components and associated proteins of complex IV (marked in red in the scatterplot) as well as proteins of complex III (marked in orange) that have been quantified in this data set were unaltered in abundance (see also Table S-11). This quantitative analysis demonstrates that COI1 deletion does not affect the biogenesis of the cytochrome c reductase (complex III), but specifically affects the biogenesis of the cytochrome c oxidase (complex IV). This is supported by results of a GO term enrichment analysis showing that exclusively components of complex IV are of reduced abundance in coi1Δ cells (Figure S-4c and Table S-9). To further demonstrate the specificity of the defect in coi1Δ, we used blue native PAGE to analyze assembled respiratory chain complexes. When mitochondria are solubilized with the mild detergent digitonin, antibodies directed against Rip1 (complex III) and Cox1 (complex IV) reveal a dramatic reduction of respiratory supercomplexes in coi1Δ, as observed by Singhal et al. (2017; Figure 4g, upper panels). Concomitantly, we observed increased levels of assembled complex III (III2) by α-Rip1 decoration in coi1Δ. To analyze the levels of individual respiratory chain complexes III and IV, we employed the detergent n-dodecyl β-d-maltoside (DDM), which is known to dissociate complex III and complex IV.59,62 We observed similar levels of complex III (α-Rip1) and a significant reduction of complex IV (α-Cox1) in coi1Δ compared to wild-type (Figure 4g, lower panels). In addition, we observed Cox1 subcomplexes (IV*) in coi1Δ, which could represent intermediates lacking Cox2. This native PAGE Western decoration confirms the results of our 2nSILAC analysis and we propose that Coi1 acts as a specific assembly factor for the cytochrome c oxidase (complex IV). COI1 deletion causes a specific reduction of the major catalytic core subunit Cox2, its partner protein Cox2663,64 and the two auxiliary subunits Cox12 and Cox13, which are not required for catalytic activity.55,65 Since the Coi1 copy number per cell is ∼10% compared to the subunits Cox2, Cox26, Cox12, and Cox13 affected by COI1 deletion,1 we speculate that Coi1 functions as assembly factor to support the efficiency of Cox2 biogenesis.
Conclusions
The 2nSILAC strategy that we describe here is a valuable addition to the quantitative proteomics toolbox. We demonstrate its feasibility by labeling the prototrophic S. cerevisiae strain BY4741 with heavy lysine and arginine under different growth conditions. Most importantly, since a large variety of yeast mutant strain collections are derivatives of BY4741, 2nSILAC allows for their direct utilization in SILAC experiments without the necessity for prior genetic manipulations, which may introduce artifacts and interfere with further analysis. Thus, it offers researchers high flexibility in the experimental design while still providing the full potential of metabolic labeling in yeast, that is, mixing of samples at the level of whole cells, to minimize experimental variations resulting in more accurate quantitative proteome data.
In this work, 2nSILAC was combined with a fast and cost-effective protocol for the purification of mitochondria from low culture volumes. The high applicability of 2nSILAC for the study of mitochondrial protein functions was demonstrated by analyzing mitochondrial gene deletion strains. The analysis of deletion cells lacking the respiratory chain complex II assembly factor Sdh5 and the prohibitin subunit Phb1 revealed the specificity and potential of the 2nSILAC methodology. In addition, we analyzed the role of Coi1, and the quantitative data of 2nSILAC suggests a specific role of Coi1 for the biogenesis of the core subunit Cox2 of the cytochrome c oxidase (respiratory chain complex IV). Further, 2nSILAC is compatible with purification strategies for any subcellular compartment and, thus, generally represents a universal approach for studying protein functions at the organelle to whole-cell level. Since 2nSILAC can directly be applied to existing yeast strain collections we believe it will help to boost systematic quantitative screens of (sub)proteome-wide gene deletion effects in many laboratories, thereby advancing research on eukaryotic protein functions.
Acknowledgments
We thank Friedel Drepper for help with data analysis and the PRIDE team for data deposition to the ProteomeXchange Consortium. This work was supported by the European Research Council (ERC) Consolidator Grant No. 648235, the Excellence Initiative of the German Federal and State Governments (EXC 294, BIOSS, and GSC-4 Spemann Graduate School) and the Deutsche Forschungsgemeinschaft (WA 1598/5-1, RTG 2202, FOR 1905, SFB 746, and SFB 1140). Work included in this study has also been performed in partial fulfillment of the requirements for the doctoral theses of S.D., M.M., and P.L. at the University of Freiburg.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b02557.
Figure S-1 (Effect of exogenous proline on the proteome of BY4741 cells), Figure S-2 (Effect of exogenous proline on the MS intensities of Pro6-containing peptides in BY4741 cells grown on different carbon sources), Figure S-3 (Metabolic incorporation of heavy lysine (Lys8) and arginine (Arg10) into proteins of BY4741 wild-type (WT) and deletion strains), Figure S-4 (Gene Ontology (GO) term enrichment analysis of proteins significantly altered in abundance in sdh5Δ (a), phb1Δ (b), and coi1Δ (c) cells), Table S-5 (Extent of heavy arginine-to-proline conversion during native SILAC of cells grown on different carbon sources calculated based on the MS intensity of Pro6-containing peptides), extended experimental procedures, and references (PDF).
Table S-1. Effect of exogenous arginine and lysine on the proteome of BY4741 yeast cells analyzed by quantitative mass spectrometry using stable isotope peptide dimethyl labeling (XLSX).
Table S-2. Effect of exogenous arginine and lysine on the proteome of W303 yeast cells analyzed by quantitative mass spectrometry using stable isotope peptide dimethyl labeling (XLSX).
Table S-3. Assessment of heavy amino acid incorporation and arginine-to-proline conversion in prototrophic yeast grown under different conditions (XLSX).
Table S-4. Effect of exogenous proline on the proteome of cells analyzed by quantitative mass spectrometry using stable isotope peptide dimethyl labeling (XLSX).
Table S-6. Protein identification and relative protein quantification data obtained in 2nSILAC and nSILAC experiments (XLSX).
Table S-7. Assessment of heavy amino acid incorporation into the proteomes of sdh5Δ, phb1Δ, coi1Δ, and wild-type cells (XLSX).
Table S-8. Quantitative changes in the mitochondrial proteome of sdh5Δ cells analyzed following the 2nSILAC strategy (XLSX).
Table S-9. Results of Gene Ontology term enrichment analysis of proteins/genes significantly altered in abundance in sdh5Δ, phb1Δ, and coi1Δ cells (XLSX).
Table S-10. Quantitative changes in the mitochondrial proteome of phb1Δ cells analyzed following the 2nSILAC strategy (XLSX).
Table S-11. Quantitative changes in the mitochondrial proteome of coi1Δ cells analyzed following the 2nSILAC strategy (XLSX).
The authors declare no competing financial interest.
Supplementary Material
References
- Morgenstern M.; Stiller S. B.; Lübbert P.; Peikert C. D.; Dannenmaier S.; Drepper F.; Weill U.; Höß P.; Feuerstein R.; Gebert M.; et al. Cell Rep. 2017, 19 (13), 2836–2852. 10.1016/j.celrep.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo J. A.; O’Connell J. D.; Everley R. A.; O’Brien J.; Gygi M. A.; Gygi S. P. J. Proteomics 2016, 148, 85–93. 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefely J. A.; Kwiecien N. W.; Freiberger E. C.; Richards A. L.; Jochem A.; Rush M. J. P.; Ulbrich A.; Robinson K. P.; Hutchins P. D.; Veling M. T.; et al. Nat. Biotechnol. 2016, 34 (11), 1191–1197. 10.1038/nbt.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.-E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1 (5), 376–386. 10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Lau H.-T.; Suh H. W.; Golkowski M.; Ong S.-E. J. Proteome Res. 2014, 13 (9), 4164–4174. 10.1021/pr500630a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.-E. Anal. Bioanal. Chem. 2012, 404 (4), 967–976. 10.1007/s00216-012-5998-3. [DOI] [PubMed] [Google Scholar]
- Ong S.-E.; Kratchmarova I.; Mann M. J. Proteome Res. 2003, 2 (2), 173–181. 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- Bendall S. C.; Hughes C.; Stewart M. H.; Doble B.; Bhatia M.; Lajoie G. a. Mol. Cell. Proteomics 2008, 7 (9), 1587–1597. 10.1074/mcp.M800113-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. K.; Liao L.; Kim J. Y.; Yates J. R. Nat. Methods 2009, 6 (3), 184–185. 10.1038/nmeth0309-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicho C. C.; de Lima Alves F.; Chen Z. a.; Rappsilber J.; Sawin K. E. Mol. Cell. Proteomics 2010, 9 (7), 1567–1577. 10.1074/mcp.M110.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lössner C.; Warnken U.; Pscherer A.; Schnölzer M. Anal. Biochem. 2011, 412 (1), 123–125. 10.1016/j.ab.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Oeljeklaus S.; Schummer A.; Suppanz I.; Warscheid B. Methods Mol. Biol. 2014, 1188, 23–46. 10.1007/978-1-4939-1142-4_3. [DOI] [PubMed] [Google Scholar]
- Piechura H.; Oeljeklaus S.; Warscheid B. Methods Mol. Biol. 2012, 893, 201–221. 10.1007/978-1-61779-885-6_14. [DOI] [PubMed] [Google Scholar]
- Gruhler A.; Olsen J. V.; Mohammed S.; Mortensen P.; Færgeman N. J.; Mann M.; Jensen O. N. Mol. Cell. Proteomics 2005, 4 (3), 310–327. 10.1074/mcp.M400219-MCP200. [DOI] [PubMed] [Google Scholar]
- Martin-Perez M.; Villén J. Anal. Chem. 2015, 87 (7), 4008–4014. 10.1021/acs.analchem.5b00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. R.; Marton M. J.; Jones A. R.; Roberts C. J.; Stoughton R.; Armour C. D.; Bennett H. A.; Coffey E.; Dai H.; He Y. D.; et al. Cell 2000, 102 (1), 109–126. 10.1016/S0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Giaever G.; Chu A. M.; Ni L.; Connelly C.; Riles L.; Véronneau S.; Dow S.; Lucau-Danila A.; Anderson K.; André B.; et al. Nature 2002, 418 (6896), 387–391. 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Huh W.-K.; Falvo J. V.; Gerke L. C.; Carroll A. S.; Howson R. W.; Weissman J. S.; O’Shea E. K. Nature 2003, 425 (6959), 686–691. 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P.; Coelho P. S.; Roemer T.; Agarwal S.; Kumar A.; Jansen R.; Cheung K. H.; Sheehan A.; Symoniatis D.; Umansky L.; et al. Nature 1999, 402 (6760), 413–418. 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- Yofe I.; Weill U.; Meurer M.; Chuartzman S.; Zalckvar E.; Goldman O.; Ben-Dor S.; Schütze C.; Wiedemann N.; Knop M.; et al. Nat. Methods 2016, 13 (4), 371–378. 10.1038/nmeth.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth D. J.; Saleem R. A.; Rogers R. S.; Mirzaei H.; Boyle J.; Aitchison J. D. J. Am. Soc. Mass Spectrom. 2010, 21 (8), 1417–1422. 10.1016/j.jasms.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich F.; Christiano R.; Walther T. C. Mol. Cell. Proteomics 2013, 12 (7), 1995–2005. 10.1074/mcp.M112.025742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneva I. N.; Longworth J.; Sudbery P. E.; Dickman M. J. Proteomics 2018, 18 (5–6), 1700278. 10.1002/pmic.201700278. [DOI] [PubMed] [Google Scholar]
- Christiano R.; Nagaraj N.; Fröhlich F.; Walther T. C. Cell Rep. 2014, 9 (5), 1959–1965. 10.1016/j.celrep.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. Nat. Biotechnol. 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. J. Proteome Res. 2011, 10 (4), 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Feller A.; Dubois E.; Ramos F.; Piérard A. Mol. Cell. Biol. 1994, 14 (10), 6411–6418. 10.1128/MCB.14.10.6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauniaux J. C.; Urrestarazu L. A.; Wiame J. M. J. Bacteriol. 1978, 133 (3), 1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abadjieva A.; Pauwels K.; Hilven P.; Crabeel M. J. Biol. Chem. 2001, 276 (46), 42869–42880. 10.1074/jbc.M103732200. [DOI] [PubMed] [Google Scholar]
- Crabeel M.; Abadjieva A.; Hilven P.; Desimpelaere J.; Soetens O. Eur. J. Biochem. 1997, 250 (2), 232–241. 10.1111/j.1432-1033.1997.0232a.x. [DOI] [PubMed] [Google Scholar]
- Grenson M.; Mousset M.; Wiame J. M.; Bechet J. Biochim. Biophys. Acta, Gen. Subj. 1966, 127 (2), 325–338. 10.1016/0304-4165(66)90387-4. [DOI] [PubMed] [Google Scholar]
- Ljungdahl P. O.; Daignan-Fornier B. Genetics 2012, 190 (3), 885–929. 10.1534/genetics.111.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sychrova H.; Chevallier M. R. Yeast 1994, 10 (5), 653–657. 10.1002/yea.320100509. [DOI] [PubMed] [Google Scholar]
- Regenberg B.; Düring-Olsen L.; Kielland-Brandt M. C.; Holmberg S. Curr. Genet. 1999, 36 (6), 317–328. 10.1007/s002940050506. [DOI] [PubMed] [Google Scholar]
- Grenson M.; Hou C.; Crabeel M. J. Bacteriol. 1970, 103 (3), 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu M.; Sekito T.; Akiyama K.; Ohsumi Y.; Kakinuma Y. J. Biol. Chem. 2005, 280 (6), 4851–4857. 10.1074/jbc.M412617200. [DOI] [PubMed] [Google Scholar]
- Middelhoven W. Biochim. Biophys. Acta, Gen. Subj. 1964, 93, 650–652. 10.1016/0304-4165(64)90349-6. [DOI] [PubMed] [Google Scholar]
- Brandriss M. C.; Magasanik B. J. Bacteriol. 1980, 143 (3), 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois E.; Hiernaux D.; Grenson M.; Wiame J. M. J. Mol. Biol. 1978, 122 (4), 383–406. 10.1016/0022-2836(78)90417-5. [DOI] [PubMed] [Google Scholar]
- Dubois E.; Messenguy F. Mol. Gen. Genet. 1997, 253 (5), 568–580. 10.1007/s004380050359. [DOI] [PubMed] [Google Scholar]
- Yoon S.; Govind C. K.; Qiu H.; Kim S.; Dong J.; Hinnebusch A. G. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (32), 11713–11718. 10.1073/pnas.0404652101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralser M.; Kuhl H.; Ralser M.; Werber M.; Lehrach H.; Breitenbach M.; Timmermann B. Open Biol. 2012, 2 (8), 120093–120093. 10.1098/rsob.120093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandriss M. C. J. Bacteriol. 1979, 138 (3), 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandriss M. C.; Falvey D. A. J. Bacteriol. 1992, 174 (11), 3782–3788. 10.1128/jb.174.11.3782-3788.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H.-X.; Khalimonchuk O.; Schraders M.; Dephoure N.; Bayley J.-P.; Kunst H.; Devilee P.; Cremers C. W. R. J.; Schiffman J. D.; Bentz B. G.; et al. Science 2009, 325 (5944), 1139–1142. 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J.; Jeong M.-Y.; Na U.; Winge D. R. J. Biol. Chem. 2012, 287 (48), 40670–40679. 10.1074/jbc.M112.405704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach J. M.; Yimit A.; Lee A. Y.; Riffle M.; Costanzo M.; Jaschob D.; Hendry J. A.; Ou J.; Moffat J.; Boone C.; et al. Nat. Cell Biol. 2012, 14 (9), 966–976. 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. P.; Stepanova E.; Everley R. A.; Paulo J. A.; Gygi S. P. Mol. Cell. Proteomics 2015, 14 (9), 2454–2465. 10.1074/mcp.M114.045849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T.; Model K.; Langer T. Mol. Biol. Cell 2005, 16 (1), 248–259. 10.1091/mbc.e04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T.; Langer T. Curr. Biol. 2017, 27 (13), R629–R631. 10.1016/j.cub.2017.04.030. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Chiang W.-C.; Sumpter R.; Mishra P.; Levine B. Cell 2017, 168 (1–2), 224–238.e10. 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger K. H.; Yaffe M. P. Mol. Cell. Biol. 1998, 18 (7), 4043–4052. 10.1128/MCB.18.7.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S.; Collins S. R.; Cassidy-Stone A.; Hummel E.; DeVay R. M.; Lackner L. L.; Westermann B.; Schuldiner M.; Weissman J. S.; Nunnari J. J. Cell Biol. 2011, 195 (2), 323–340. 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans L. G.; de Jong L.; Artal Sanz M.; Coates P. J.; Berden J. A.; Back J. W.; Muijsers A. O.; van der Spek H.; Grivell L. A. EMBO J. 2000, 19 (11), 2444–2451. 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taanman J. W.; Capaldi R. A. J. Biol. Chem. 1993, 268 (25), 18754–18761. [PubMed] [Google Scholar]
- Singhal R. K.; Kruse C.; Heidler J.; Strecker V.; Zwicker K.; Düsterwald L.; Westermann B.; Herrmann J. M.; Wittig I.; Rapaport D. Mol. Biol. Cell 2017, 28 (20), 2609–2622. 10.1091/mbc.e17-02-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strogolova V.; Furness A.; Robb-McGrath M.; Garlich J.; Stuart R. A. Mol. Cell. Biol. 2012, 32 (8), 1363–1373. 10.1128/MCB.06369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers M.; Balleininger M.; Vukotic M.; Römpler K.; Bareth B.; Juris L.; Dudek J. FEBS Lett. 2014, 588 (17), 2985–2992. 10.1016/j.febslet.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Römpler K.; Müller T.; Juris L.; Wissel M.; Vukotic M.; Hofmann K.; Deckers M. J. Biol. Chem. 2016, 291 (45), 23769–23778. 10.1074/jbc.M116.734665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-C.; Taylor E. B.; Dephoure N.; Heo J.-M.; Tonhato A.; Papandreou I.; Nath N.; Denko N. C.; Gygi S. P.; Rutter J. Cell Metab. 2012, 15 (3), 348–360. 10.1016/j.cmet.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukotic M.; Oeljeklaus S.; Wiese S.; Vögtle F. N.; Meisinger C.; Meyer H. E.; Zieseniss A.; Katschinski D. M.; Jans D. C.; Jakobs S.; et al. Cell Metab. 2012, 15 (3), 336–347. 10.1016/j.cmet.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Schägger H.; Pfeiffer K. EMBO J. 2000, 19 (8), 1777–1783. 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker V.; Kadeer Z.; Heidler J.; Cruciat C.-M.; Angerer H.; Giese H.; Pfeiffer K.; Stuart R. A.; Wittig I. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863 (7), 1643–1652. 10.1016/j.bbamcr.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko M.; Wuttke J.-M.; Römpler K.; Schmidt B.; Neifer K.; Juris L.; Wissel M.; Rehling P.; Deckers M. Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863 (7), 1624–1632. 10.1016/j.bbamcr.2016.04.007. [DOI] [PubMed] [Google Scholar]
- LaMarche A. E.; Abate M. I.; Chan S. H.; Trumpower B. L. J. Biol. Chem. 1992, 267 (31), 22473–22480. [PubMed] [Google Scholar]
- Messenguy F.; Dubois E. Gene 2003, 316, 1–21. 10.1016/S0378-1119(03)00747-9. [DOI] [PubMed] [Google Scholar]
- Park S. S.; Wu W. W.; Zhou Y.; Shen R. F.; Martin B.; Maudsley S. J. Proteomics 2012, 75 (12), 3720–3732. 10.1016/j.jprot.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.