

Periodontitis is an inflammatory disease caused by polymicrobial biofilms. The periodontal pathogen Aggregatibacter actinomycetemcomitans displays two proteinaceous surface structures, the fimbriae and the nonfimbrial extracellular matrix binding protein A (EmaA), as observed by electron microscopy.
KEYWORDS: adhesins, autotransporter proteins, biofilms, periodontitis
ABSTRACT
Periodontitis is an inflammatory disease caused by polymicrobial biofilms. The periodontal pathogen Aggregatibacter actinomycetemcomitans displays two proteinaceous surface structures, the fimbriae and the nonfimbrial extracellular matrix binding protein A (EmaA), as observed by electron microscopy. Fimbriae participate in biofilm biogenesis and the EmaA adhesins mediate collagen binding. However, in the absence of fimbriae, A. actinomycetemcomitans still retains the potential to form robust biofilms, suggesting that other surface macromolecules participate in biofilm development. Here, isogenic mutant strains lacking EmaA structures, but still expressing fimbriae, were observed to have reduced biofilm potential. In strains lacking both EmaA and fimbriae, biofilm mass was reduced by 80%. EmaA enhanced biofilm formation in different strains, independent of the fimbriation state or serotype. Confocal microscopy revealed differences in cell density within microcolonies between the EmaA positive and mutant strains. EmaA-mediated biofilm formation was found to be independent of the glycosylation state and the precise three-dimensional conformation of the protein, and thus this function is uncorrelated with collagen binding activity. The data suggest that EmaA is a multifunctional adhesin that utilizes different mechanisms to enhance bacterial binding to collagen and to enhance biofilm formation, both of which are important for A. actinomycetemcomitans colonization and subsequent infection.
INTRODUCTION
Aggregatibacter actinomycetemcomitans is a Gram-negative bacterium associated with several forms of periodontal disease (1, 2). The major consequence of these inflammatory diseases is tooth loss, which results from the destruction of the supporting connective tissue and resorption of the underlying bone (3). A. actinomycetemcomitans is also capable of causing infections at a variety of distal sites by hematologic spreading of bacteria from the oral cavity, e.g., infectious endocarditis (4, 5). These bacteria adhere to tissue components and, as the name suggests, aggregate to form a tenacious biofilm crucial for disease.
Bacterial adhesion to a variety of substrates is mediated by both fimbrial and nonfimbrial adhesins (6). A. actinomycetemcomitans fimbriae are classified as bundle-forming type IVb-like fimbriae composed of repeating subunits of a 6.5-kDa Flp protein protruding up to a few microns from the cell surface (7). These fibril-like appendages are the foundation of the tenacious biofilm and of the unique star- or cigar-shaped colony morphology associated with this bacterium (8). Fimbriae are suggested to mediate generalized attachment to host tissues and abiotic surfaces, including hydroxylapatite, plastic, and glass (9). In contrast, nonfimbrial adhesins typically mediate adhesion to specific biotic surfaces, including cells and extracellular matrix (ECM) proteins (10–12).
Several nonfimbrial adhesins have been characterized in A. actinomycetemcomitans and shown to be involved with the binding of the bacterium to either human oral epithelial cells (Aae [13, 14] and Omp100/ApiA [15, 16]) or collagen (extracellular matrix adhesin protein A, EmaA [10, 17], and Omp100/ApiA [15]). These nonfimbrial adhesins are classified as type V secreted proteins, or autotransporters. In these proteins, the carboxyl terminus or the translocator domain in conjunction with the BAM (β-barrel assembly machinery) complex forms a pore in the outer membrane (18). The translocator domain (formed by a single [type Va] or three [trimeric autotransporters, type Vc] polypeptide chains [19]) catalyzes the transport of the remaining protein (passenger domain) through the pore, facilitating protein folding and exposure of the functional domain to the extracellular environment. The passenger domains of trimeric autotransporters are typically divided into three distinct regions: an N-terminal head, a neck, and a stalk (20).
Trimeric autotransporter proteins (TAAs) are found encoded in the genome of a wide variety of Gram-negative bacterial pathogens. These proteins are generally associated with bacterial adherence to either host epithelial cells or ECM proteins (21), though several are suggested to play multiple roles. The Yersinia enterocolitica YadA adhesin, which mediates adherence to epithelial cells and ECM proteins, is the prototypic protein and is crucial for intestinal colonization and serum resistance (22). Proteins specific for the binding to epithelial cells include NadA and NhhA from Neisseria meningitidis (23, 24), Hia and Hsf from Haemophilus influenzae (25, 26), and UspA1 and UspA2 from Moraxella catarrhalis (27, 28). Other TAAs are more promiscuous for binding to a variety of substrates and involvement in cell aggregation: UpaG from uropathogenic Escherichia coli (29), SadA from Salmonella enterica serovar Typhimurium, (30), and EhaG from enterohemorrhagic Escherichia coli (31). Lastly, there are adhesins that are specific for ECM proteins, which include BadA from Bartonella henselae, the largest of the TAAs, consisting of 3,000 amino acids (32), and EmaA from A. actinomycetemcomitans, known to be specific for binding to collagen (10).
The collagen binding adhesin EmaA from A. actinomycetemcomitans is composed of three 202-kDa monomers that form antenna-like structures protruding up to 150 nm from the bacterial surface (33). EmaA mediates the binding of the bacteria to collagen fibrils of damaged heart valves in an animal model of infective endocarditis (17). The collagen binding activity is associated with specific sequences located at the amino terminal region of the protein and the precise three-dimensional (3-D) conformation of the structures is a prerequisite for activity (34, 35). All serotypes of A. actinomycetemcomitans harbor an emaA gene; however, there is heterogeneity in the sequence that is correlated with the serotype of the bacterium. Serotype b and c strains express the cognate 202-kDa protein, whereas serotype a and d strains have sequence differences in the collagen binding domain (up to 25%) and a 279-amino-acid deletion in the stalk-like domain, which results in a 173-kDa protein variant (36).
EmaA is a glycoprotein, and glycosylation is suggested to occur in the periplasmic space of the bacterium (37). The composition of the sugars is hypothesized to be similar to the O-polysaccharide (O-PS) of the lipopolysaccharide (LPS) and glycosylation is catalyzed, at least in part, by the O-antigen ligase (WaaL) associated with the LPS biosynthetic pathway (38). In serotype b A. actinomycetemcomitans, the O-PS is composed of a trio of repeating sugars, d-fucose, l-rhamnose, and d-N-acetylgalactosamine (39). Specifically, the rhamnose epimerase gene rmlC is essential for O-PS production in serotype b, as well as for posttranslational modification of EmaA (38). Importantly, glycosylation is necessary for EmaA collagen binding activity (37).
In this investigation, we present evidence for an additional function attributed to the presence of EmaA structures on the surface of the bacterium. We demonstrate a role for EmaA in biofilm formation in different strains, independent of fimbriation state or serotype. The inactivation of emaA resulted in a significant decrease in biofilm formation. We have determined that the activity is associated with the amino terminus of the protein, which is also required for collagen binding. However, a single amino acid change in the protein sequence (G162S) that inactivates collagen adhesion and does not perturb the overall EmaA structure, but only the precise 3-D conformation of the adhesin, does not affect biofilm formation. In addition, the contribution of EmaA to biofilm formation is independent of the glycosylation state of the protein and therefore uncorrelated with collagen binding activity. Our studies suggest that EmaA is a multifunctional adhesin that promotes binding to collagen and biofilm formation and thus may play a key role in pathogenesis.
RESULTS
Visualization of surface structures of A. actinomycetemcomitans.
Two surface structures are associated with the distinctive rugose outer membrane of A. actinomycetemcomitans, as observed in transmission electron micrographs from negatively stained whole-mount bacterial preparations (Fig. 1). Fimbriae are observed as long, thick bundles composed of individual fimbriae that are approximately 6 to 7 nm in diameter and up to several microns in length (40) (black arrows, Fig. 1A). In contrast, the EmaA structures form antenna-like appendages that are narrower and smaller in length (3 to 5 nm in diameter and up to 150 nm in length) (white arrows, Fig. 1). Fimbriated strains, which are typically isolated from biological samples, may lose these long appendages upon consecutive rounds of subculturing during planktonic growth (Fig. 1B). The loss of fimbriae does not noticeably change the appearance of the rugose outer membrane; however, the EmaA appendages become easily visible and the most prominent structures extending outward from the bacterial surface.
FIG 1.

Electron micrographs of whole-mount bacterial preparations of A. actinomycetemcomitans strains. Images of negatively stained preparations of a serotype b fimbriated strain (VT1257) (A) and a spontaneous mutant nonfimbriated strain (KM733) (B) of A. actinomycetemcomitans are shown. The trimeric structures of EmaA are indicated by white arrows and fimbriae are labeled by black arrows. Scale bar, 100 nm.
EmaA contributes to biofilm biogenesis.
Fimbriae are known to influence biofilm formation (41). However, to our knowledge, biofilm formation activity of genetically related fimbriated and nonfimbriated strains has not been investigated. Therefore, multiple experimental strains were tested for biofilm formation activity in static single-species biofilm assays. In all cases, the mass of biofilm formed by the fimbriated strain was much greater than the mass produced by the genetically comparable nonfimbriated strain, as depicted in Fig. 2A. The fimbriated serotype b strain VT1257 produced a biofilm mass approximately 60% (59.4% ± 12.6%) greater than that of the corresponding nonfimbriated strain; however, the genetically similar nonfimbriated strain still formed a robust biofilm. This residual activity led us to speculate that other surface structures might contribute to biofilm formation.
FIG 2.

Quantification of the contribution of fimbriae and EmaA to biofilm formation by A. actinomycetemcomitans. Standard static biofilm assays were performed in triplicate and quantified by determining the absorbance at 562 nm. (A) Comparison of the mass of the 3-day biofilm formed by the serotype b fimbriated strain (VT1257) and that formed by the spontaneous nonfimbriated strain (KM733). (B) Biofilm formation activities of two different fimbriated strains, one serotype b and one serotype c. Results for serotype b (VT1257, panel B, left) (wild type), an isogenic emaA mutant strain (mutant), and an in trans emaA complement of the mutant strain (complement) are shown. Results for a serotype c strain (D11S-1, panel B, right), which does not express EmaA (wild type), and the transformation of D11S-1 with emaA in trans (complement) are also shown. A minimum of three biological replicates were performed for each strain. Error bars represent the standard deviations of the mean. The statistical significance is indicated (**, P < 0.01; *, P < 0.05).
To investigate the involvement of EmaA in biofilm formation, insertional emaA isogenic mutants in a fimbriated strain (VT1257) and a nonfimbriated serotype b strain (KM733) were generated. All emaA mutant strains generated did not express any EmaA, as demonstrated by immuno-dot blot analyses (data not shown). The absence of EmaA on the surface resulted in a decrease in the mass of the biofilm formed, 15.0% ± 4.8% for the fimbriated strain (Fig. 2B) and 72.7% ± 3.4% for the nonfimbriated emaA mutant strain (Fig. 3). The wild-type biofilm phenotype was restored by complementation of the emaA gene in trans. Further evidence for the role of EmaA in biofilm formation was a gain-of-function phenotype observed in a fimbriated serotype c strain (D11S-1) that does not express the protein. In trans complementation with emaA resulted in a 22.9% ± 16.7% increase in biofilm cell mass (Fig. 2B) and a >100% increase in a nonfimbriated version of this strain (Fig. 3). Fimbriae exert a major influence on biofilm formation in A. actinomycetemcomitans; however, our data demonstrate that EmaA also influences biofilm formation in a manner independent of the fimbriation status of the strain.
FIG 3.

Conserved role of EmaA in biofilm formation in multiple A. actinomycetemcomitans strains. Representative standard static biofilm assays of nonfimbriated strains were performed in triplicate and quantified by absorbance at 562 nm. Results for wild-type (black), emaA mutant (light gray), and emaA complement (dark gray) strains are shown. VT1257 and HK1651, serotype b; ATCC 29523 and VT1218, serotype a; ATCC 33384 and D11S-1, serotype c. The ATCC 29523 and D11S-1 strains do not express EmaA. A minimum of three biological replicates were performed for each strain. The statistical significance is indicated (*, P < 0.05).
The association of EmaA with biofilm biogenesis is present in multiple strains.
A conserved role for EmaA in biofilm biogenesis was investigated among additional laboratory and/or more clinically related isolates of A. actinomycetemcomitans. These strains correspond to serotypes a, b, and c, and either express EmaA on the surface or do not produce structures due to a mutation in the gene sequence. emaA mutants were generated in strains expressing EmaA on the surface, and all strains were analyzed for biofilm formation activity. Strains with insertionally inactivated emaA all displayed a decrease in the mass of the biofilm compared to the parent strain (Fig. 3). Complementation of these mutant strains with emaA on a replicating plasmid produced EmaA and rescued the mutant phenotype to levels equal or greater than that of the wild type. For strains that do not produce EmaA, transformation with the emaA plasmid resulted in EmaA surface structures (data not shown) and gain-in-function of biofilm formation activity compared to the original strains (Fig. 3).
The distal region of the EmaA structure is required for biofilm formation.
EmaA was originally identified as a collagen binding adhesin, and the distal region of the structure, corresponding to the amino terminus of the protein, is required for collagen binding based on deletion analysis (35). Plasmids expressing proteins corresponding to deletions of amino acids 57 to 123 or amino acids 57 to 625 were transformed into an emaA mutant serotype b strain to determine whether this region of the protein is associated with biofilm formation. Deletion of the first 123 amino acids or deletion of the complete distal region of the protein (amino acids 57 to 625) resulted in an 89.7% ± 2.1% or 73.8% ± 11.5% reduction in the mass of the biofilm, respectively, compared to the mutant strain expressing the full-length emaA gene (Fig. 4). These values are not statistically different from the mass formed by the emaA mutant strain.
FIG 4.
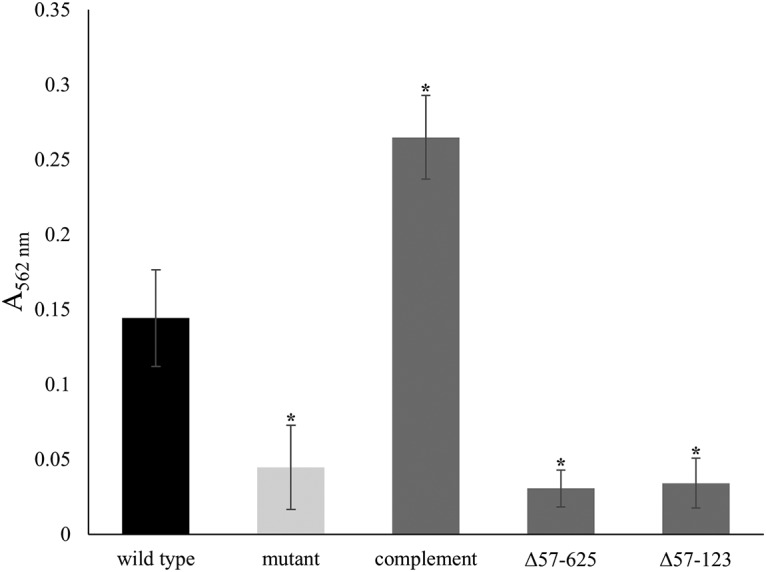
Quantification of biofilms formed by strains expressing truncations of EmaA. A representative standard static biofilm assay of nonfimbriated strains was performed in triplicate and quantified by determining the absorbance at 562 nm. The emaA mutant of the nonfimbriated, serotype b strain (VT1257) was transformed with plasmids expressing truncations in the head domain of EmaA. Δ57-625, deletion of amino acids 57 to 625, corresponding to the entire head domain; Δ57-123, deletion of amino acids 57 to 123, corresponding to subdomain I of the head domain. Both strains expressed equivalent amounts of EmaA compared to the complemented strain. A minimum of three biological replicates were performed for each strain. The statistical significance is indicated (*, P < 0.05).
EmaA collagen binding activity is uncorrelated with biofilm formation.
A single point mutation in the emaA nucleic acid sequence changes the amino acid at position 162 from a glycine to a serine (G162S) and results in a strain that displays a complete EmaA surface structure but is deficient in collagen binding (35). To determine whether this functional constraint of EmaA is applicable to biofilm formation, the emaA mutant strain used in this study was transformed with the G162S-expressing plasmid and assayed for collagen binding and biofilm formation (Fig. 5A). The strain expressing the nonfunctional EmaA is deficient in collagen binding (Fig. 5A, left) and generated a biofilm with a mass similar to the emaA mutant strain complemented with the native gene and in excess of the wild-type strain (Fig. 5A, right).
FIG 5.

Quantification of collagen binding and biofilm formation by strains expressing modified EmaA proteins. The results of representative assays of nonfimbriated strains are shown. (A) Comparison of collagen binding activity (left) and biofilm assay (right) of nonfimbriated VT1257 (wild type), emaA mutant (mutant), emaA mutant transformed with a plasmid expressing wild-type full-length EmaA (complement), and emaA mutant transformed with a plasmid expressing a G162S point mutation EmaA (G162S). (B) Biofilm assay of the O-PS-deficient strain. The results for nonfimbriated VT1257 (wild type), rmlC mutant rhamnose epimerase gene-inactivated (rmlC), and rmlC mutant strains transformed with full-length emaA (rmlC/emaA+) are shown. A minimum of three biological replicates were performed for each strain. The statistical significance is indicated (*, P < 0.05).
The presence of glycan moieties and of enzymes associated with the LPS biosynthetic pathway affects EmaA protein stability and collagen binding activity (37). We investigated the necessity of this posttranslational modification in biofilm development by overexpressing emaA in a strain with the rhamnose epimerase gene rmlC inactivated. The overexpression of emaA in this background resulted in the synthesis of EmaA equivalent to the wild-type strain (data not shown). In the biofilm assay, overexpression of EmaA in this glycosylation-deficient strain exhibited biofilm mass levels not statistically different from the wild type (Fig. 5B). Taken together, these data suggest that the contribution of EmaA to A. actinomycetemcomitans biofilm formation is uncorrelated with collagen binding activity.
EmaA modulates biofilm architecture.
Confocal microscopy was used to visualize the biofilm architecture of the serotype b fimbriated wild-type and emaA mutant strains (Fig. 6A). The biofilm surface area remained largely unchanged between the two strains; however, the calculated volume of the biofilms formed by the emaA mutant strain was increased compared to the wild-type strain (Table 1). A comparison of the height of the biofilm formed by these strains, as observed by a 90° rotation of the confocal z-stacks, demonstrated a substantial difference (Fig. 6B and C). The height of the emaA mutant strain biofilm was observed to be consistently higher than the wild type. Since the surface area of the biofilms remained the same, the change in height would account for the increase in the volume of the biofilm. Interestingly, the fluorescence intensity of microcolonies per z-slice was greatest in the strains expressing emaA (Fig. 7), suggesting an increase in cell density within the biofilm formed by the parent strain. The number of small microcolonies associated with the parent strain was also reduced in the biofilm formed by the mutant strain. Similar results were observed for the nonfimbriated wild-type and mutant strains (data not shown). The absence of EmaA in the outer membrane does not influence the uptake of the fluorescent dye used in these experiments (data not shown).
FIG 6.

Architecture of fimbriated biofilms formed by wild-type and emaA mutant strains of A. actinomycetemcomitans. Representative images show the microcolony formation of fimbriated serotype b strain VT1257 biofilm. (A) Static images of confocal z-stacks. (Left panel) Wild-type strain; (right panel) emaA mutant strain. (B) The z-stack was rotated by 90° around the x axis to visualize the height of each biofilm. (Left panel) Wild type (white); (right panel) mutant (red). (C) Merged view of rotated z-stacks (wild type, white; mutant, red). Scale bars, 30 µm.
TABLE 1.
Quantification of biofilm architecture
| Strain | Avg ± SDa |
Surface/vol (μm2 μm−3) |
|
|---|---|---|---|
| Biofilm vol (μm3) | Biofilm surface area (μm2) | ||
| Wild type | 106,236.04 ± 50,113.87 | 156,767.39 ± 31,606.26 | 1.48 |
| emaA mutantb | 134,786.83 ± 46,048.55 | 174,609.50 ± 50,644.52 | 1.81 |
The results are averages of three separate experiments.
emaA mutant statistical significance: P < 0.1.
FIG 7.

Confocal microscopy of fimbriated biofilms formed by wild-type and emaA mutant strains of A. actinomycetemcomitans. Representative confocal micrographs show 24-h fimbriated, serotype b strain VT1257 biofilms. (A) Wild type. (B) emaA mutant. Micrographs were artificially colored: redder tones indicate higher captured fluorescence intensity. Scale bars, 30 µm. (C) Average integrated fluorescence intensities of serotype b strain VT1257 (wild type) and isogenic emaA mutant strain (mutant) expressed in arbitrary units. The statistical significance is indicated (*, P < 0.05).
DISCUSSION
Biofilm formation is a multistep process initiated by the attachment of bacterial cells to a substratum. The adhesion process is mediated by surface appendages and can involve both physiochemical and electrostatic interactions (6). Although flagella, structures typically associated with cellular motility, have been implicated in early attachment (42), fimbriae and nonfimbrial adhesins are typically associated with this process (7, 43). Both of these surface appendages exhibit diversity in terms of protein composition and mode of secretion (21, 44). Fimbriae are typically composed of repeating protein monomers built upon one another to extend the structures microns from the bacterial surface. In contrast, nonfimbrial adhesins are composed of either one or three identical polypeptide chains, which may extend hundreds of nanometers from the cell surface (33). The difference in length suggests that the adhesion process is sequential, with fimbrial interaction occurring first, followed by nonfimbrial adhesin interaction. The latter specifically mediates cell-cell interaction, also termed autoaggregation, or may promote nonreversible interactions (45).
Fimbriae play an important role in the attachment of A. actinomycetemcomitans to different substrates, as well as cell aggregation (46). The data presented in this study, however, suggest that the nonfimbrial adhesin, EmaA, also contributes to A. actinomycetemcomitans attachment and aggregation. A decrease in the mass of the formed biofilm was demonstrated in emaA mutant fimbriated strains, and this decrease was even more apparent in comparisons of nonfimbriated strains. Complementation of these strains (fimbriated and nonfimbriated) with emaA plasmids reverted the phenotype back to levels equivalent to or in excess of the parent strains (likely due to the effect of plasmid copy number). The effect of EmaA on biofilm formation was also observed in multiple strains within the same serotype and across three of the seven known serotypes (i.e., serotypes a, b, and c). In strains lacking EmaA, transformation with the emaA plasmid demonstrated a gain of function in biofilm formation. The mass of the biofilm was increased compared to the parent strain transformed with the empty vector. Together, these studies suggest that the presence of EmaA on the cell surface enhances biofilm formation and that this function is conserved among A. actinomycetemcomitans strains.
EmaA structures are composed of three identical protein monomers forming a stalk and a head domain (35). The distal region of the structures, corresponding to amino acids 57 to 625, is associated with the ascribed collagen binding activity (34, 35). We have determined that the head domain is required for biofilm formation. Complete or partial removal of the head region of the structure decreases activity to levels comparable with the emaA mutant strain, suggesting that the head domain of EmaA is involved in biofilm formation, whereas no activity is associated with the remaining stalk structure.
The crucial role played by the head domain in both collagen binding and biofilm formation would suggest that these two functions are correlated. However, analysis of collagen binding mutants in biofilm assays demonstrated that the functions are uncorrelated. Both the G162S EmaA substitution mutant and a glycosylation-deficient mutant exhibited no biofilm formation defect. The G162S mutation results in a strain presenting adhesins with the overall structural integrity of the functional domain intact, albeit with a slightly altered subdomain structure; however, the strain displays reduced collagen binding activity (34, 35). Expression of the same construct in the emaA mutant strain used in this study only slightly affected biofilm formation compared to controls. Inactivation of the rhamnose epimerase rmlC, an essential rhamnose biosynthetic gene involved with LPS biosynthesis, results in bacteria expressing decreased amounts of surface EmaA and changes in the electrophoretic mobility of the monomers (37, 38). Associated with these changes in EmaA is a decrease in the collagen binding activity of the bacteria (37). The rmlC mutant strain in this study transformed with the plasmid expressing emaA (to equalize the amount of EmaA found on the surface) displayed no difference in the mass of biofilm formed compared to control cells. A glycosylation-independent mechanism of biofilm formation for EmaA is similar to that observed for the classical (type Va) Escherichia coli autotransporters (45, 47, 48). AIDA and TibA are glycoproteins where this posttranslational modification is required for adherence to human cells but not necessary for enhanced biofilm formation or cell aggregation. Our data suggest that the biofilm forming activity is less sensitive to structural changes in the EmaA molecule than is collagen binding and that EmaA glycosylation, although required for collagen binding, is not implicated in biofilm formation.
Confocal microscopy revealed a noticeable decrease in the relative fluorescent signal intensity of the microcolonies formed by the emaA mutant strain compared to the parent strain, suggesting a greater density or number of bacteria constituting the microcolonies of the emaA-positive strain. The greater cell density correlates well with the difference in mass observed between the two strains in the crystal violet biofilm assay. Interestingly, although the surface area remained constant, the calculated volume of the emaA mutant microcolonies was approximately 50% greater than that formed by cells derived from the parent strain. This observation implied a difference in the height of the microcolonies formed by these two strains as observed in a view perpendicular to the biofilm surface. The data support the hypothesis that strains lacking EmaA on the surface result in more loosely organized cells within the biofilm, suggesting that EmaA plays an important role in mediating cell-to-cell interactions. The presentation of EmaA structures on the surface of the bacteria may contribute to overcoming repulsive forces and allows for more intimate contact between individual bacteria. Therefore, EmaA may be necessary for enhanced biofilm formation or cell aggregation akin to the E. coli autotransporter proteins AIDA and TibA.
Our data suggest that there is a hierarchical order in the interactive forces of the surface structures in the biofilm formation process. Fimbriae, in A. actinomycetemcomitans, should be considered the primary structures associated with biofilm formation, while EmaA plays a secondary role. The contribution of EmaA in biofilm formation, however, becomes prominent in the absence of fimbriae. Other surface macromolecules are suggested to contribute to biofilm formation. The O-PS of LPS has also been implicated in adhesion to abiotic surfaces based on data collected using the serotype a strain ATCC 29523, which lacks fimbriae and EmaA (49). We have observed, however, little to no change in biofilm formation in the wild-type/O-PS mutant pairs of the serotype b strain used in this study (KM733 and KM799, Fig. 5B), which is nonfimbriated but expresses EmaA. Our data support the observation from another study using strain pairs derived from the serotype f strain CU1000N, which is fimbriated but does not express EmaA. These serotype f strains showed no difference in adherence (50). The absence of either fimbriae or EmaA results in a strain with modest biofilm-forming activity (Fig. 3), and the absence of O-PS in the ATCC 25923 strain may change the surface charge of the bacterium to further reduce electrostatic interactions of the bacterium with the substratum. Alternatively, the observed differences among these strains may be due to heterogeneity in the genomes between these strains (51) and/or the relative abundance and binding affinities of additional macromolecules in contributing to bacterial adhesion.
In this study, we have identified an alternative surface structure involved in biofilm formation of A. actinomycetemcomitans. We have demonstrated that the trimeric autotransporter protein EmaA mediates biofilm formation in multiple A. actinomycetemcomitans strains, and this activity is independent of the molecular form of the adhesin. Both collagen binding and biofilm formation activity are dependent on the same sequences of the protein or region of the structure. However, the structural constraints and glycosylation required for collagen binding are not necessary for biofilm formation. These observations suggest that EmaA is a multifunctional adhesin that uses different mechanisms for collagen adhesion and biofilm biogenesis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 2. All A. actinomycetemcomitans strains were grown from frozen stocks on solid TSBYE medium (0.3% tryptic soy broth, 0.6% yeast extract, 1.5% agar; Becton Dickinson, Franklin Lakes, NJ) in a humidified 10% CO2 atmosphere at 37°C. A single colony was used as the inoculum for all experiments. Fimbriated strains were maintained exclusively on solid TSBYE medium to avoid the potential loss of fimbriation (52, 53); nonfimbriated strains were grown statically in TSBYE broth. Plasmids were maintained in A. actinomycetemcomitans strains by incorporation of either 1.0 µg/ml chloramphenicol or 50 μg/ml kanamycin in the growth medium. E. coli strains were grown in LB medium (1.0% tryptone, 0.5% yeast extract, 0.5% NaCl; Becton Dickinson) at 37°C in ambient air with agitation. Strains containing plasmids were maintained at the following antibiotic concentrations: 100 µg/ml ampicillin, 20 µg/ml chloramphenicol, 50 µg/ml spectinomycin, or 50 µg/ml kanamycin.
TABLE 2.
Bacterial strains and plasmids
| Name | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5αλpir |
endA1 hadR17(r− m+) supE44 thi-1 recA gyrA1(Nalr) relA1 Δ(lacIZYA-argF)U169 deoR
[ϕ80dlacΔ(lacZ)M15] λpir, for shuttle plasmids |
60 |
| XL10G |
endA1 glnV44 recA1 thi-1 gyrA96 relA1 lac Hte Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173
Tetr F′[proAB lacIqZΔM15 Tn10(Tetr Amy Cmr)] |
Agilent Technologies |
| KM482 | DAP auxotroph strain with chromosomal λpir, for conjugation | Andrew Goodman, Yale |
| A. actinomycetemcomitans | ||
| VT1257 | Fimbriated clinical isolate, serotype b | Maria Saarela, IDH, Finland |
| KM667 | Isogenic emaA mutant of VT1257; Specr | This study |
| KM733 | Nonfimbriated variant of VT1257 | This study |
| KM714 | Isogenic emaA mutant of KM733; Specr | This study |
| VT1519 | Nonfimbriated variant of HK1651; serotype b | This study |
| KM785 | Isogenic emaA mutant of VT1519; Specr | This study |
| VT1218 | Fimbriated clinical isolate; serotype a | Maria Saarela, IDH, Finland |
| KM354 | Nonfimbriated variant of VT1218 | This study |
| KM762 | Isogenic emaA mutant of KM354; Specr | This study |
| VT1281 | Nonfimbriated laboratory strain; serotype a | ATCC 29523 |
| KM281 | Fimbriated D11S-1; serotype c | Casey Chen, USC |
| KM316 | Nonfimbriated variant of D11S-1; serotype c | This study |
| KM397 | Nonfimbriated laboratory strain; serotype c | ATCC 33384 |
| KM799 | Isogenic rmlC mutant of KM733; Specr | This study |
| Plasmids | ||
| pGEM | TA cloning vector; Ampr | Promega |
| pKM2 | E. coli and A. actinomycetemcomitans shuttle vector; Cmr | 61 |
| pKM11 | pKM2 containing full-length serotype B emaA | 38 |
| pKM48 | Serotype b emaA mutant corresponding to Δ57-625; Kanr | 35 |
| pKM60 | Serotype b emaA mutant corresponding to Δ57-123; Kanr | 35 |
| pKM89 | Serotype b emaA with G162S point mutation; Kanr | 35 |
| pKM753 | pKM2 containing full-length serotype A emaA | This study |
| pKM524 | Full-length emaA mutant construct on mobilizable plasmid; Kan/Specr | 10 |
| pKM758 | Intermediate-length emaA mutant construct on mobilizable plasmid; Kan/Specr | This study |
| pKM797 | rmlC mutant construct on mobilizable plasmid; Kan/Specr | This study |
Cmr, chloramphenicol resistance; Kanr, kanamycin resistance; Tetr, tetracycline resistance; Specr, spectinomycin resistance; Ampr, ampicillin resistance; Nalr, nalidixic acid resistance.
Generation of nonfimbriated variants.
Fimbriated A. actinomycetemcomitans spontaneously converts to a nonfimbriated state upon serial subculturing in broth cultures (52, 53). The frequency of this mutation rate is strain dependent (unpublished data). Nonfimbriated variants used in this study were generated by serial passage in a borosilicate glass tube containing TSBYE broth. The fimbriated bacteria adhered to the tube wall, leaving the medium with little to no turbidity. After a 24-h growth period, the broth was decanted and replaced with fresh media, and this process was repeated until the clear culture medium became turbid, indicating that the bacteria have lost the ability to produce fimbriae. All of the strains used in this study were characterized for expression of EmaA, as described previously (35). Bacteria from the medium were plated on TSBYE and a single colony was selected, grown, and frozen at –80°C in 10% dimethyl sulfoxide.
Construction of emaA and rmlC mutant strains.
One of two forms of emaA are present in A. actinomycetemcomitans strains (36). Serotypes b and c encode the prototypic full-length, 202-kDa protein, whereas serotypes a and d express a 173-kDa protein. emaA insertional mutants of serotype b and c strains were generated using a mobilizable plasmid for conjugation as described previously (10). For serotype a strains, the emaA gene (accession number DQ991439) and promoter region were amplified by PCR using isolated genomic DNA as the template and primers CBP1-5′up (5′-ACATGCATGCAACAAATCGCCGTCATCGCC-3′) and 3′EmaAEcoRV (5′-CAGGATATCGAATAAGCGCATTTTACCA-3′). The amplicon was gel purified and cloned into pGEM T-Easy (Promega, Madison, WI) and verified by sequencing at the Advanced Genome Technologies Core facility at the University of Vermont. To generate an insertional mutant, DNA coding for the aad9 gene conferring resistance to spectinomycin (54) was inserted into the plasmid following treatment with BamHI and HindIII restriction endonucleases, resulting in a 4,408-bp internal deletion of the gene. After transformation and antibiotic selection, the plasmid was isolated, and the corresponding sequence was released by restriction with EcoRI and ligated with the mobilization plasmid, as described previously (10). The plasmids containing both forms of emaA were transformed into a diaminopimelic acid-deficient (DAP) strain of E. coli (55), which was used as the donor strain. Transconjugants were selected on TSBYE-spectinomycin agar plates and verified to be emaA mutants by colony PCR and immuno-dot blot analyses (35).
Inactivation of rmlC followed the protocol stated above using the primers rmlC_5′comp (ATGAAAGTTATTGATAC) and rmlC_3′comp (AAATTTTACCGTTTCTGCC), with aad9 inserted at the HpaI restriction site within the DNA sequence. The loss of O-PS was verified using anti-A. actinomycetemcomitans antisera previously demonstrated to be reactive to the O-PS (37).
All mutants used in this study displayed no observable growth defects or unexpected phenotypic changes from the parental strains.
Biofilm assay.
Biofilm assays were based on the method of Merritt et al. (56). A. actinomycetemcomitans strains were initially grown from frozen stocks on TSBYE agar. For fimbriated strains, bacteria were collected by scraping an agar plate in the presence of 1.5 ml of TSBYE. The cell suspension was vortexed rigorously for 30 s, and the tube incubated for 10 min to allow large clumps to settle to the bottom. A portion of the suspension was removed, and the concentration adjusted to an optical density of 0.05 at a wavelength of 495 nm (ca. 107 CFU/ml). Portions (200 μl) of each strain were inoculated into sterile 96-well microtiter plates (Nunc, Roskilde, Denmark) and grown for 72 h. After 24 and 48 h, 100 µl of spent medium was removed and replaced with fresh medium to allow for continuous growth. For nonfimbriated strains, single colonies were inoculated into 5 ml of TSBYE and grown overnight. The cultures were diluted 1:10 and grown to an absorbance of 0.3 at a wavelength of 495 nm. A 200-µl aliquot of a 1:1,000 dilution (ca. 104 CFU/ml) was added to the 96-well plates, and the cultures were grown for 24 h. After the growth interval, the supernatants were aspirated, and the remaining nonadherent cells were removed by three consecutive washes with phosphate-buffered saline (PBS; 136.9 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, 1.46 mM KH2PO4, 0.46 mM MgCl2 [pH 7.4]; Sigma-Aldrich, St. Louis, MO). Biofilms were stained with 0.1% crystal violet in water for 20 min, washed three times with PBS, and solubilized using a 2:1 solution of water-glacial acetic acid. The relative biofilm mass of each strain was quantified by absorbance at 562 nm using an ELx800 plate reader (BioTek, Winooski, VT). A two-tailed Student t test was used to identify significant differences (P < 0.05). A minimum of three independent experiments was performed for each strain in triplicate.
Collagen binding assay.
The binding of whole bacteria to collagen was assayed using an enzyme-linked immunosorbent assay format with human type V collagen (Sigma) as the substrate and polyclonal antibodies isolated and purified from serum of rabbits immunized with whole bacteria (57) following the protocol described by Yu et al. (35).
TEM.
A. actinomycetemcomitans cells were visualized by transmission electron microscopy (TEM) based on the method described by Azari et al. (58). Briefly, appropriate strains were streaked for isolation on solid media. Colonies were directly transferred to carbon-coated grids and stained using Nano-W (NanoProbes, Yaphank, NY). Images were collected on a 2,048-by-2048-pixel charge-coupled camera with a 14-µm pixel size (TVIPS, Gaunting, Germany) at 52,000 nominal magnification using a Tecnai 12 electron microscope operating at 100 kV (FEI, Portland, OR).
Confocal microscopy.
Fimbriated A. actinomycetemcomitans biofilms were grown in glass-bottom petri dishes (MatTek, Ashland, MA) as described above for 24 h. After growth, the supernatants were removed by aspiration, and nonadherent cells were removed by three washes with Tris-buffered saline (TBS; 20 mM Tris, 150 mM NaCl [pH 7.4]; Sigma). Biofilms were stained with SYTO9 (Invitrogen, Carlsbad, CA) in TBS at 5 µM for 30 min. The staining solution was aspirated, and any unbound stain was removed by four washes with TBS. Images were recorded at the University of Vermont Microscopy Imaging Center. For morphological measurements, a Zeiss LSM 510 META confocal microscope (Zeiss, Oberkochen, Germany) with a Plan-Apochromat 63× objective and an excitation wavelength of 488 nm was used. For fluorescence intensity measurements, a Nikon A1R-ER point scanning confocal microscope (Nikon, Tokyo, Japan) was used. For both image sets, random fields were selected, z-slices were acquired at increments of 0.37 µm, and z-stacks were generated. Volume, surface area, and integrated fluorescence intensities of the bacterial architectures were quantified using the Volocity software package (Perkin-Elmer, Waltham, MA). 3-D images were generated using Chimera (59). All experiments were performed in triplicate. For comparisons between strains, a Student t test was used, with significance defined as P < 0.1.
The overall average integrated fluorescence intensity per z-stack was calculated by thresholding each z-slice per stack to include pixel intensity between 80 and 4,095 (to exclude background). Integrated intensity was then calculated per colony in each z-slice as the sum of the intensity within the colony divided by the colony area in that z-slice. An integrated intensity per z-slice was taken by summing all the integrated intensities of colonies that met the threshold requirements in each slice. The average integrated intensity per z-stack was calculated by averaging the integrated intensities for each slice. Since four z-stacks were captured per sample, averaging the integrated intensities for each yields an average integrated intensity per sample.
ACKNOWLEDGMENTS
We thank Jake Tristano and Nicole Bouffard for technical assistance.
Imaging work was supported by National Institutes of Health (NIH) awards 1S10OD025030-01 and 1S10RR019246 from the National Center for Research Resources. The research was supported by grant 5R01DE024554 from NIH/National Institutes for Dental Craniofacial Research to K.P.M. and T.R.
REFERENCES
- 1.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 2.Zambon JJ, Slots J, Genco RJ. 1983. Serology of oral Actinobacillus actinomycetemcomitans and serotype distribution in human periodontal disease. Infect Immun 41:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haubek D, Ennibi OK, Poulsen K, Benzarti N, Baelum V. 2004. The highly leukotoxic JP2 clone of Actinobacillus actinomycetemcomitans and progression of periodontal attachment loss. J Dent Res 83:767–770. doi: 10.1177/154405910408301006. [DOI] [PubMed] [Google Scholar]
- 4.Paturel L, Casalta JP, Habib G, Nezri M, Raoult D. 2004. Actinobacillus actinomycetemcomitans endocarditis. Clin Microbiol Infect 10:98–118. doi: 10.1111/j.1469-0691.2004.00794.x. [DOI] [PubMed] [Google Scholar]
- 5.Das M, Badley AD, Cockerill FR, Steckelberg JM, Wilson WR. 1997. Infective endocarditis caused by HACEK microorganisms. Annu Rev Med 48:25–33. doi: 10.1146/annurev.med.48.1.25. [DOI] [PubMed] [Google Scholar]
- 6.Berne C, Ducret A, Hardy GG, Brun YV. 2015. Adhesins involved in attachment to abiotic surfaces by Gram-negative bacteria. Microbiol Spectr 3:MB-0018-2015. doi: 10.1128/microbiolspec.MB-0018-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kachlany SC, Planet PJ, Desalle R, Fine DH, Figurski DH, Kaplan JB. 2001. flp-1, the first representative of a new pilin gene subfamily, is required for nonspecific adherence of Actinobacillus actinomycetemcomitans. Mol Microbiol 40:542–554. doi: 10.1046/j.1365-2958.2001.02422.x. [DOI] [PubMed] [Google Scholar]
- 8.Inouye T, Ohta H, Kokeguchi S, Fukui K, Kato K. 1990. Colonial variation and fimbriation of Actinobacillus actinomycetemcomitans. FEMS Microbiol Lett 57:13–17. [DOI] [PubMed] [Google Scholar]
- 9.Fine DH, Furgang D, Schreiner HC, Goncharoff P, Charlesworth J, Ghazwan G, Fitzgerald-Bocarsly P, Figurski DH. 1999. Phenotypic variation in Actinobacillus actinomycetemcomitans during laboratory growth: implications for virulence. Microbiology 145:1335–1347. doi: 10.1099/13500872-145-6-1335. [DOI] [PubMed] [Google Scholar]
- 10.Mintz KP. 2004. Identification of an extracellular matrix protein adhesin, EmaA, which mediates the adhesion of Actinobacillus actinomycetemcomitans to collagen. Microbiology 150:2677–2688. doi: 10.1099/mic.0.27110-0. [DOI] [PubMed] [Google Scholar]
- 11.Tahir YE, Kuusela P, Skurnik M. 2000. Functional mapping of the Yersinia enterocolitica adhesin YadA: identification of eight NSVAIG-S motifs in the amino-terminal half of the protein involved in collagen binding. Mol Microbiol 37:192–206. doi: 10.1046/j.1365-2958.2000.01992.x. [DOI] [PubMed] [Google Scholar]
- 12.St Geme JW III, Grass S. 1998. Secretion of the Haemophilus influenzae HMW1 and HMW2 adhesins involves a periplasmic intermediate and requires the HMWB and HMWC proteins. Mol Microbiol 27:617–630. doi: 10.1046/j.1365-2958.1998.00711.x. [DOI] [PubMed] [Google Scholar]
- 13.Rose JE, Meyer DH, Fives-Taylor PM. 2003. Aae, an autotransporter involved in adhesion of Actinobacillus actinomycetemcomitans to epithelial cells. Infect Immun 71:2384–2393. doi: 10.1128/IAI.71.5.2384-2393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fine D, Velliyagounder K, Furgang D, Kaplan J. 2005. The Actinobacillus actinomycetemcomitans autotransporter adhesin Aae exhibits specificity for buccal epithelial cells from humans and old world primates. Infect Immun 73:1947–1953. doi: 10.1128/IAI.73.4.1947-1953.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asakawa R, Komatsuzawa H, Kawai T, Yamada S, Goncalves RB, Izumi S, Fujiwara T, Nakano Y, Suzuki N, Uchida Y, Ouhara K, Shiba H, Taubman MA, Kurihara H, Sugai M. 2003. Outer membrane protein 100, a versatile virulence factor of Actinobacillus actinomycetemcomitans. Mol Microbiol 50:1125–1139. doi: 10.1046/j.1365-2958.2003.03748.x. [DOI] [PubMed] [Google Scholar]
- 16.Yue G, Kaplan JB, Furgang D, Mansfield KG, Fine DH. 2007. A second Aggregatibacter actinomycetemcomitans autotransporter adhesin exhibits specificity for buccal epithelial cells in humans and Old World primates. Infect Immun 75:4440–4448. doi: 10.1128/IAI.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang G, Kitten T, Munro CL, Wellman GC, Mintz KP. 2008. EmaA, a potential virulence determinant of Aggregatibacter actinomycetemcomitans in infective endocarditis. Infect Immun 76:2316–2324. doi: 10.1128/IAI.00021-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauri A, Soprova Z, Wickström D, de Gier J, Van der Schors R, Smit A, Jong W, Luirink J. 2009. The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155:3982–3991. doi: 10.1099/mic.0.034991-0. [DOI] [PubMed] [Google Scholar]
- 19.Leyton D, Rossiter A, Henderson I. 2012. From self sufficiency to dependence: mechanisms and factors important for autotransporter biogenesis. Nat Rev Microbiol 10:213–225. doi: 10.1038/nrmicro2733. [DOI] [PubMed] [Google Scholar]
- 20.Roggenkamp A, Ackermann N, Jacobi CA, Truelzsch K, Hoffmann H, Heesemann J. 2003. Molecular analysis of transport and oligomerization of the Yersinia enterocolitica adhesin YadA. J Bacteriol 185:3735–3744. doi: 10.1128/JB.185.13.3735-3744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson IR, Nataro JP. 2001. Virulence functions of autotransporter proteins. Infect Immun 69:1231–1243. doi: 10.1128/IAI.69.3.1231-1243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schutz M, Weiss EM, Schindler M, Hallstrom T, Zipfel PF, Linke D, Autenrieth IB. 2010. Trimer stability of YadA is critical for virulence of Yersinia enterocolitica. Infect Immun 78:2677–2690. doi: 10.1128/IAI.01350-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarselli M, Serruto D, Montanari P, Capecchi B, Adu-Bobie J, Veggi D, Rappuoli R, Pizza M, Aricò B. 2006. Neisseria meningitidis NhhA is a multifunctional trimeric autotransporter adhesin. Mol Microbiol 61:631–644. doi: 10.1111/j.1365-2958.2006.05261.x. [DOI] [PubMed] [Google Scholar]
- 24.Capecchi B, Adu-Bobie J, Di Marcello F, Ciucchi L, Masignani V, Taddei A, Rappuoli R, Pizza M, Aricò B. 2005. Neisseria meningitidis NadA is a new invasin which promotes bacterial adhesion to and penetration into human epithelial cells. Mol Microbiol 55:687–698. doi: 10.1111/j.1365-2958.2004.04423.x. [DOI] [PubMed] [Google Scholar]
- 25.Surana NK, Cutter D, Barenkamp SJ, St Geme JW III. 2004. The Haemophilus influenzae Hia autotransporter contains an unusually short trimeric translocator domain. J Biol Chem 279:14679–14685. doi: 10.1074/jbc.M311496200. [DOI] [PubMed] [Google Scholar]
- 26.Laarmann S, Cutter D, Juehne T, Barenkamp SJ, St Geme JW III. 2002. The Haemophilus influenzae Hia autotransporter harbours two adhesive pockets that reside in the passenger domain and recognize the same host cell receptor. Mol Microbiol 46:731–743. doi: 10.1046/j.1365-2958.2002.03189.x. [DOI] [PubMed] [Google Scholar]
- 27.Cope LD, Lafontaine ER, Slaughter CA, Hasemann CA Jr, Aebi C, Henderson FW, McCracken GH Jr, Hansen EJ. 1999. Characterization of the Moraxella catarrhalis uspA1 and uspA2 genes and their encoded products. J Bacteriol 181:4026–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aebi C, Maciver I, Latimer JL, Cope LD, Stevens MK, Thomas SE, McCracken GH Jr, Hansen EJ. 1997. A protective epitope of Moraxella catarrhalis is encoded by two different genes. Infect Immun 65:4367–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valle J, Mabbett AN, Ulett GC, Toledo-Arana A, Wecker K, Totsika M, Schembri MA, Ghigo JM, Beloin C. 2008. UpaG, a new member of the trimeric autotransporter family of adhesins in uropathogenic Escherichia coli. J Bacteriol 190:4147–4161. doi: 10.1128/JB.00122-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raghunathan D, Wells TJ, Morris FC, Shaw RK, Bobat S, Peters SE, Paterson GK, Jensen KT, Leyton DL, Blair JM, Browning DF, Pravin J, Flores-Langarica A, Hitchcock JR, Moraes CT, Piazza RM, Maskell DJ, Webber MA, May RC, MacLennan CA, Piddock LJ, Cunningham AF, Henderson IR. 2011. SadA, a trimeric autotransporter from Salmonella enterica serovar Typhimurium, can promote biofilm formation and provides limited protection against infection. Infect Immun 79:4342–4352. doi: 10.1128/IAI.05592-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Totsika M, Wells TJ, Beloin C, Valle J, Allsopp LP, King NP, Ghigo JM, Schembri MA. 2012. Molecular characterization of the EhaG and UpaG trimeric autotransporter proteins from pathogenic Escherichia coli. Appl Environ Microbiol 78:2179–2189. doi: 10.1128/AEM.06680-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu YY, Franz B, Truttmann MC, Riess T, Gay-Fraret J, Faustmann M, Kempf VA, Dehio C. 2013. Bartonella henselae trimeric autotransporter adhesin BadA expression interferes with effector translocation by the VirB/D4 type IV secretion system. Cell Microbiol 15:759–778. doi: 10.1111/cmi.12070. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz T, Lenox C, Radermacher M, Mintz KP. 2006. Novel surface structures are associated with the adhesion of Actinobacillus actinomycetemcomitans to collagen. Infect Immun 74:6163–6170. doi: 10.1128/IAI.00857-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Azari F, Radermacher M, Mintz K, Ruiz T. 2012. Correlation of the amino-acid sequence and the 3D structure of the functional domain of EmaA from Aggregatibacter actinomycetemcomitans. J Struct Biol 177:439–446. doi: 10.1016/j.jsb.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu C, Ruiz T, Lenox C, Mintz KP. 2008. Functional mapping of an oligomeric autotransporter adhesin of Aggregatibacter actinomycetemcomitans. J Bacteriol 190:3098–3109. doi: 10.1128/JB.01709-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang G, Ruiz T, Barrantes-Reynolds R, Mintz KP. 2007. Molecular heterogeneity of EmaA, an oligomeric autotransporter adhesin of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Microbiology 153:2447–2457. doi: 10.1099/mic.0.2007/005892-0. [DOI] [PubMed] [Google Scholar]
- 37.Tang G, Ruiz T, Mintz KP. 2012. O-polysaccharide glycosylation is required for stability and function of the collagen adhesin EmaA of Aggregatibacter actinomycetemcomitans. Infect Immun 80:2868–2877. doi: 10.1128/IAI.00372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang G, Mintz KP. 2010. Glycosylation of the collagen adhesin EmaA of Aggregatibacter actinomycetemcomitans is dependent upon the lipopolysaccharide biosynthetic pathway. J Bacteriol 192:1395–1404. doi: 10.1128/JB.01453-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry MB, MacLean LL, Gmur R, Wilson ME. 1996. Characterization of the O-polysaccharide structure of lipopolysaccharide from Actinobacillus actinomycetemcomitans serotype b. Infect Immun 64:1215–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosan B, Slots J, Lamont RJ, Listgarten MA, Nelson GM. 1988. Actinobacillus actinomycetemcomitans fimbriae. Oral Microbiol Immunol 3:58–63. doi: 10.1111/j.1399-302X.1988.tb00082.x. [DOI] [PubMed] [Google Scholar]
- 41.Fine DH, Furgang D, Kaplan J, Charlesworth J, Figurski DH. 1999. Tenacious adhesion of Actinobacillus actinomycetemcomitans strain CU1000 to salivary-coated hydroxyapatite. Arch Oral Biol 44:1063–1076. doi: 10.1016/S0003-9969(99)00089-8. [DOI] [PubMed] [Google Scholar]
- 42.Serra DO, Richter AM, Klauck G, Mika F, Hengge R. 2013. Microanatomy at cellular resolution and spatial order of physiological differentiation in a bacterial biofilm. mBio 4:e00103-13. doi: 10.1128/mBio.00103-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoiczyk E, Roggenkamp A, Reichenbecher M, Lupas A, Heesemann J. 2000. Structure and sequence analysis of Yersinia YadA and Moraxella UspAs reveal a novel class of adhesins. EMBO J 19:5989–5999. doi: 10.1093/emboj/19.22.5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C. 1999. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000 20:136–167. doi: 10.1111/j.1600-0757.1999.tb00161.x. [DOI] [PubMed] [Google Scholar]
- 45.Benz I, Schmidt MA. 2001. Glycosylation with heptose residues mediated by the aah gene product is essential for adherence of the AIDA-I adhesin. Mol Microbiol 40:1403–1413. doi: 10.1046/j.1365-2958.2001.02487.x. [DOI] [PubMed] [Google Scholar]
- 46.Inoue T, Shingaki R, Sogawa N, Sogawa CA, Asaumi J, Kokeguchi S, Fukui K. 2003. Biofilm formation by a fimbriae-deficient mutant of Actinobacillus actinomycetemcomitans. Microbiol Immunol 47:877–881. doi: 10.1111/j.1348-0421.2003.tb03454.x. [DOI] [PubMed] [Google Scholar]
- 47.Benz I, Schmidt MA. 2002. Never say never again: protein glycosylation in pathogenic bacteria. Mol Microbiol 45:267–276. doi: 10.1046/j.1365-2958.2002.03030.x. [DOI] [PubMed] [Google Scholar]
- 48.Cote JP, Charbonneau ME, Mourez M. 2013. Glycosylation of the Escherichia coli TibA self-associating autotransporter influences the conformation and the functionality of the protein. PLoS One 8:e80739. doi: 10.1371/journal.pone.0080739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujise O, Wang Y, Chen W, Chen C. 2008. Adherence of Aggregatibacter actinomycetemcomitans via serotype-specific polysaccharide antigens in lipopolysaccharides. Oral Microbiol Immunol 23:226–233. doi: 10.1111/j.1399-302X.2007.00416.x. [DOI] [PubMed] [Google Scholar]
- 50.Kaplan JB, Meyenhofer MF, Fine DH. 2003. Biofilm growth and detachment of Actinobacillus actinomycetemcomitans. J Bacteriol 185:1399–1404. doi: 10.1128/JB.185.4.1399-1404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kittichotirat W, Bumgarner R, Asikainen S, Chen C. 2011. Identification of the pangenome and its components in 14 distinct Aggregatibacter actinomycetemcomitans strains by comparative genomic analysis. PLoS One 6:e22420. doi: 10.1371/journal.pone.0022420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Chen C. 2005. Mutation analysis of the flp operon in Actinobacillus actinomycetemcomitans. Gene 351:61–71. doi: 10.1016/j.gene.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Liu A, Chen C. 2005. Genetic basis for conversion of rough-to-smooth colony morphology in Actinobacillus actinomycetemcomitans. Infect Immun 73:3749–3753. doi: 10.1128/IAI.73.6.3749-3753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lukomski S, Hoe NP, Abdi I, Rurangirwa J, Kordari P, Liu M, Dou SJ, Adams GG, Musser JM. 2000. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infect Immun 68:535–542. doi: 10.1128/IAI.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Babic A, Guerout AM, Mazel D. 2008. Construction of an improved RP4 (RK2)-based conjugative system. Res Microbiol 159:545–549. doi: 10.1016/j.resmic.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 56.Merritt JH, Kadouri DE, O'Toole GA. 2005. Growing and analyzing static biofilms. Curr Protoc Microbiol Chapter 1:Unit 1B1. doi: 10.1002/9780471729259.mc01b01s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mintz KP, Fives-Taylor PM. 1999. Binding of the periodontal pathogen Actinobacillus actinomycetemcomitans to extracellular matrix proteins. Oral Microbiol Immunol 14:109–116. doi: 10.1034/j.1399-302X.1999.140206.x. [DOI] [PubMed] [Google Scholar]
- 58.Azari F, Nyland L, Yu C, Radermacher M, Mintz KP, Ruiz T. 2013. Ultrastructural analysis of the rugose cell envelope of a member of the Pasteurellaceae family. J Bacteriol 195:1680–1688. doi: 10.1128/JB.02149-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 60.Mintz K, Brissette C, Fives-Taylor P. 2002. A recombinase A-deficient strain of Actinobacillus actinomycetemcomitans constructed by insertional mutagenesis using a mobilizable plasmid. FEMS Microbiol Lett 206:87–92. doi: 10.1111/j.1574-6968.2002.tb10991.x. [DOI] [PubMed] [Google Scholar]
- 61.Gallant CV, Sedic M, Chicoine EA, Ruiz T, Mintz KP. 2008. Membrane morphology and leukotoxin secretion are associated with a novel membrane protein of Aggregatibacter actinomycetemcomitans. J Bacteriol 190:5972–5980. doi: 10.1128/JB.00548-08. [DOI] [PMC free article] [PubMed] [Google Scholar]