

Streptococcus sanguinis, an abundant and benign inhabitant of the oral cavity, is an important etiologic agent of infective endocarditis (IE), particularly in people with predisposing cardiac valvular damage. Although commonly isolated from patients with IE, little is known about the factors that make any particular S. sanguinis isolate more virulent than another or, indeed, whether significant differences in virulence exist among isolates.
KEYWORDS: infective endocarditis, Streptococcus, biofilms, genomics, manganese, viridans, virulence
ABSTRACT
Streptococcus sanguinis, an abundant and benign inhabitant of the oral cavity, is an important etiologic agent of infective endocarditis (IE), particularly in people with predisposing cardiac valvular damage. Although commonly isolated from patients with IE, little is known about the factors that make any particular S. sanguinis isolate more virulent than another or, indeed, whether significant differences in virulence exist among isolates. In this study, we compared the genomes of a collection of S. sanguinis strains comprised of both oral isolates and bloodstream isolates from patients diagnosed with IE. Oral and IE isolates could not be distinguished by phylogenetic analyses, and we did not succeed in identifying virulence genes unique to the IE strains. We then investigated the virulence of these strains in a rabbit model of IE using a variation of the Bar-seq (barcode sequencing) method wherein we pooled the strains and used Illumina sequencing to count unique barcodes that had been inserted into each isolate at a conserved intergenic region. After we determined that several of the genome sequences were misidentified in GenBank, our virulence results were used to inform our bioinformatic analyses, identifying genes that may explain the heterogeneity in virulence. We further characterized these strains by assaying for phenotypes potentially contributing to virulence. Neither strain competition via bacteriocin production nor biofilm formation showed any apparent relationship with virulence. Increased cell-associated manganese was, however, correlated with blood isolates. These results, combined with additional phenotypic assays, suggest that S. sanguinis virulence is highly variable and results from multiple genetic factors.
INTRODUCTION
Streptococcus sanguinis is a bacterium with a dual nature, having roles in both health and disease. With regards to health, S. sanguinis plays an important role in establishment of the oral biofilm. Studies show that when S. sanguinis colonization precedes that of Streptococcus mutans, a causative agent of dental caries (1), the acquisition of S. mutans is delayed (2). More recent studies using molecular methods have likewise demonstrated an association between S. sanguinis and health with regard to caries (3, 4). This benefit is in direct opposition to the role of S. sanguinis as an opportunistic pathogen and causative agent of infective endocarditis (IE). IE is a disease characterized by bacterial colonization of heart valves or endocardium that leads to inflammation of the tissues. Due to the location of S. sanguinis in the oral cavity, introduction into the bloodstream is a regular hazard and can occur as a result of practices such as oral surgery (5), eating, regular oral hygiene (6–8), and poor oral care habits (9). For most people, this transient bacteremia does not result in disease; however, in people with preexisting heart damage, S. sanguinis can adhere to exposed sterile “vegetations,” composed of platelets and fibrin at the site of damage, and proliferate (10). Continued proliferation and dissemination of these masses can lead to fatal outcomes, such as congestive heart failure and stroke, respectively (11).
Recent advances in our understanding of the mechanisms by which S. sanguinis contributes to health and disease have relied primarily on a small number of bacterial strains. For the oral cavity, recent studies have utilized strain SK150 as a reference to explore bacterial composition and interactions within biofilms (12–14). Likewise, the literature reflects a preference for strains such as SK36, which is the first S. sanguinis strain to have its complete genome sequence published (15) and the type strain (ATCC 10556; also known as SK1 and NCTC 7863), to advance our understanding of S. sanguinis IE virulence, investigating traits such as binding to sterile vegetations by adhering to proteins similar to those found in the oral cavity (16, 17), evading the host immune response (18–20), and utilizing a high-affinity manganese uptake system (21) for oxidative stress defense (22) and deoxyribonucleotide synthesis (23, 24).
Although these studies have provided a critical foundation for our understanding of S. sanguinis physiology, a single strain cannot encompass the full genotypic or phenotypic range of a species. As one example, recent work in Streptococcus pneumoniae, a close relative of S. sanguinis, has highlighted how a small interrogative strain pool can lead to oversimplifications of bacterial functions and host relationships (25–27). Fortunately, a number of S. sanguinis strains are available with corresponding information, including anatomical origin of isolation (oral cavity of normal subjects or blood of IE patients), multilocus sequence analysis (MLSA) (28), and draft genome sequences (NCBI). Therefore, the incorporation of increasing numbers of strains into research studies is not only preferable for understanding the properties of the S. sanguinis species as a whole but also attainable.
We set out here to subject a collection of 20 S. sanguinis strains, comprised of oral and IE isolates, to a battery of bioinformatic, virulence, and phenotypic analyses to investigate factors that contribute to the ability of individual S. sanguinis strains to cause IE. Using a pooled inoculum approach known as Bar-seq, we determined that strain virulence varied over roughly 4 orders of magnitude and was not correlated with strain source; therefore, virulence in this species is likely multifactorial. Of additional note, we identified several serious errors in GenBank, such that four strains have been assigned incorrect whole-genome sequences, and a fifth strain, which was suggested to have been sequenced, lacks a whole-genome sequence.
RESULTS
Genomic overview.
We began with a bioinformatic analysis of 20 S. sanguinis strains: the oral isolate SK36, which we had sequenced previously (15), and 19 other strains that were draft sequenced as part of the Human Microbiome Project. Nine of these strains were oral isolates, with the remaining 10 isolated from IE patients (Table 1). As is typical, the IE isolates were from blood, except perhaps SK1, whose exact source is unknown apart from being isolated from an IE patient. The draft sequences were downloaded from GenBank as whole-genome shotgun contigs (WGS sequences). The sequence denoted SK1059 was dropped from the bioinformatic analyses for reasons described below. The only complete genome sequence was that of SK36, which contains 2,388,435 bp of DNA with a GC content of 43.4%. The average number of scaffolds for the remaining 18 unfinished genomes was 6, and based on these scaffolds, their genomes ranged in size from 2.28 to 2.42 Mb DNA with GC contents ranging from 42.8% to 43.4%. SK340 is annotated as containing the most protein-coding sequences (2,298) and SK49 having the least (2,166), whereas SK36 has 2,270.
TABLE 1.
Strains used in this study
| Strain name | Repository IDa | Sourceb | Country | Reference(s) |
|---|---|---|---|---|
| SK1/ATCC 10556 | CCUG 35770 | Human subacute bacterial endocarditis | Unknown | 28, 74, 75 |
| SK36 | CCUG 55078, BAA-1455 | Dental plaque | Denmark | 15, 28, 74–76 |
| SK49 | CCUG 59319 | Dental plaque | Denmark | 28, 74, 76 |
| SK72 | CCUG 27744 | Dental plaque | Greenland | 28, 74, 75, 77 |
| SK115 | CCUG 59320 | Dental plaque | Greenland | 28, 74, 77 |
| SK150 | CCUG 25605 | Dental plaque | Denmark | 28, 74, 75, 78 |
| SK160 | CCUG 35766 | Dental plaque | Denmark | 28, 74, 75, 78 |
| SK330 | CCUG 59322 | Dental plaque | USA | 28 |
| SK340 | CCUG 59323 | Dental plaque | USA | 28 |
| SK353 | CCUG 59324 | Dental plaque | Japan | 28 |
| SK355 | CCUG 59325 | Dental plaque | Japan | 28 |
| SK405 | CCUG 59326 | Blood; endocarditis | Denmark | 28 |
| SK408 | CCUG 59327 | Blood; endocarditis | Denmark | 28 |
| SK678 | CCUG 59328 | Blood; endocarditis | Denmark | 28 |
| SK1056 | CCUG 59329 | Blood; endocarditis | Denmark | 28 |
| SK1057 | CCUG 59330 | Blood; endocarditis | Denmark | 28 |
| SK1058 | CCUG 59331 | Blood; endocarditis | Denmark | 28 |
| SK1059 | CCUG 59332 | Blood; endocarditis | Denmark | 28 |
| SK1087 | CCUG 59333 | Blood; endocarditis | Denmark | 28 |
| VMC66 | HM-275 | Blood; endocarditis | USA | 79 |
Accession numbers for strains are archived at the Culture Collection of the University of Gothenburg (CCUG), the American Type Culture Collection (ATCC and BAA), or BEI Resources (HM).
All SK strains were obtained from Mogens Kilian, Aarhus University, Denmark, ATCC 10556 was obtained from the ATCC, and VMC66 was isolated previously by our group.
PGAP in the gene family (GF) mode (29) was used to explore the pangenomes, core genomes, and dispensable genomes of the 19 S. sanguinis strains. Analysis of the 19 S. sanguinis genomes by the GF method indicates an open pangenome, suggesting that analysis of additional strains would result in an ever-increasing pangenome size (Fig. 1). Our analysis identified 4,051 clusters of orthologous alleles. This figure is lower than the 5,100 orthologous clusters found by Zheng et al. (30) when analyzing the pangenome of 27 S. sanguinis strains with the same strategy (PGAP using GF method). The lesser number is consistent with expectations for an open pangenome, which is defined as increasing to infinity as the number of strains increases (31). The value of 1,499 core clusters (defined as clusters shared by all strains) identified in our analysis (Fig. 1; see also Fig. S1 in the supplemental material) is again somewhat lower than that of Zheng et al. (30), who found 1,739 core genes in 27 strains of S. sanguinis (30). The 1,499 core clusters identified in our analysis contained 68% of all SK36 genes. In addition to the core genome, we identified the gene clusters unique to each strain (Fig. S1).
FIG 1.
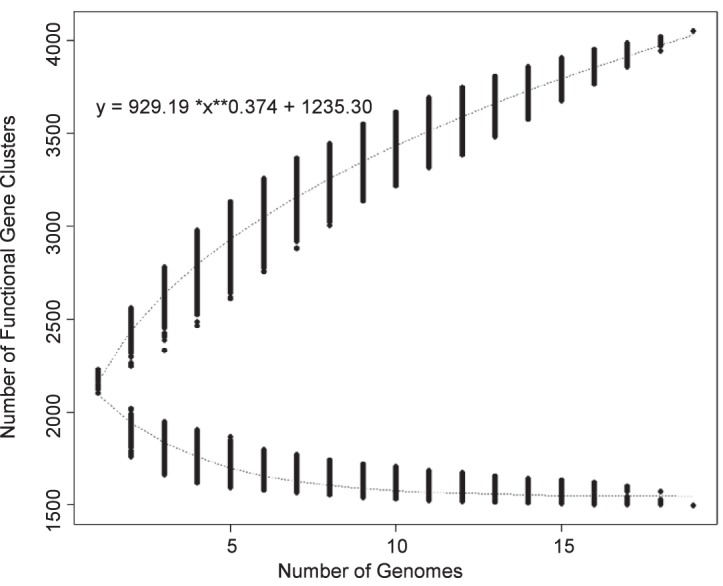
Cluster analysis of 19 genomes by PGAP in GF mode. Upper curve, pangenome calculation; lower curve, core genome calculation.
Phylogenetic analysis.
We were interested in determining whether endocarditis isolates would be phylogenetically distinct from oral isolates. Figure 2A shows a neighbor-joining phylogram based on a pangenome analysis in which relatedness reflects the presence or absence of shared genes. The phylogram shows one major clade, labeled 1, and two smaller clades, 2 and 3. Clade 1 is comprised of five IE isolates, while clades 2 and 3 contain isolates from both sources. Thus, blood and oral isolates were not fully distinguished by a pangenomic approach; however, the pangenome-based tree does show half of the blood isolates clustering into a single well-defined clade.
FIG 2.

Phylograms computed by PGAP using the neighbor-joining method of clustering. Clades are labeled and accentuated by darker branches emanating from the clade’s most basal node. Red indicates blood isolates, and blue denotes oral isolates. (A) Pangenome-based phylogram created using a gene distance matrix approach built upon gene presence or absence in each strain. (B) SNP-based phylogram created from analysis of core genes lacking paralogs.
The single-nucleotide polymorphism (SNP)-based neighbor-joining phylogram shown in Fig. 2B is derived from mutation and insertion/deletion (indel) variations within core gene clusters that lack paralogs, and the relationship depicted shows two major clades. Interestingly, clade 1 is very similar to clade 1 in the pangenome-based tree, in that it is comprised of the same blood isolates as those described above plus one oral isolate. Clade 2 is very similar to clade 3 of the pangenome-based tree.
Identification of putative virulence genes and examination in an endocarditis model.
Comparison of the genomes of strains isolated from the oral cavity versus those obtained from the blood of individuals with IE indicated the presence of several genes more commonly found in the blood isolates that could explain the progression to IE (Table S1). Of these genes, a subset of genes, including a putative lipoprotein gene (HMPREF9390_RS07860) and an ∼12-kb genomic island (HMPREF9390_RS08365 through HMPREF9390_RS08320) containing a tellurite resistance gene, was chosen for further investigation.
The presence of the tellurite resistance genes was of particular interest, as deletion of a cluster of tellurite resistance genes, yceGH in Bacillus anthracis, generated strains that were more susceptible to reactive oxygen species and cathelicidin-mediated killing and were attenuated in murine models of infection (32). We considered that the genomic island could contribute to IE virulence as well. The putative lipoprotein gene was selected because it was one of only a few genes exclusive to the blood isolates. Additionally, our laboratory and others have identified lipoproteins as being important for streptococcal virulence (21, 22, 33).
Two strains were chosen for investigating the relationship of these genes to IE virulence in a rabbit model of endocarditis. The first, SK36, is a commonly utilized reference strain and oral isolate lacking both loci, whereas SK405, a blood isolate, possesses both loci. For virulence testing, we have typically used strains marked with different antibiotic resistance genes, including JFP36, a derivative of SK36 that contains an erythromycin resistance gene (ermB) inserted into the hypothetical gene SSA_0169 (22, 23, 34). Several phenotypes, including virulence, were shown to be unaffected by insertion of this or other antibiotic resistance cassettes at this site (34). Use of this site was not possible for this study, however, as no allele of SSA_0169 was found in SK405. Anticipating that additional strains might be included in future studies, a locus that was conserved in all isolates at our disposal was sought. Additional desired characteristics included a location between convergent genes, flanked by a terminator sequence, whose manipulation would not result in any apparent phenotypic effects. A conserved intergenic region (CIR) between SSA_1217, encoding a hypothetical protein, and SSA_1218, encoding DNA repair protein RadC (Fig. 3), appeared to fulfill our requirements.
FIG 3.

Selection of a conserved intergenic region (CIR) for insertion of exogenous DNA into S. sanguinis strains. The spectinomycin resistance cassette is denoted aad9. The stem and loop structure represents the terminator sequence.
We inserted a spectinomycin resistance cassette containing the aad9 gene (34, 35) and its native promoter into SK36 by overlap extension PCR (36) and transformation (37) (Fig. 3). We first examined growth after 24 h in pooled rabbit serum at an initial O2 concentration of 12%, a condition representative of arterial blood flow found at the site of an aortic vegetation (38) and previously determined to reproduce results seen in vivo for mutants sensitive to oxidative stress (22). Insertion of aad9 at the site did not affect growth (data not shown). We therefore proceeded to test this mutant in our in vivo rabbit model of endocarditis. The SK36 derivative with the ermB gene in the SSA_0169 locus (34) and the newly created SK36 derivative with aad9 inserted at the CIR were coinoculated into rabbits that had been catheterized to induce aortic valve damage. As shown in Fig. 4, recovery of the spectinomycin-resistant (Scr) mutant was indistinguishable from that of the Emr strain, demonstrating the suitability of the site for genetic manipulations. To verify that four differently marked strains could be tested in the same animals and to reduce vertebrate animal use, two differently marked deletion mutants of the ssaACB operon were also included. A variant of these mutants, with an insertion in the ssaB gene, is attenuated for virulence in this model (21, 22). All four strains produced the expected results, as neither the antibiotic insertion site (SSA_0169 versus CIR) nor the choice of resistance gene (ermB versus aad9 or tetM versus aphA-3) affected the outcome.
FIG 4.
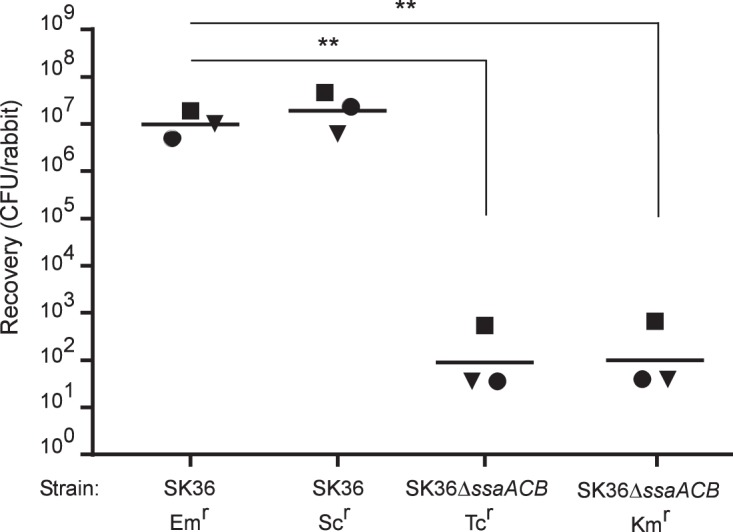
Examination of SK36 derivatives in a rabbit model of IE. Rabbits were coinoculated with the strains indicated. Like shapes indicate values obtained from the same rabbit in a single experiment. Geometric means are indicated by horizontal bars. Significance was determined by repeated-measures ANOVA with Tukey-Kramer post hoc test. **, P < 0.001.
We next proceeded to construct SK405 mutant strains. An Scr version of SK405 comparable to the SK36 derivative was created by insertion of aad9 (34) into the CIR. Two mutants of SK405 were also created in which the tel genomic island or lipoprotein gene was replaced by aphA-3, encoding kanamycin resistance (34), or tetM, encoding tetracycline resistance (34). As with the Scr SK36 strain, growth of Scr SK405 was indistinguishable from that of wild-type (WT) SK405 in rabbit serum (data not shown). Coinoculation of the previously utilized Emr strain of SK36 (34) alongside Scr SK405, the tel mutant, and the lipoprotein mutant into the rabbit model of IE did not indicate a role for either locus in IE virulence (Fig. 5). Interestingly, the SK405 reference strain and both of its mutants were significantly more virulent than the marked SK36 strain in this model.
FIG 5.
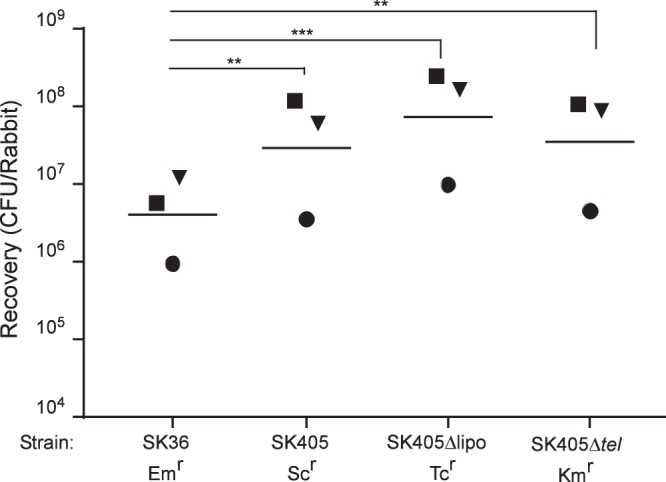
Examination of SK36 and SK405 derivatives in a rabbit model of IE. Rabbits were coinoculated with the strains indicated. Like shapes indicate values obtained from the same rabbit in a single experiment. Geometric means are indicated by horizontal bars. Significance was determined by repeated-measures ANOVA with Tukey-Kramer post hoc test. **, P < 0.01; ***, P < 0.001.
Examination of strain identity and biologically relevant phenotypes.
We next considered that our failure to identify virulence factors from our comparison of oral and IE isolates might be because the IE isolates were not uniformly more virulent than the oral isolates. Therefore, we decided to examine all of our strains in our in vivo model. Because we had more strains than antibiotic resistance markers, we modified a previous technique, called Bar-seq, that assesses mutant fitness through sequencing of transposon insertions containing random and unique barcode (BC) sequences (39). We chose to utilize the previously identified CIR for integration into each strain an 879-bp cassette that contained a common upstream (P1) primer binding site, a unique 10-bp BC identifier, and the aad9 gene to allow for selection of transformed strains and also to provide a common binding site for the downstream (P2) primer, allowing for later PCR amplification of the barcodes (Fig. 6).
FIG 6.
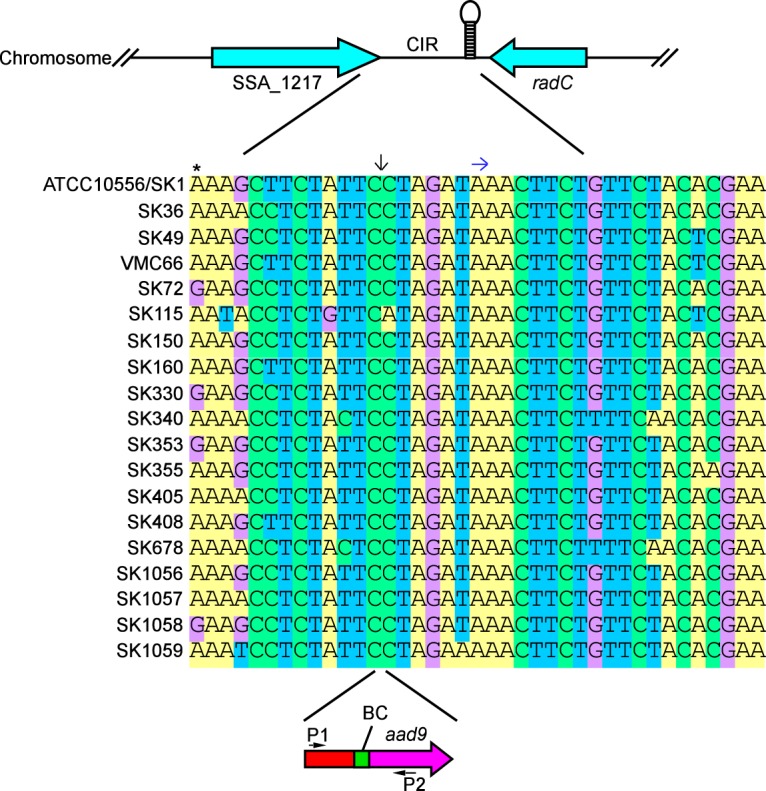
Use of a conserved intergenic region for barcode insertion. Stem and loop structure indicates terminator sequence. An asterisk indicates the last bp of stop codon in SSA_1217. Downward arrow indicates barcode (BC) insertion site. Right arrow indicates start of terminator sequence. P1 and P2 designate primers used to amplify DNA from inoculum cells and from harvested rabbit vegetations for Illumina sequencing.
We had set out to create barcoded versions of all 20 strains; however, we were unable to include three, SK1, SK72, and SK1087, each for a different reason. SK1 was not in our collection when the study was initiated, and although we later obtained ATCC 10556, which is thought to be identical to SK1 (40), it was not available in time for the Bar-seq experiment. Transformation of SK72 was unsuccessful for reasons we could not determine, as every gene shown previously to be required for transformation of SK36 (41) was present and intact in the SK72 draft genome sequence. Finally, SK1087 was excluded because we were unable to amplify its CIR for reasons described below. Concurrent with unsuccessful amplification of SK1087, creation of other marked strains brought to our attention that the sequences of the strain-specific regions flanking the barcode insertion sites in some of the strains were inconsistent with expectations based on the WGS sequences in GenBank. Closer inspection revealed that when the cassette-flanking regions from one of our strains did not match its expected sequence, it did match the expected sequence of a different strain. Thus, it appeared that either our strains were misidentified or the GenBank WGS sequences were.
To clarify these findings, we took advantage of the fact that seven housekeeping genes were sequenced from all of the strains except VMC66 as part of a multilocus sequence analysis (MLSA) study published previously (28). Although these sequences apparently were never submitted to GenBank, they were made available for download from the authors’ website (http://viridans.emlsa.net/). We used the sequences of each of the seven genes from each of the 19 available strains as a query for a BLASTN search of the WGS database, as well as the nr database for SK36. The results, presented in File S1 in the supplemental material, indicated that the MLSA sequences from the same strains that were called into question by our study were also inconsistent with the WGS sequence identities listed in GenBank. Moreover, our CIR-flanking sequences agreed with the strain identity suggested by the MLSA sequences whenever the MLSA sequences disagreed with the GenBank sequences. As another test, we amplified selected MLSA genes from the strains in our possession that were in question so that we could compare them directly to the published MLSA and GenBank sequences. This comparison indicated that our strains matched the identities of the strains analyzed in the MLSA study in all cases. As a final test, we obtained new stocks of three of the strains in question directly from Mogens Kilian (Aarhus University, Denmark), who originally provided all of the SK strains to us, to the Human Microbiome Project, and to the MLSA study. Once again, sequences from these stocks matched the sequence data obtained from our earlier stocks and the MLSA study. Our findings are summarized in Fig. 7. In brief, we conclude that in the GenBank database the sequences for SK678 and SK1058 are reversed, the sequence identified as belonging to SK1087 actually belongs to SK1059, the sequence identified as SK1059 actually belongs to SK340, as does the sequence identified as SK340 (meaning that there are two versions of the SK340 sequence in GenBank), and no sequence exists in GenBank for strain SK1087. This last conclusion explains our earlier inability to amplify the region flanking the CIR from SK1087 and, therefore, our inability to insert a barcode for this strain. Thus, at this point we had created 17 BC strains, comprised of 9 oral isolates and 8 blood isolates, and strains SK1, SK72, and SK1087 were excluded from all further experimentation. The results in all figures and tables have been adjusted to reflect our current WGS sequence identifications.
FIG 7.
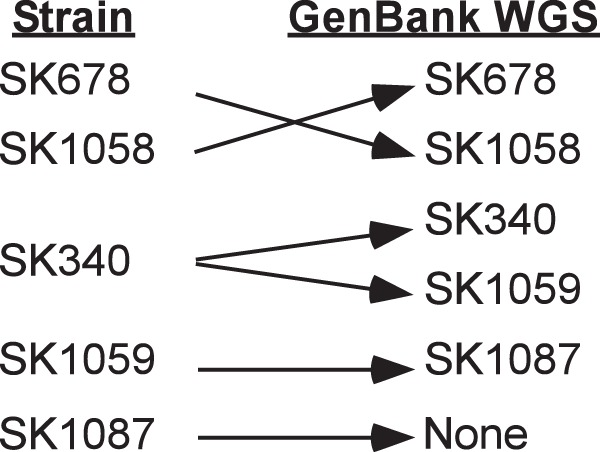
Misidentified GenBank WGS sequences as determined by our analysis.
As had been done previously with marked SK36 and SK405, the new BC strains were assessed for growth in rabbit serum alongside their respective parent strains. Growth of the BC strains was found to be unaffected by barcode insertion, as all strains grew indistinguishably from their respective parent strains (data not shown).
Pooled virulence analysis by Bar-seq.
We were now ready to test the BC strains in our rabbit IE model using a Bar-seq approach. The premise for Bar-seq is to monitor strain fitness by quantifying barcodes in harvested tissues (output) versus those in an equally distributed inoculum (input) using Illumina short-read sequencing (39). As indicated in File S2 and Fig. 8A, each strain was detected at near-equal levels in the inoculum pool by barcode enumeration. This result was in agreement with the results obtained by dilution plating of each strain performed prior to pooling (data not shown). In contrast, there was wide variability (∼4 logs) in the levels of recovery of the individual strains from the infected vegetations, with 3 strains present in greater abundance in the output samples than in the inocula (Fig. 8B) and the remainder exhibiting the opposite behavior. Interestingly, two of the three strains (SK405, SK678, and SK160) that increased in relative abundance were blood isolates; however, the overall results revealed no clear association between strain source (blood of IE patient versus oral cavity) and virulence (P = 0.1234 by unpaired t test).
FIG 8.
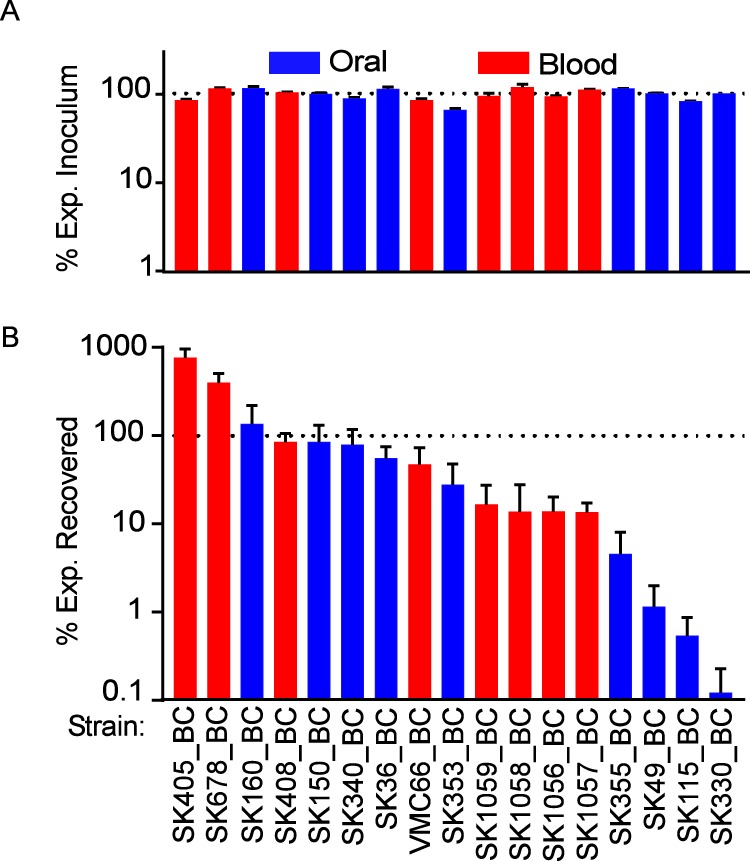
Examination of marked strains in a rabbit model of IE. Six rabbits were coinoculated with 17 barcoded strains in one of two duplicate pools. Blue and red indicate oral and blood isolates, respectively. % Exp., percent expected. (A) The % Exp. Inoculum for each strain was determined by dividing the BC counts for each strain by the total BC counts for that inoculum and then multiplying by 17. (B) The % Exp. Recovered was determined similarly by the equation (% each strain in the total recovered counts for each rabbit)/(% each strain in the total inoculum counts). The dashed line represents 1/17 of total bacterial burden in each inoculum (A) and percent abundance in each rabbit (B) relative to its abundance in the corresponding inoculum. There was no significant association between strain source (blood of IE patient versus oral cavity) and virulence (P = 0.1234 by unpaired t test).
We then reassessed the genomes of our strains by comparing the more virulent strains to the less virulent to determine if a genetic difference could explain the differences. Results shown in Table S2 indicate the presence of 30 genes exclusive to the more virulent strains; however, 10 of the genes are in the tel locus assessed previously (Fig. 5), and few of the remaining genes have known ties to virulence. Additional studies will be required to determine their significance.
Phenotypic assays.
We next wanted to determine whether there were any phenotypic properties that could readily explain the results obtained in the Bar-seq analysis shown in Fig. 8. One possibility was that the variation in recovery observed was due to competition between the strains at the site of infection due to bacteriocin production (42). We performed a bioinformatic search for bacteriocin genes using the program BAGEL (43). We found a single putative bacteriocin, an apparent lantibiotic encoded by HMPREF9391_RS05365 in strain SK408 homologous to the streptin of Streptococcus pyogenes M1GAS (44). Examination of Fig. 8 suggests that this strain is among the more virulent, but it is impossible to determine from this analysis whether this bacteriocin is being produced, whether other bacteriocins were missed, or whether any of the strains was being inhibited by any bacteriocin. To address these questions, we next assayed bacteriocin production using a method previously employed by others (45, 46), wherein each strain is stabbed into an agar plate and the following day subjected to an overlay containing an indicator strain. Our analysis revealed no inhibition by any S. sanguinis strain of any other S. sanguinis strain (data not shown); however, all strains displayed sensitivity to one or more bacteriocins produced by Streptococcus mutans UA159 (data not shown).
Previous work has shown that in vitro biofilm formation does not correlate with virulence for S. sanguinis SK36 derivatives (47); however, for other streptococcal species, disruptions in biofilm formation or structure result in attenuation of virulence (48, 49). To determine whether there is a link between biofilms and virulence among the additional S. sanguinis strains in our collection, strains were grown in biofilm medium supplemented with either sucrose (50) or glucose (51) to model oral (52, 53) and cardiac conditions, respectively. As shown in Fig. 9, there was no correlation between biofilm formation and virulence for strains grown under either condition (for sucrose, P = 0.2392 by Pearson correlation; for glucose, P = 0.5686 by Pearson correlation). Additionally, there was no correlation in either assay between strain origin and biofilm formation (for sucrose, P = 0.6493 by unpaired t test; for glucose, P = 0.3029 by unpaired t test).
FIG 9.

S. sanguinis biofilm production. Strains were grown in biofilm medium supplemented with either 1% sucrose (A) or 1% glucose (B) and grown under anaerobic or aerobic conditions, respectively. Biofilm formation was determined by absorbance at 560 nm. Blue indicates oral isolates, while red indicates blood isolates. Results are from four experiments. There was no significant correlation between biofilm formation and virulence, as determined by Pearson correlation, for results shown in panel A (P = 0.2392) or B (P = 0.5686). There was no significant relationship between biofilm formation and strain origin, as determined by unpaired t test, for results shown in panel A (P = 0.6493) or B (P = 0.3029).
A final area of investigation was metal acquisition. Work in our laboratory and others has established that metal deficiency can have dire effects on streptococcal physiology and virulence (22, 54). Our bioinformatic analysis indicated that all of the strains in our collection have the manganese transport system encoded by ssaACB (22), with some strains also having NRAMP-family transporters (55). We therefore expected that none of the strains would be deficient for manganese to the same extent as transport mutants examined in past studies (21, 22, 56). Nevertheless, we wondered whether manganese levels would vary among strains and, if so, whether levels would correlate with virulence. To test this, strains were grown aerobically in brain heart infusion (BHI) and subjected to inductively coupled plasma-optical emission spectrometry (ICP-OES). As shown in Fig. 10A, there was no correlation between cellular manganese content and virulence (P = 0.9350 by Pearson correlation). Interestingly, though, the blood isolates were found to have significantly higher manganese levels than their oral counterparts (P = 0.0124 by unpaired t test), which is more readily appreciated when the strains are sorted by manganese level (Fig. 10B).
FIG 10.
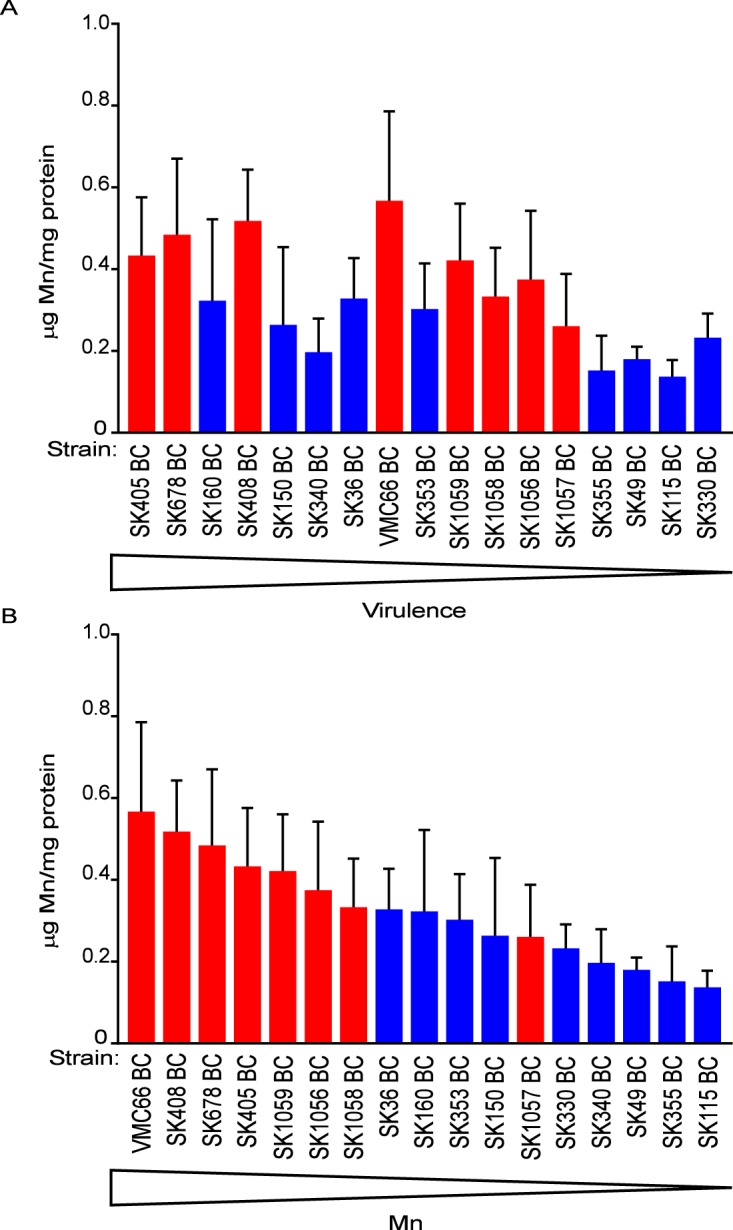
Cell-associated manganese content of S. sanguinis strains. Strains were grown in 12% O2 and analyzed by ICP-OES, displayed in order of decreasing virulence (A) or decreasing Mn content (B). Means and standard deviations from four independent experiments are shown. (A) There was no significant correlation between virulence and Mn content, as determined by Pearson correlation (P = 0.9350). (B) There was significant correlation between strain origin and Mn content, as determined by unpaired t test (P = 0.0124).
DISCUSSION
In this study, we utilized bioinformatic analyses to interrogate a collection of S. sanguinis strains. The availability of this genetic information combined with experimental data from a rabbit model of endocarditis allowed for a targeted investigation of S. sanguinis-induced IE. In short, we could address three questions. (i) Are some strains more virulent than others? (ii) If so, are IE isolates generally more virulent than oral isolates? (iii) If there are variations in virulence among strains, can we attribute these variations to specific genetic differences?
To address these questions while minimizing animal use, we developed a method that allowed us to test pools larger than the four to five strains that can be employed with the unique antibiotic markers available to most investigators. To this end, we identified a conserved intergenic region and then inserted into it a unique barcode sequence. We successfully generated a pool of 17 marked strains that we first validated with in vitro serum growth assays and then coinoculated into rabbits with induced cardiac damage. Our inclusion of barcode-flanking sequences that were identical in all strains, so as to guard against differences in efficiency of the primer binding or extension steps of the subsequent barcode amplification reactions, appeared successful, as evidenced by the near-equal recovery of each strain’s barcode from duplicate inoculum pools. Therefore, we were able to confidently quantify virulence of individual strains by counting each unique barcode in the cardiac homogenates.
Strain recovery from the infected heart valves appeared quite variable, covering almost 4 logs, with no correlations between virulence and strain source, i.e., blood of IE patients or oral cavity. Although our sample size was small, generalization of these results suggests that most oral isolates are capable of causing IE rather than virulence being restricted to rare strains. A similar conclusion was reached previously by other investigators who performed phylogenetic analyses of oral and IE isolates using data derived from multilocus sequence typing (57). This is, perhaps, not surprising. IE is a relatively rare disease, with a reported incidence in the United States of 1.7 to 11.6 cases per 100,000 people per year (58). A previous study found that cardiac risk factors for IE were far more important than risk factors related to bacteremia in determining IE causation (59). Thus, even a highly virulent strain is presumably unlikely to cause IE in most people. However, our study took the additional step of assessing virulence in an animal model. When we consider these results, we see that the most virulent oral isolate in our assay, SK160, clusters phylogenetically with the three most virulent IE isolates, SK405, SK678, and SK408. This suggests that there are more virulent lineages within the species. It is interesting that these strains are contained within the same clade in both the pangenome-based and SNP-based trees. The former tree reflects differences in gene content, while the latter reflects the evolution of core or shared genes. Thus, the former tree suggests that it was reasonable to look for genes found in the most virulent strains that are absent from the least virulent ones; however, the latter tree suggests that virulence differences result instead from variations in alleles of genes common to all the strains. This is one explanation for the paucity of obvious candidate virulence genes identified in Table S2 in the supplemental material.
Considering the variation in strain recovery from the infected animals, we questioned whether coinoculation affected our results. One possible explanation for the variation was a bottleneck effect, whereby so few cells initiate an infection that differences in recovery are due to random chance rather than differences in virulence (60). This seemed unlikely, given that this was uncommon in a previous signature-tagged mutagenesis study with strain SK36 that employed pools of 40 strains in the same rabbit model (61). Indeed, despite substantial differences in the number of barcodes recovered from each animal, the percentages of the total number of sequence reads belonging to each of the 17 strains were remarkably similar from one animal to the next, as is apparent from the data in File S2 and the error bars in Fig. 8B. Thus, variation is unlikely to be explained by random chance.
We next considered whether differential recovery of strains might be explained by interstrain competition at the site of infection. Given that S. sanguinis is well known for its genetic competence (62), we chose to examine bacteriocin production. Many species have evolved mechanisms to kill cells that are not competent, thereby creating a source of available DNA, increasing the genetic plasticity of the competent population, and perhaps also controlling social cheaters (63). In streptococci, this is often achieved through the production of bacteriocins (42, 64, 65). No strains exhibited killing in our bacteriocin assay and all the strains were susceptible to inhibition by S. mutans UA159, which has been shown to be due to bacteriocins (66). Thus, we see no evidence for differential bacteriocin production or sensitivity among the S. sanguinis strains. We also considered whether lysogenized phages play a role in IE competition, either by benefiting host strains through horizontal transfer of virulence-related genes or by hindering bacteria through lysis resulting from induction. To address this possibility, we searched for integrated phages using the program PHASTER (67). Although 35 instances of up to 9 different phages, ranging in size from 7.5 to 51.6 kb, were identified in the genomes of each of the 19 strains, with 31 of the 35 occurring in the 17 strains included in the Bar-seq experiment (Table S3), there was no obvious association of any phage with strain recovery from the Bar-seq analysis. Finally, comparison of results in the 17-strain Bar-seq experiment (Fig. 8) with the 4-strain experiment shown in Fig. 5 reveals that, at least for SK405 relative to SK36, the differences in recovery were similar in both studies. Although other forms of intraspecies competition cannot be excluded, these results together suggest that virulence outcomes obtained for the strains in our collection are unlikely to be explained solely by competition effects.
The disparity in strain virulence combined with observations that the more virulent strains were found within a clade led us to question whether other commonly investigated phenotypes, biofilm formation and cell-associated manganese, also exhibit a similar level of strain variation and whether those phenotypes correlate with an enhanced ability to cause IE. Using conditions that promote biofilm formation in the oral cavity (sucrose as a carbon source) or that are more similar to that in the bloodstream (with glucose as a carbon source), we demonstrated that, similar to the case for virulence, strains were heterogeneous for biofilm formation. Additionally, neither condition had a correlation, positive or negative, with the virulence outcomes shown in Fig. 8 or with strain origin. Interestingly, although there was no significant correlation between biofilm formation and strain origin or virulence, wide variability in biofilm formation was observed, particularly in glucose. These results may have implications for oral health, particularly with regard to the potential development of S. sanguinis strains as probiotics, as strains that are better able to establish a biofilm might better suppress the attachment of cariogenic species.
There have been numerous publications on the importance of metal homeostasis for cellular health. For the manganese-centric streptococci (68), loss of manganese import systems has been shown to cause deficiencies in oxidative stress resistance and virulence (22). However, to our knowledge, studies comparing differences in manganese content among WT strains of the same species have not been reported. In S. sanguinis our ICP-OES analysis indicates that, much like the virulence and biofilm outcomes, metal content is variable among the strains in our collection, with some strains having approximately 3 times more manganese than others. However, compared to previously published data, even the strains with the lowest levels of manganese still possess ∼20-fold more manganese than the manganese transporter-deficient mutant of SK36 (22), providing further evidence for the manganese-centric biology of S. sanguinis. Although there was no correlation with virulence, a final observation from Fig. 10 is that the blood isolates are associated with higher manganese content. The significance of this finding is unclear.
For the streptococcal community, the presence of a complete genome sequence for SK36 (15) and draft sequences of additional S. sanguinis strains is a valuable asset, particularly when genomic sequences are paired with phenotypic characterization. The current study clarifies the former and adds to the latter. With the availability of this compilation of genetic information we are able to delve more quickly and deeply into the nature of S. sanguinis and related species. Indeed, this study exploited this information to identify and validate an intergenic region useful for in vivo virulence analyses, thereby providing a new route for inclusion of diverse S. sanguinis isolates in future studies.
While generating marked strains, it came to our attention that some of the S. sanguinis WGS sequences are misidentified in GenBank. We confirmed our findings through multiple analyses, as indicated above. We have contacted NCBI in an effort to resolve this issue. At the time of this submission, these apparent errors have not been addressed by NCBI and thus are documented only within this publication.
We are not aware of any erroneous findings that have been published as a result of these sequence identification errors; however, we believe the implications of incorrect strain identification require serious consideration. Incorrect sequence information can negatively impact processes at all experimental levels, from basic primer design for PCR to more advanced bioinformatic comparisons. These misidentifications not only waste the time and funds of individual research groups but also tend to persist within sequence databases, in some cases taking years to be resolved (69, 70). This allows further propagation of incorrect information and continued misallocation of resources. Given the advances in next-generation sequencing technologies that have arisen since these sequences were originally determined (e.g., SMRT sequencing [71]), the best solution may be to resequence these strains, producing complete rather than draft sequences and employing distinct nomenclature.
MATERIALS AND METHODS
Strains.
Streptococcus sanguinis strains used in this study are listed in Table 1. The complete genome sequence of S. sanguinis strain SK36 and the latest RefSeq draft assemblies for the nineteen remaining strains were downloaded from NCBI’s ftp site on 13 December 2017.
Sequence processing and bioinformatic analysis.
Nucleotide, protein, and corresponding protein function files required for PGAP input were created from available RefSeq assembly files, which were downloaded from NCBI. Files were formatted using custom Python scripts and UNIX commands. Nucleotide files were generated from Assembly_cds_from_genomic.fna files, protein files were generated from Assembly_protein.faa files, and function files were created from the Assembly_feature_table.txt files. Minor alterations were made to draft assembly files to meet requirements of the PGAP program for exact translation from nucleotide to protein sequences. A summary of alterations can be found in Table S4 in the supplemental material.
The GF method of PGAP, version 1.12 (29), using default settings, was employed to analyze the pangenome and core genomes of S. sanguinis strains, detect variation in coding sequences, and generate single-nucleotide polymorphism (SNP) and pangenome-based neighbor-joining trees. Briefly, PGAP’s GF mode combines the total protein sequences (marked by gene name) of each strain and performs BLASTALL. The filtered BLAST result is then clustered by the MCL algorithm. For each sequence pair in a cluster, the global match region is at least 50% of the longer sequence, with an identity of at least 50%, a minimum score of at least 50, and an E value no greater than 1 × 10−8. Pangenome- and SNP-based neighbor-joining trees were visualized using version v1.16.1 of the UGENE software package (72). The PHASTER program (67) was used to analyze S. sanguinis strains for the presence of phage-like elements within the complete genome of SK36 and the RefSeq scaffold genomic.fna files of the other strains.
Bacteriocin analysis was performed using BAGEL4 (43). RefSeq assembly accession numbers were used as the input, with the exception of SK1, which was not accepted by the program. For that strain, FASTA files for SK1 scaffolds 1, 2, 3, and 4 were used as the input.
Selection of a conserved chromosomal insertion site.
Several programs were utilized to identify a conserved site among our strains, including UGENE for identification of convergent genes, MEGA 6 and BLASTN for alignment and identification of conservation among strains, respectively, and ARNOLD for identification of putative terminator sequences.
Strain construction.
All mutant strains were generated using overlap-extension PCR (37) with the primers listed in Table S5. In all cases, left and right flanking PCR products were generated that contained sequences specific to the strain into which the fragment would be transformed, adjoined to sequences complementary to the DNA to be inserted so as to generate the overlap necessary for the overlap extension step. For the knockout mutants, these flanking sequences were on either side of the gene(s) to be deleted. For SK405-Spcr and subsequent barcoded mutants, the flanking sequences were located in a conserved region between two convergent genes identified using MEGA 6 and BLASTN. For creation of the barcoded mutants, the primer was modified to include both an additional conserved sequence and a unique 10-bp barcode (Table S5). All fragments were purified using a Qiagen MinElute PCR purification kit before proceeding to the overlap extension step. The final PCR mixture employed 10 ng of each fragment as the template and Q5 high-fidelity master mix (NEB, Ipswich, MA). The resulting product was gel purified using the Qiagen MinElute gel extraction kit and then transformed into the appropriate strain as described previously (61), except for the barcode introduction, in which case strains were incubated with DNA for 5 h. Successful transformation was confirmed by selective growth on BHI agar plates containing appropriate antibiotics and by subsequent sequencing of the insert and flanking DNA.
Strains and growth conditions.
For growth studies, all strains were inoculated from frozen single-use aliquots at a thousand-fold dilution into BHI broth (BD) and grown utilizing the Anoxomat Mark II jar-filling system (AIG, Inc.) at 37°C and 6% O2 (6% O2, 7% H2, 7% CO2, and 80% N2) for 18 h. The next day the cultures were diluted and plated onto BHI supplemented with 1.5% (wt/vol) agar and incubated at 37°C for 24 h in 0% O2 (0.2% O2, 9.9% H2, 9.9% CO2, and 80% N2) with additional O2 removal by palladium catalyst. These cultures were also used to inoculate pooled rabbit serum (Gibco) at a 106-fold dilution that was then incubated at 37°C at 12% O2 (12% O2, 4.3% H2, 4.3% CO2, and 80% N2). After 24 h, 1 ml of the serum cultures was removed, sonicated, diluted, plated onto BHI agar, and incubated in 0.2% O2, 9.9% H2, 9.9% CO2, and 80% N2 for 1 day. Bacterial enumeration was determined by counting colonies from plated cultures.
Confirmation of strain identity by sequencing.
All strains created in this study were verified by sequencing of regions corresponding to the DNA introduced into each strain. DNA sequencing was performed by Eurofins, Inc., and sequences were assembled and compared using the SeqMan program (DNASTAR, Inc.). In the case of strains whose identity was unclear, amplification of some or all of the following MLSA genes was performed using primers suggested previously: pyk, pfl, map, sod, and ppaC (28). The amplicons then were sequenced. The sequences generated were compared to S. sanguinis MLSA sequences (28) that were downloaded from www.eMLSA.net and converted to a BLASTN database and also to S. sanguinis sequences in the WGS database using BLASTN.
Rabbit endocarditis model.
Specific-pathogen-free New Zealand White rabbits (2 to 4 kg; RSI Biotechnology) were utilized in the previously described endocarditis model (22, 23) to assess virulence. Prior to surgery, the rabbits were sedated and anesthetized. Minor damage to the endocardium was induced by insertion of a 19-gauge catheter to reach the aortic valve by way of the right carotid artery. Each catheter was trimmed and sutured in place, and the incision was closed with staples. The following afternoon, all strains to be inoculated into animals were cultured in BHI for overnight growth in 6% O2. For all studies except the barcoded strain study, sample preparation, inoculation, and quantification of bacteria from harvested tissue were performed as described previously (22). For the barcoded strains, duplicate inoculum pools were prepared. In both cases, 2 ml of each strain was removed the next morning from each overnight culture. The first milliliter was sonicated, diluted in phosphate-buffered saline (PBS), and plated to determine the relative abundance of each strain in the inoculum. The second milliliter for each strain was combined into a 50-ml conical tube and vortexed. The combined cells were washed twice in PBS and diluted to give a concentration of ∼108 CFU/ml. A portion of the inoculum pool was withheld for DNA purification and amplification using primers specific for the conserved region and spc cassette. Rabbits were inoculated with 0.5 ml of the pooled cells via the peripheral ear vein. Approximately 20 h after inoculation, rabbits were sacrificed via intravenous injection of Euthasol. Catheter placement was verified upon necropsy, and harvested cardiac vegetations were placed into PBS and homogenized. From these homogenates, DNA was isolated as described for the inoculum.
DNA isolation.
DNA was isolated from 1 ml of each pooled inoculum and 1 ml homogenized vegetations using the Qiagen DNeasy blood and tissue kit by following the protocol for Gram-positive organisms. Briefly, each sample was pelleted by centrifugation, resuspended in 180 μl enzymatic lysis buffer (20 mm Tris-Cl, 2 mm EDTA, 1.2% [vol/vol] Triton X-100) supplemented with lysozyme (20 mg/ml) and mutanolysin (150 U/ml), and incubated at 37°C for 1 h. Twenty-five microliters of proteinase K was added and samples were incubated overnight at 37°C. The next day samples were vortexed vigorously, 4 µl RNase A (100 mg/ml) was added, and the samples were incubated for 2 min at room temperature. Samples were vortexed for 15 s, and 200 μl buffer AL was added, followed by vortexing. One hundred microliters ethanol (96 to 100%) was added and samples were vortexed. Subsequent column purification was performed by following the manufacturer’s instructions.
Barcode sequencing and quantitation.
After DNA isolation from both the inoculum and homogenate, samples were amplified utilizing primers listed in Table S5, generating products with Illumina adapters. These amplicons were purified as described before and sent for multiplexed paired-end sequencing on a MiSeq instrument (Illumina) with a Phi-X spike. Barcode counts were determined using the Geneious R10 (version 10.2.3) program. Within the program, reads were trimmed using BBDuk and then sorted using the “separate reads by barcode” tool. Inoculum levels of each strain were determined as total number of reads per strain divided by total number of reads from all strains. Output values were each normalized to the inoculum by dividing the percent relative abundance of each strain in the output by the percent relative abundance in the corresponding inoculum.
Bacteriocin assays.
Bacteriocin assays were performed using a modification of the procedure described by Perry et al. (73). Cultures were grown for 18 h in BHI in 6% O2. The following day all strains were stabbed into 18-replicate BHI agar plates using pipette tips dipped into the overnight cultures. The stabbed plates were incubated anaerobically at 37°C for 24 h. New cultures grown for 18 h in BHI at 6% O2 were diluted 20-fold into fresh BHI and incubated until an optical density at 600 nm (OD600) of 0.2 was reached. Five milliliters of each diluted culture was separately added to 10 ml of 1% molten low-melting-point agarose, gently mixed, and then poured on top of a stabbed plate. After solidification of the overlays, plates were incubated overnight in 6% O2. Plates were removed and bacteriocin production was recorded as presence or absence of zones of inhibition. S. mutans strain UA159 was used as a positive control of bacteriocin production. The assay was performed three times for each strain.
Biofilm formation.
Strains were inoculated into BHI at a 1,000-fold dilution and grown in 6% O2 overnight at 37°C. The following day, cultures were used to inoculate 96-well plates containing biofilm medium (BM) (50) with 1% added sucrose or glucose at a 1:100 dilution. Plates were incubated in either 0% or 12% O2 overnight at 37°C. Planktonic cells were then removed and the wells were allowed to dry. The wells were washed with 200 µl of distilled water and then stained by addition of 50 µl of 0.4% crystal violet (CV) for 30 min at room temperature. The CV was removed by pipette and the biofilms were washed twice with 200 µl of distilled water. The biofilms were solubilized in 300 µl of 30% acetic acid for 30 min, which was then diluted 1:8 and assayed for absorbance at 560 nm.
ICP-OES.
For each strain, 10 ml of BHI containing appropriate antibiotics was inoculated at a 1,000-fold dilution and incubated overnight at 37°C and 6% O2. Two additional 50-ml conical tubes containing 38 ml BHI were also incubated under the same conditions for each strain. The next day, 3 ml of the overnight culture was used to inoculate each 38-ml BHI tube. Inoculated cultures were placed back in the incubator at 12% O2. After 3 h, cells were harvested by centrifugation at 3,740 × g for 10 min at 4°C. The supernatant was discarded, 5 ml cold, Chelex-treated (Bio-Rad) PBS was added, and the sample was resuspended. Centrifugation and resuspension of cells in 5 ml Chelex-treated PBS were repeated as before. The two 5-ml tubes were combined into a single 15-ml metal-free conical tube (VWR). At this point, 1 ml was removed for protein determination using the Pierce bicinchoninic acid (BCA) protein assay kit (ThermoFisher). The remaining sample was centrifuged as before and the supernatant discarded. The pellet was resuspended by adding 4.5 ml Chelex-treated deionized water, followed by 1.5 ml of concentrated (67 to 70%) trace-metal-grade nitric acid. The mixture was then placed into modified polytetrafluoroethylene (TFM) digestion vessels in a Multiwave Go microwave digestion system (Anton Paar) and digested at 20 × 105 Pa using a modification of the Organic B template, increasing the temperature of the first hold step to 120°C and prolonging the second hold to 180°C for 20 min. After cooling, each sample was transferred to a new 15-ml metal-free conical tube. The vessel was rinsed with 1 ml Chelex-treated water, which was then added to the sample in the 15-ml metal-free tube. Samples were diluted 1:3 in Chelex-treated water to yield a final acid concentration of 5%. Samples were analyzed on an Agilent 5110 ICP-VDV OES using a CMS-5 metal standard (Inorganic Ventures, Inc.) for reference and a Pb internal standard (MSPB-10PPM; Inorganic Ventures). Final values were determined by normalizing the sample concentrations to the protein amounts obtained by BCA assay.
Statistics.
All statistical tests were performed using InStat (GraphPad Software, Inc.). Significance was determined by paired t test or paired analysis of variance (ANOVA) as indicated in the figure legends. For ANOVA, a post hoc test was employed when differences among groups were indicated as significant. P values of ≤0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mogens Kilian for kindly providing S. sanguinis strains. We gratefully acknowledge the advice and support of Joseph Turner, VCU Department of Chemistry, and the assistance of Michael Yeager with identification of the conserved chromosomal insertion site, Swati Pandey with deletion of the tel locus, and Rachel Korba with ICP-OES analysis.
This work was supported by grant R01AI114926 from NIAID (T.K.) and funds from the Department of Oral and Craniofacial Molecular Biology.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00703-18.
REFERENCES
- 1.Loesche WJ. 1986. Role of Streptococcus mutans in human dental decay. Microbiol Rev 50:353–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caufield PW, Dasanayake AP, Li Y, Pan Y, Hsu J, Hardin JM. 2000. Natural history of Streptococcus sanguinis in the oral cavity of infants: evidence for a discrete window of infectivity. Infect Immun 68:4018–4023. doi: 10.1128/IAI.68.7.4018-4023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker MR, Paster BJ, Leys EJ, Moeschberger ML, Kenyon SG, Galvin JL, Boches SK, Dewhirst FE, Griffen AL. 2002. Molecular analysis of bacterial species associated with childhood caries. J Clin Microbiol 40:1001–1009. doi: 10.1128/JCM.40.3.1001-1009.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, Romero H, Simón-Soro A, Pignatelli M, Mira A. 2012. The oral metagenome in health and disease. ISME J 6:46–56. doi: 10.1038/ismej.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinane DF, Riggio MP, Walker KF, MacKenzie D, Shearer B. 2005. Bacteraemia following periodontal procedures. J Clin Periodontol 32:708–713. doi: 10.1111/j.1600-051X.2005.00741.x. [DOI] [PubMed] [Google Scholar]
- 6.Moreillon P, Que YA. 2004. Infective endocarditis. Lancet 363:139–149. doi: 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- 7.Silver JG, Martin AW, McBride BC. 1977. Experimental transient bacteraemias in human subjects with varying degrees of plaque accumulation and gingival inflammation. J Clin Periodontol 4:92–99. doi: 10.1111/j.1600-051X.1977.tb01888.x. [DOI] [PubMed] [Google Scholar]
- 8.Silver JG, Martin AW, McBride BC. 1979. Experimental transient bacteraemias in human subjects with clinically healthy gingivae. J Clin Periodontol 6:33–36. doi: 10.1111/j.1600-051X.1979.tb02288.x. [DOI] [PubMed] [Google Scholar]
- 9.Kholy KE, Genco RJ, Van Dyke TE. 2015. Oral infections and cardiovascular disease. Trends Endocrinol Metab 26:315–321. doi: 10.1016/j.tem.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Moreillon P, Que YA, Bayer AS. 2002. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect Dis Clin North Am 16:297–318. doi: 10.1016/S0891-5520(01)00009-5. [DOI] [PubMed] [Google Scholar]
- 11.Bashore TM, Cabell C, Fowler JV. 2006. Update on infective endocarditis. Curr Probl Cardiol 31:274–352. doi: 10.1016/j.cpcardiol.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Huang X, Palmer SR, Ahn S-J, Richards VP, Williams ML, Nascimento MM, Burne RA. 2016. A highly arginolytic Streptococcus species that potently antagonizes Streptococcus mutans. Appl Environ Microbiol 82:2187–2201. doi: 10.1128/AEM.03887-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlafer S, Raarup MK, Meyer RL, Sutherland DS, Dige I, Nyengaard JR, Nyvad B. 2011. pH landscapes in a novel five-species model of early dental biofilm. PLoS One 6:e25299. doi: 10.1371/journal.pone.0025299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen Z, Yates D, Ahn S-J, Burne R. 2010. Biofilm formation and virulence expression by Streptococcus mutans are altered when grown in dual-species model. BMC Microbiol 10:111. doi: 10.1186/1471-2180-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, Hendricks S, Wang Y, Chaplin MD, Akan D, Paik S, Peterson DL, Macrina FL, Buck GA. 2007. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol 189:3166–3175. doi: 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bensing BA, Khedri Z, Deng L, Yu H, Prakobphol A, Fisher SJ, Chen X, Iverson TM, Varki A, Sullam PM. 2016. Novel aspects of sialoglycan recognition by the Siglec-like domains of streptococcal SRR glycoproteins. Glycobiology 26:1222–1234. doi: 10.1093/glycob/cww042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Douglas CWI. 2005. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol 129:101–109. doi: 10.1111/j.1365-2141.2005.05421.x. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi M, Terao Y, Ogawa T, Takahashi T, Hamada S, Kawabata S. 2006. Role of Streptococcus sanguinis sortase A in bacterial colonization. Microbes Infect 8:2791–2796. doi: 10.1016/j.micinf.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Camargo TM, Stipp RN, Alves LA, Harth-Chu EN, Höfling JF, Mattos-Graner RO. 2018. Novel two-component system of Streptococcus sanguinis affecting functions associated with viability in saliva and biofilm formation. Infect Immun 86:e00942-17. doi: 10.1128/IAI.00942-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morita C, Sumioka R, Nakata M, Okahashi N, Wada S, Yamashiro T, Hayashi M, Hamada S, Sumitomo T, Kawabata S. 2014. Cell wall-anchored nuclease of Streptococcus sanguinis contributes to escape from neutrophil extracellular trap-mediated bacteriocidal activity. PLoS One 9:e103125. doi: 10.1371/journal.pone.0103125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das S, Kanamoto T, Ge X, Xu P, Unoki T, Munro CL, Kitten T. 2009. Contribution of lipoproteins and lipoprotein processing to endocarditis virulence in Streptococcus sanguinis. J Bacteriol 191:4166–4179. doi: 10.1128/JB.01739-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crump KE, Bainbridge B, Brusko S, Turner LS, Ge X, Stone V, Xu P, Kitten T. 2014. The relationship of the lipoprotein SsaB, manganese, and superoxide dismutase in Streptococcus sanguinis virulence for endocarditis. Mol Microbiol 92:1243–1259. doi: 10.1111/mmi.12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhodes DV, Crump KE, Makhlynets O, Snyder M, Ge X, Xu P, Stubbe J, Kitten T. 2014. Genetic characterization and role in virulence of the ribonucleotide reductases of Streptococcus sanguinis. J Biol Chem 289:6273–6287. doi: 10.1074/jbc.M113.533620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makhlynets O, Boal AK, Rhodes DV, Kitten T, Rosenzweig AC, Stubbe J. 2014. Streptococcus sanguinis class Ib ribonucleotide reductase: high activity with both iron and manganese cofactors and structural insights. J Biol Chem 289:6259–6272. doi: 10.1074/jbc.M113.533554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amin Z, Harvey RM, Wang H, Hughes CE, Paton AW, Paton JC, Trappetti C. 2015. Isolation site influences virulence phenotype of serotype 14 Streptococcus pneumoniae strains belonging to multilocus sequence type 15. Infect Immun 83:4781–4790. doi: 10.1128/IAI.01081-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnston JW, Briles DE, Myers LE, Hollingshead SK. 2006. Mn2+-dependent regulation of multiple genes in Streptococcus pneumoniae through PsaR and the resultant impact on virulence. Infect Immun 74:1171–1180. doi: 10.1128/IAI.74.2.1171-1180.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendriksen WT, Bootsma HJ, van Diepen A, Estevao S, Kuipers OP, de Groot R, Hermans PWM. 2009. Strain-specific impact of PsaR of Streptococcus pneumoniae on global gene expression and virulence. Microbiology 155:1569–1579. doi: 10.1099/mic.0.025072-0. [DOI] [PubMed] [Google Scholar]
- 28.Bishop C, Aanensen D, Jordan G, Kilian M, Hanage W, Spratt B. 2009. Assigning strains to bacterial species via the internet. BMC Biol 7:3. doi: 10.1186/1741-7007-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. 2012. PGAP: pan-genomes analysis pipeline. Bioinformatics 28:416–418. doi: 10.1093/bioinformatics/btr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng W, Tan MF, Old LA, Paterson IC, Jakubovics NS, Choo SW. 2017. Distinct biological potential of Streptococcus gordonii and Streptococcus sanguinis revealed by comparative genome analysis. Sci Rep 7:2949. doi: 10.1038/s41598-017-02399-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guimarães LC, Florczak-Wyspianska J, de Jesus LB, Viana MVC, Silva A, Ramos RTJ, de Castro Soares C. 2015. Inside the pan-genome–methods and software overview. Curr Genomics 16:245–252. doi: 10.2174/1389202916666150423002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franks SE, Ebrahimi C, Hollands A, Okumura CY, Aroian RV, Nizet V, McGillivray SM. 2014. Novel role for the yceGH tellurite resistance genes in the pathogenesis of Bacillus anthracis. Infect Immun 82:1132–1140. doi: 10.1128/IAI.01614-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacs-Simon A, Titball RW, Michell SL. 2011. Lipoproteins of bacterial pathogens. Infect Immun 79:548–561. doi: 10.1128/IAI.00682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner LS, Das S, Kanamoto T, Munro CL, Kitten T. 2009. Development of genetic tools for in vivo virulence analysis of Streptococcus sanguinis. Microbiology 155:2573–2582. doi: 10.1099/mic.0.024513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin B, Prudhomme M, Alloing G, Granadel C, Claverys JP. 2000. Cross-regulation of competence pheromone production and export in the early control of transformation in Streptococcus pneumoniae. Mol Microbiol 38:867–878. doi: 10.1046/j.1365-2958.2000.02187.x. [DOI] [PubMed] [Google Scholar]
- 36.Horton RM. 1995. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol 3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
- 37.Ge X, Xu P. 2012. Genome-wide gene deletions in Streptococcus sanguinis by high throughput PCR. J Vis Exp doi: 10.3791/4356:e4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atkuri KR, Herzenberg LA, Niemi A-K, Cowan T, Herzenberg LA. 2007. Importance of culturing primary lymphocytes at physiological oxygen levels. Proc Natl Acad Sci U S A 104:4547–4552. doi: 10.1073/pnas.0611732104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, Roth FP, Giaever G, Nislow C. 2009. Quantitative phenotyping via deep barcode sequencing. Genome Res 19:1836–1842. doi: 10.1101/gr.093955.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poulsen K, Reinholdt J, Jespersgaard C, Boye K, Brown TA, Hauge M, Kilian M. 1998. A comprehensive genetic study of streptococcal immunoglobulin A1 proteases: evidence for recombination within and between species. Infect Immun 66:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez AM, Callahan JE, Fawcett P, Ge X, Xu P, Kitten T. 2011. Physiological and molecular characterization of genetic competence in Streptococcus sanguinis. Mol Oral Microbiol 26:99–116. doi: 10.1111/j.2041-1014.2011.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shanker E, Federle MJ. 2017. Quorum sensing regulation of competence and bacteriocins in Streptococcus pneumoniae and mutans. Genes (Basel) 8:15. doi: 10.3390/genes8010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Heel AJ, de Jong A, Song C, Viel JH, Kok J, Kuipers OP. 2018. BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res 46:W278–W281. doi: 10.1093/nar/gky383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross KF, Ronson CW, Tagg JR. 1993. Isolation and characterization of the lantibiotic salivaricin A and its structural gene salA from Streptococcus salivarius 20P3. Appl Environ Microbiol 59:2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yonezawa H, Kuramitsu HK. 2005. Genetic analysis of a unique bacteriocin, Smb, produced by Streptococcus mutans GS5. Antimicrob Agents Chemother 49:541–548. doi: 10.1128/AAC.49.2.541-548.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawid S, Roche AM, Weiser JN. 2007. The blp bacteriocins of Streptococcus pneumoniae mediate intraspecies competition both in vitro and in vivo. Infect Immun 75:443–451. doi: 10.1128/IAI.01775-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ge X, Kitten T, Chen Z, Lee SP, Munro CL, Xu P. 2008. Identification of Streptococcus sanguinis genes required for biofilm formation and examination of their role in endocarditis virulence. Infect Immun 76:2551–2559. doi: 10.1128/IAI.00338-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawabata S, Hamada S. 1999. Studying biofilm formation of mutans streptococci. Methods Enzymol 310:513–523. doi: 10.1016/S0076-6879(99)10039-9. [DOI] [PubMed] [Google Scholar]
- 49.Shenoy AT, Brissac T, Gilley RP, Kumar N, Wang Y, Gonzalez-Juarbe N, Hinkle WS, Daugherty SC, Shetty AC, Ott S, Tallon LJ, Deshane J, Tettelin H, Orihuela CJ. 2017. Streptococcus pneumoniae in the heart subvert the host response through biofilm-mediated resident macrophage killing. PLoS Pathog 13:e1006582. doi: 10.1371/journal.ppat.1006582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Stone VN, Ge X, Tang M, Elrami F, Xu P. 2017. TetR family regulator brpT modulates biofilm formation in Streptococcus sanguinis. PLoS One 12:e0169301. doi: 10.1371/journal.pone.0169301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu B, Ge X, Stone V, Kong X, El-Rami F, Liu Y, Kitten T, Xu P. 2017. ciaR impacts biofilm formation by regulating an arginine biosynthesis pathway in Streptococcus sanguinis SK36. Sci Rep 7:17183. doi: 10.1038/s41598-017-17383-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowen WH. 2002. Do we need to be concerned about dental caries in the coming millennium? Crit Rev Oral Biol Med 13:126–131. doi: 10.1177/154411130201300203. [DOI] [PubMed] [Google Scholar]
- 53.Koo H, Xiao J, Klein MI. 2009. Extracellular polysaccharides matrix–an often forgotten virulence factor in oral biofilm research. Int J Oral Sci 1:229–234. doi: 10.4248/IJOS.09086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eijkelkamp BA, McDevitt CA, Kitten T. 2015. Manganese uptake and streptococcal virulence. Biometals 28:491–508. doi: 10.1007/s10534-015-9826-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nevo Y, Nelson N. 2006. The NRAMP family of metal-ion transporters. Biochim Biophys Acta 1763:609–620. doi: 10.1016/j.bbamcr.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Colomer-Winter C, Flores-Mireles AL, Baker SP, Frank KL, Lynch AJL, Hultgren SJ, Kitten T, Lemos JA. 2018. Manganese acquisition is essential for virulence of Enterococcus faecalis. PLoS Pathog 14:e1007102. doi: 10.1371/journal.ppat.1007102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Do T, Gilbert SC, Klein J, Warren S, Wade WG, Beighton D. 2011. Clonal structure of Streptococcus sanguinis strains isolated from endocarditis cases and the oral cavity. Mol Oral Microbiol 26:291–302. doi: 10.1111/j.2041-1014.2011.00618.x. [DOI] [PubMed] [Google Scholar]
- 58.Bin Abdulhak AA, Baddour LM, Erwin PJ, Hoen B, Chu VH, Mensah GA, Tleyjeh IM. 2014. Global and regional burden of infective endocarditis, 1990–2010: a systematic review of the literature. Global Heart 9:131–143. doi: 10.1016/j.gheart.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 59.Strom BL, Abrutyn E, Berlin JA, Kinman JL, Feldman RS, Stolley PD, Levison ME, Korzeniowski OM, Kaye D. 1998. Dental and cardiac risk factors for infective endocarditis–a population-based, case-control study. Ann Intern Med 129:761–769. doi: 10.7326/0003-4819-129-10-199811150-00002. [DOI] [PubMed] [Google Scholar]
- 60.Mecsas J. 2002. Use of signature-tagged mutagenesis in pathogenesis studies. Curr Opin Microbiol 5:33–37. doi: 10.1016/S1369-5274(02)00282-5. [DOI] [PubMed] [Google Scholar]
- 61.Paik S, Senty L, Das S, Noe JC, Munro CL, Kitten T. 2005. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect Immun 73:6064–6074. doi: 10.1128/IAI.73.9.6064-6074.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaustad P. 2009. Genetic transformation in Streptococcus sanguis. Distribution of competence and competence factors in a collection of strains. Acta Pathol Microbiol Scand B 87B:123–128. doi: 10.1111/j.1699-0463.1979.tb02414.x. [DOI] [PubMed] [Google Scholar]
- 63.Wang M, Schaefer AL, Dandekar AA, Greenberg EP. 2015. Quorum sensing and policing of Pseudomonas aeruginosa social cheaters. Proc Natl Acad Sci U S A 112:2187–2191. doi: 10.1073/pnas.1500704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cotter PD, Hill C, Ross RP. 2005. Bacteriocins: developing innate immunity for food. Nat Rev Microbiol 3:777–788. doi: 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- 65.Steinmoen H, Teigen A, Havarstein LS. 2003. Competence-induced cells of Streptococcus pneumoniae lyse competence-deficient cells of the same strain during cocultivation. J Bacteriol 185:7176–7183. doi: 10.1128/JB.185.24.7176-7183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kreth J, Merritt J, Shi W, Qi F. 2005. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol 187:7193–7203. doi: 10.1128/JB.187.21.7193-7203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lisher JP, Giedroc DP. 2013. Manganese acquisition and homeostasis at the host-pathogen interface. Front Cell Infect Microbiol 3:91. doi: 10.3389/fcimb.2013.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stavrou AA, Mixao V, Boekhout T, Gabaldon T. 2018. Misidentification of genome assemblies in public databases: The case of Naumovozyma dairenensis and proposal of a protocol to correct misidentifications. Yeast 35:425–429. doi: 10.1002/yea.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ballester A-R, Marcet-Houben M, Levin E, Sela N, Selma-Lázaro C, Carmona L, Wisniewski M, Droby S, González-Candelas L, Gabaldón T. 2015. Genome, transcriptome, and functional analyses of Penicillium expansum provide new insights into secondary metabolism and pathogenicity. Mol Plant Microbe Interact 28:232–248. doi: 10.1094/MPMI-09-14-0261-FI. [DOI] [PubMed] [Google Scholar]
- 71.Roberts RJ, Carneiro MO, Schatz MC. 2013. The advantages of SMRT sequencing. Genome Biol 14:405. doi: 10.1186/gb-2013-14-6-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Okonechnikov K, Golosova O, Fursov M, UGENE Team . 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- 73.Perry JA, Jones MB, Peterson SN, Cvitkovitch DG, Levesque CM. 2009. Peptide alarmone signalling triggers an auto-active bacteriocin necessary for genetic competence. Mol Microbiol 72:905–917. doi: 10.1111/j.1365-2958.2009.06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kilian M, Mikkelsen L, Henrichsen J. 1989. Taxonomic study of viridans streptococci: description of Streptococcus gordonii sp. nov. and emended descriptions of Streptococcus sanguis (White and Niven 1946), Streptococcus oralis (Bridge and Sneath 1982), and Streptococcus mitis (Andrewes and Horder 1906). Int J Syst Bacteriol 39:471–484. doi: 10.1099/00207713-39-4-471. [DOI] [Google Scholar]
- 75.Hsu SD, Cisar JO, Sandberg AL, Kilian M. 1994. Adhesive properties of viridans streptococcal species. Microb Ecol Health Dis 7:125–137. doi: 10.3109/08910609409141342. [DOI] [Google Scholar]
- 76.Kilian M, Holmgren K. 1981. Ecology and nature of immunoglobulin A1 protease-producing streptococci in the human oral cavity and pharynx. Infect Immun 31:868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mikkelsen L, Jensen SB, Schiott CR, Loe H. 1981. Classification and prevalences of plaque streptococci after two years oral use of chlorhexidine. J Periodontal Res 16:646–658. doi: 10.1111/j.1600-0765.1981.tb01003.x. [DOI] [PubMed] [Google Scholar]
- 78.Nyvad B, Kilian M. 1987. Microbiology of the early colonization of human enamel and root surfaces in vivo. Scand J Dent Res 95:369–380. [DOI] [PubMed] [Google Scholar]
- 79.Kitten T, Munro CL, Zollar NQ, Lee SP, Patel RD. 2012. Oral streptococcal bacteremia in hospitalized patients: taxonomic identification and clinical characterization. J Clin Microbiol 50:1039–1042. doi: 10.1128/JCM.06438-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.