

ABSTRACT
Introduction: V600E BRAF mutation is an established driver mutation in a variety of tumors. Vemurafenib is a selective inhibitor of the BRAF V600E kinase, known to be highly effective in BRAF V600E-positive metastatic melanoma. As a single agent, vemurafenib is relatively ineffective in other V600E-positive malignancies.
Case 1: A 72 year old man with metastatic CRC who failed several previous lines of chemotherapy. Genetic analysis of 315 cancer-related genes (Foundation Medicine, FMI) revealed a BRAF V600E mutation. The patient was treated with vemurafenib resulting in a partial response of 18 months. Genetic analysis following development of resistance revealed a new mutation in KRAS-G12R.
Case 2: V600E mutation was identified in a 59 year old woman with metastatic PTC refractory to radioiodine therapy. The patient was treated with vemurafenib resulting in a partial response lasting 43 months. Genetic analysis following development of resistance revealed a new mutation in NRAS-Q61K.
The presented cases demonstrated the development of rare RAS mutations as a genetic mechanism of acquired BRAF inhibitor resistance. This observation is strongly supported by the analysis of a large database consisting of 712 BRAF V600E-positive melanoma samples showing higher rates of BRAF V600E and RAS mutations co-occurrence in metastatic lesions compared to local tumors (OR = 3.8, p = 0.035). This enrichment is likely a result of the development of RAS mutations following treatment with BRAF inhibitors.
Discussion: We report two cases showing extreme response to vemurafenib, which could not be predicted prior to treatment commencement. Genetic testing demonstrated a resistant mechanism not previously reported in CRC or PTC patients, namely an acquired mutation of RAS. This is supported by an analysis of a large cohort of BRAF V600E-positive melanomas.
Further studies are needed in order to identify predictive markers for response to vemurafenib and to explore novel strategies to overcome RAS-mediated resistance.
Keywords: RAF V600E, RAS, vemurafenib, colon cancer, thyroid cancer
Introduction
The BRAF V600E mutation confers constitutive activation of the MAPK pathway and thereby drive tumor cells growth1,2. It is an established oncogenic driver observed in approximately half of melanomas1 and is also present in approximately 40% of papillary thyroid cancer (PTC) and 10% of colorectal cancer (CRC)3,4.
Vemurafenib is a small-molecule tyrosine kinase inhibitor specific to the ATP-binding domain of BRAF V600E5. Vemurafenib is an approved therapy for BRAF V600E-positive metastatic melanoma and has been shown to improve survival in these patients6. As a single agent, vemurafenib is considered to be relatively ineffective in other BRAF V600E-positive malignancies. A phase II study demonstrated only a 5% response rate with vemurafenib monotherapy in patients with metastatic BRAF V600E-positive CRC7. A phase IB study illustrated a 35% response rate with a triple combination therapy consisting of vemurafenib, cetuximab and irinotecan8. Recently a phase II trial showed a 35% response rate with vemurafenib monotherapy in a cohort of 26 patients with BRAF V600E-positive PTC refractory to radioactive iodine9.
Resistance to vemurafenib develops in all patients with metastatic melanoma and studies in melanoma patients have identified numerous acquired resistance mechanisms including mutations in KRAS or NRAS10,11, BRAF V600E/K amplification11,12, alternate splicing of BRAF11,13, MEK1/2 mutations11,14,and non-MAPK pathway alterations11. Mechanisms mediating vemurafenib resistance in other tumors have not been well characterized. Studies in cell lines suggest that resistance to vemurafenib in CRC might be mediated through feedback activation of epidermal growth factor receptor (EGFR) signaling15,16 and that resistance of thyroid cancer cells may coincided with the spontaneous acquisition of a KRAS G12D activating mutation17. To our knowledge mechanisms mediating acquired resistance to vemurafenib in CRC and PTC patients have yet to be established.
We report here an unusual long-term response to vemurafenib in two patients with CRC and PTC harboring the BRAF V600E mutation, followed by acquired resistance due to the appearance of uncommon mutations in RAS proteins.
Report of the cases
Case 1: A 60 year old man underwent right hemicolectomy in 2005 due to stage 3 colonic adenocarcinoma and received six months of oxaliplatin-based adjuvant therapy. In 2007, a single abdominal wall metastasis appeared and was resected. Three years later a single lung metastasis was resected. In 2012 another recurrence presenting with a large symptomatic abdominal metastasis. The patient was treated with various modalities including systemic chemotherapy with fluorouracil and irinotecan, underwent surgery and received radiation therapy. After a few months he began to suffer from severe abdominal pain and a CT scan revealed recurrence of the disease in the abdomen and bones. Genetic analysis of a biopsy from the abdominal wall metastasis (Foundation Medicine, FMI) revealed several alterations, including BRAF V600E mutation (Table 1). On August 2013 treatment with vemurafenib 960 mg twice daily was commenced. His pain resolved within days and CA19.9 levels decreased from 6293 u/ml to 1120 u/ml within 6 weeks and an additional decrease 207u/ml was measured at18 weeks (Figure 1). Response lasted 18 months, until February 2015. Upon recurrence, the metastasis was re-biopsied and a repeat genetic testing revealed a new mutation in KRAS-G12R (Table 1). The patient was then treated with FOLFOX-bevacizumab regimen and responded for an additional 12 months. A liquid biopsy (Foundation ACT) taken at the time of disease progression revealed the presence of both mutations (Table 1).
Table 1.
Genomic alterations identified by genetic analysis before treatment with vemurafenib and following development of resistance.
| 1st biopsy: before vemurafib treatment | 2nd biopsy: following disease progression on vemurafenib | cfDNA analysis: following a year of chemotherapy | |
|---|---|---|---|
| Patient 1 CRC |
BRAF V600E FBXW7 E121 TP53 R213 MCL1 amplification |
BRAF V600E FBXW7 E121 TP53 R213 KRAS G12R CCND3 amplification |
BRAF V600E, L505H TP53 R213, R282W KRAS G12R FLT3 amplification |
| Patient 2 PTC |
BRAF V600E TERT promotor – 124C> T |
BRAF V600E TERT promotor – 124C> T NRAS Q61K CDKN2A D108N p14ARF F122Q CDKN2C K136 RB1 loss exons 3–27 |
Figure 1.
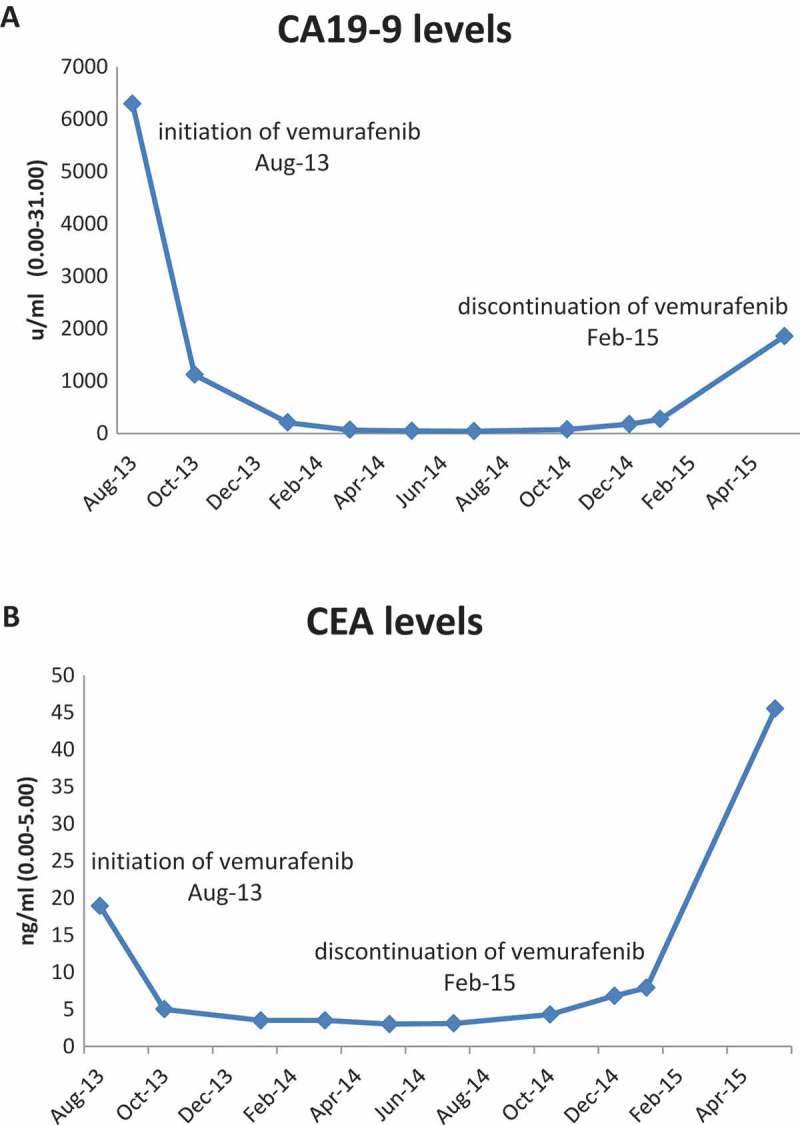
Tumor markers levels of patient 1 following vemurafenib treatment. (A) CA19-9 levels, (B) CEA levels.
Case 2: A 47 year old woman underwent total thyroidectomy and neck dissection in 2005. Pathology showed PTC with extension of the tumor to the larynx. The patient received radioiodine therapy. In 2006 she underwent a second neck dissection followed by radioiodine therapy due to recurrence in cervical nodes and in 2010 a third neck operation was conducted and followed by radioiodine therapy due to recurrence in the neck and lungs. Between 2011–2013 the patient was treated with sorafenib until the disease progressed. At that time the patient reported on cough and snoring. Genetic analysis conducted on the tumor obtained from the third operation revealed BRAF V600E mutation (Table 1). On October 2013 treatment with vemurafenib 960 mg twice daily was initiated. Her symptoms resolved within few weeks and CT scan conducted on December 2013 showed response of the lung metastases (Figure 2). The response lasted 43 months and by May 2017 the disease progressed again. Another biopsy was taken from a cervical mass and genetic analysis revealed a new mutation in NRAS-Q61K (Table 1).
Figure 2.
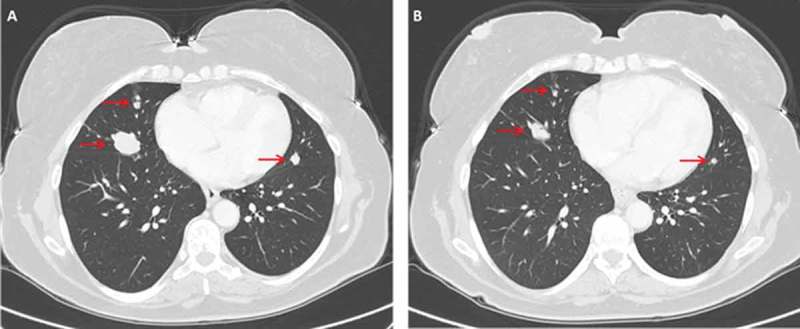
Chest CT of patient 2 before and after initiation of vermurafenib treatment. (A) Arrows indicate presence of lung metastases. (B) The same regions are denoted by arrows on follow up imaging performed 2 month later.
FMI Database analyses: In order to test the hypothesis that RAS pathway alterations mediate resistance to BRAF inhibition, we searched the Foundation One clinical database for tumors harboring BRAF V600E and an oncogenic RAS pathway alterations (alterations in KRAS, NRAS, and HRAS). Because melanomas are frequently tested for V600E alterations and treated with vemurafiniab, we focused our analysis on melanomas. Of 3,399 melanomas, we identified 712 harboring a BRAF V600E alteration. Of these, 190 could be assigned as local/primary based on site of biopsy and 213 could be assigned as distant metastases. While co-occurrence of BRAF V600E and RAS mutations was noted in only 1.5% (3) of primary tumors, it was observed in 5.6% (12) of metastatic lesions (OR = 3.8, p = 0.035).
Discussion
We report an unusually long response to vemurafenib in two CRC and PTC patients. While vemurafenib treatment is associated with high response rates among patients with BRAF V600E-positive metastatic melanoma6, only a minority of the patients with other BRAF V600E-positive malignancies will respond7,9,18,19, and a major limitation for the use of vemurafenib in these tumors is the lack of predictive markers. We were not able to identify any clinical or genomic predictive factors in our patients. The targeted genomic analysis conducted on our patients covered only 315 cancer related genes. It is possible that a wider genomic analysis will be able to identify such markers. We are currently conducting a whole genome analyses on the tumor samples of our patients. Yet, it is also possible that the response is associated with non-genomic alterations. In that case, the use of other platforms (i.e. transcriptomics, epigenomics) may be required.
The presented cases indicate development of RAS mutations as a genetic mechanism of acquired BRAF inhibitor resistance in BRAF V600E-positive CRC and PTC. This observation is strongly supported by the analysis of a large database consisting of 712 samples showing higher rates of BRAF V600E and RAS mutations co-occurrence in metastatic lesions compared to local tumors (OR = 3.8, p = 0.035). This enrichment is likely a result of the development of RAS mutations following treatment with BRAF inhibitors. Furthermore, this observation is supported by studies in cell lines, indicating the development of RAS mutations following long-term exposure to vemurafenib17.
An activating mutation of RAS protein is a well-established primary oncogenic driver in up to 30% of human tumors20, with the most frequent activating point mutations identified in KRAS (about 85% of total), followed by NRAS (~ 15%) and HRAS (< 1%)21. The incidence of mutated RAS gene varies strongly among the different tumor types. The highest incidence is found in pancreatic adenocarcinoma, in which over 90% of the tumors harbor activating mutations of KRAS codon 1222. Mutant KRAS is found in almost half of CRCs, and codon 12 and 13 are two hotspots, which account for about 95% of all mutation types, with approximately 80% occurring in codon 12 (mostly G12D and G12V) and 15% in codon 13 (mostly G13D). Other mutations in codons 61, 146 and 154 occur less frequently in CRC, accounting for 5% of all mutation type23,24. The incidence of RAS mutations in thyroid tumors varies among the different subtypes, ranging from 50% of anaplastic thyroid carcinoma to 10% of PTC25. Thyroid cancers are unique in that they have been associated with all three mutant isoforms of the RAS gene, although NRAS Q61K mutation have been suggested as more prominent, especially in follicular thyroid carcinoma26,27. Interestingly, the RAS mutations observed in our patients following vemurafenib treatment (KRAS G12R and NRAS Q61K), serving as a mechanism of acquired resistance, are rarely observed as primary driver mutations. A recent comprehensive analysis of KRAS mutations activity in a zebra fish model indicated differential activities for the various mutations28. While the more common mutations seem to have a higher tumorigenic capacity, it is possible that activities required for tumor initiation are different from those required for resistance. This hypothesis must be further explored.
In conclusion, we report here on two cases demonstrating extreme response to vemurafenib, followed by the emergence of rare RAS mutations suggesting mediating acquired resistance. Further studies are needed in order to identify predictive markers for response to vemurafenib and to explore novel strategies to overcome RAS-mediated resistance.
Disclosure statement
The authors have nothing to disclose
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Davis N, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Project CG, Jones CM, Marshall CJ, Springer CJ, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004; 116:855–867. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 3.Yokota T, Ura T, Shibeta N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104(5):856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakayama H, Yoshida A, Nakamura Y, Hayashi H, Miyagi Y, Wada N, Rino Y, Masuda M, Imada T.. Clinical significance of BRAF (V600E) mutation and Ki-67 labeling index in papillary thyroid carcinomas. Anticancer Res. 2007;27(5B):3645–3649. [PubMed] [Google Scholar]
- 5.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Burton EA, Zhang C, Zhang Y, Nolop K, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Flaherty KT, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New England J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Saltz L, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–4038. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, Overman MJ, Piha-Paul S, Subbiah V, Kopetz S, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6(12):1352–1365. doi: 10.1158/2159-8290.CD-16-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, Sherman SI, Sherman EJ. Vemurafenib in patients with BRAF V600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(9):1272–1282. doi: 10.1016/S1470-2045(16)30166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee M-K, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51(18):2792–2799. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi H, Moriceau G, Kong X, Lee M-K, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012; 3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E). Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 17.Danysh BP, Rieger EY, Sinha DK, Evers CV, Cote GJ, Cabanillas ME, Hofmann M-C. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget. 2016;7(21):30907. doi: 10.18632/oncotarget.v7i21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ, Nolop KB, Saltz L. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(15_suppl):3534. doi: 10.1200/jco.2010.28.15_suppl.3534. [DOI] [Google Scholar]
- 20.Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93(14):1062–1074. [DOI] [PubMed] [Google Scholar]
- 21.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689. [PubMed] [Google Scholar]
- 22.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, et al. MR. COSMIC 2005. Br J Cancer. 2006;94(2):318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neumann J, Zeindl-Eberhart E, Kirchner T, Jung A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathology-Research Pract. 2009;205(12):858–862. doi: 10.1016/j.prp.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Howell GM, Hodak SP, Yip L. RAS mutations in thyroid cancer. Oncologist. 2013;18(8):926–932. doi: 10.1634/theoncologist.2013-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, Tallini G, Tg K, Nikiforov YE. RAS point mutations and PAX8-PPARγ rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metabolism. 2003;88(5):2318–2326. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- 27.Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TW, Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol (Oxf). 1999;50(4):529–535. [DOI] [PubMed] [Google Scholar]
- 28.Park JT, Johnson N, Liu S, Levesque M, Wang YJ, Ho H, Huso D, Maitra A, Parsons MJ, Prescott JD, et al. Differential in vivo tumorigenicity of diverse KRAS mutations in vertebrate pancreas: A comprehensive survey. Oncogene. 2015;34(21):2801–2806. doi: 10.1038/onc.2014.223. [DOI] [PMC free article] [PubMed] [Google Scholar]