

Complement is a critical component of antimicrobial immunity. Various complement regulatory proteins prevent host cells from being attacked.
KEYWORDS: C4b-binding protein, complement, pneumococcal surface protein, Streptococcus pneumoniae
ABSTRACT
Complement is a critical component of antimicrobial immunity. Various complement regulatory proteins prevent host cells from being attacked. Many pathogens have acquired the ability to sequester complement regulators from host plasma to evade complement attack. We describe here how Streptococcus pneumoniae adopts a strategy to prevent the formation of the C3 convertase C4bC2a by the rapid conversion of surface bound C4b and iC4b into C4dg, which remains bound to the bacterial surface but no longer forms a convertase complex. Noncapsular virulence factors on the pneumococcus are thought to facilitate this process by sequestering C4b-binding protein (C4BP) from host plasma. When S. pneumoniae D39 was opsonized with human serum, the larger C4 activation products C4b and iC4b were undetectable, but the bacteria were liberally decorated with C4dg and C4BP. With targeted deletions of either PspA or PspC, C4BP deposition was markedly reduced, and there was a corresponding reduction in C4dg and an increase in the deposition of C4b and iC4b. The effect was greatest when PspA and PspC were both knocked out. Infection experiments in mice indicated that the deletion of PspA and/or PspC resulted in the loss of bacterial pathogenicity. Recombinant PspA and PspC both bound serum C4BP, and both led to increased C4b and reduced C4dg deposition on S. pneumoniae D39. We conclude that PspA and PspC help the pneumococcus to evade complement attack by binding C4BP and so inactivating C4b.
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) is the leading cause of community-acquired pneumonia, claiming more than a million lives every year, and the majority of these fatalities occur in developing countries (1, 2). It is also a major cause of otitis media, septicemia, and meningitis. Children under five, the elderly, and immunocompromised individuals are at particularly high risk of pneumococcal infection (3–5). Complement-mediated opsonophagocytosis is a vital component of the host response to pneumococcal infections (6–8).
Complement is triggered via three pathways: the classical, lectin, and alternative pathways (9). Activation of the classical pathway starts when C1q recognizes the Fc region of IgG or IgM bound to specific antigen. Binding of C1q to its target initiates the activation of the serine proteases C1r and C1s. Activated C1s then cleaves C4 into C4a and C4b. The larger C4b fragment binds to the activating surface via its newly exposed reactive thioester group. C4b subsequently binds C2, which is also cleaved by C1s creating the C3 convertase (C4b2a) (10).
The lectin pathway is activated when recognition molecules, including mannose-binding lectin (MBL), collectin-11, heterotrimers of collectin-11 and collectin-10, and three different members of the ficolin family, bind to carbohydrate moieties on the pathogen surface (11). The binding of these lectins activates the MBL-associated serine proteases MASP-1, -2, and -3. Like the classical pathway enzyme C1s, MASP-2 cleaves both C4 and C4b bound C2 to generate the C3 convertase (C4b2a) complex (12).
Unlike the classical and the lectin pathways, the initiation of the alternative activation pathway of complement does not involve specific recognition complexes. Spontaneous low-level hydrolysis of C3 leads to deposition of C3b on cell surfaces. Zymogen factor B binds to the immobilized C3b and is subsequently cleaved by factor D to form C3bBb, the alternative pathway C3 convertase (13, 14).
All three pathways result in the formation of a C3 convertase, which cleaves the abundant plasma protein C3 into C3a and C3b. C3b is the major opsonin of the complement system and also provides new sites for the binding of factor B, leading to amplification of complement activation via the alternative pathway. Binding of additional C3b to the C3 convertases results in the formation of C5 convertases, which cleave C5 into C5a and C5b. C5a is a potent anaphylatoxin, with important roles in inflammation and chemotaxis, whereas C5b initiates the terminal pathway, leading to the formation of the membrane attack complex (MAC) (15–17).
Host tissues are protected from complement attack by a variety of complement regulators, including C4BP, factor H, C1 inhibitor, and other surface-bound regulators and inhibitory proteins (9). C4BP is a vital fluid phase regulator of the complement system. It binds to C4b and serves as a cofactor for the factor I-mediated conversion of C4b into inactive fragments, thus limiting the half-life of the classical and lectin pathway C3 convertase (C4b2a). On host cells, the membrane cofactor protein (MCP; CD46) serves the same function as C4BP, acting as an alternative cofactor for factor I. The breakdown of C4b by factor I is sequential: an initial cleavage leaves surface bound iC4b, which is opsonic, but cannot form an active C3 convertase. Cleavage at a second site releases a large fragment, C4c, and leaves a small fragment of the C4 α-chain, C4dg, covalently bound to the surface (18–20).
Many pathogens have developed strategies to avoid complement activation on their surfaces, frequently by capturing host complement regulatory proteins. The resistance of Neisseria gonorrhoeae to serum killing is mainly due to its ability to sequester host C4BP (21–23). Streptococcus pyogenes, Candida albicans, and Haemophilus influenzae were also reported to bind C4BP (24–27).
Streptococcus pneumoniae has a thick and rigid cell wall that resists MAC-mediated lysis. Therefore, opsonization with C4 and C3 is the key weapon with which complement fights S. pneumoniae (28). S. pneumoniae has evolved different strategies to resist complement attack. The pneumococcal polysaccharide capsule impairs C3 deposition on the pneumococcal surface, thus reducing opsonization, resulting in decreased phagocytosis. It also masks cell wall-bound IgM from complement C1q, affecting classical pathway activation (6, 29, 30). Pneumolysin, the major toxin of S. pneumoniae, indirectly contributes to complement evasion by diverting complement activation away from the pneumococcal surface (31, 32). Pneumococcal surface protein C (PspC) triggers factor I-mediated cleavage of C3b by binding to host factor H, which decays and inactivates the alternative pathway C3 convertase and limits C5 convertase formation on the bacterium (33, 34). Pneumococcal surface protein A (PspA), another surface protein, is present in all S. pneumoniae serogroups (35) and is reported to inhibit complement activation on the surface of the bacteria (36). Recently, PspA was shown to inhibit C-reactive protein and limit complement deposition via the classical pathway (37). Moreover, PspA and PspC have been reported to inhibit C4 deposition on the pneumococcal surface, resulting in impaired activation of the classical pathway (38–40).
We investigated here the mechanism that effectively restricts the deposition of C4b on the pneumococcal surface. We demonstrate the roles of PspA and PspC in this process by using PspA–, PspC–, and PspA–/C– strains of S. pneumoniae and show to what extent the presence or absence of these two surface proteins prevents C4b deposition to S. pneumoniae.
RESULTS
When C4 is activated in close proximity to the surface of a pathogen, the larger C4b fragment binds to the surface via its newly exposed reactive thioester group. C4b then binds C2, which is activated by C1s, building a C3 convertase (C4b2a). The activity of this C3 convertase is primarily controlled by the factor I-mediated degradation of C4b into iC4b then C4c and C4dg, which is the smallest C4 fragment that remains covalently bound to the surface. Factor I requires C4BP or MCP as a cofactor. We have previously shown that when wild-type S. pneumoniae D39 is opsonized with serum, the larger activation products, C4b and iC4b, are virtually undetectable on the bacterial surface, whereas C4dg is present in abundance (7). These observations, and reports by others (40, 41), led us to the hypothesis that S. pneumoniae sequesters C4BP from host serum to inactivate C4b and that the likely receptors for C4BP are PspA and/or PspC.
To test this hypothesis, we generated PspA–, PspC–, and PspA–/C– knockout strains of D39 and compared the binding of C4BP with the deposition of C4 degradation fragments, on the mutant and parent strains.
In a preliminary experiment, we demonstrated that the Psp knockout strains used in this study have limited virulence in vivo. When mice were infected intranasally with 2.5 × 106 CFU of D39, 90% succumbed to the infection after 96 h. Mortality was reduced to 30% in the PspA– strain and 50% in the PspC– strain, while the PspA–/C– knockout strain proved to be avirulent (see Fig. S1 in the supplemental material).
Serotype variations and encapsulation are not associated with the lack of C4b deposition on the pneumococcal surface.
Deposition of C4b was assessed on eleven encapsulated strains of S. pneumoniae, covering six serotypes. Formalin-fixed wild-type S. pneumoniae samples were immobilized on microtiter plates, incubated with serial dilutions of normal human serum (NHS), and assayed for C4b deposition. No C4b was detected on any of the strains tested, whatever the serotype. C4b was abundantly deposited on Escherichia coli and mannan, which were used as positive controls (Fig. 1A). Eight nonencapsulated strains were also tested, none of which showed any C4b deposition (Fig. 1B). Thus, the factors that determine resistance to C4b deposition are conserved across strains and serotypes, have nothing to do with the capsule, and are therefore likely to reside in the cell wall.
FIG 1.

(A and B) C4b deposition is undetectable on different strains and serotypes of encapsulated (A) and nonencapsulated (B) S. pneumoniae. Microtiter plates were coated with formalin-fixed bacteria and incubated with serial dilutions of human serum. C4 was detected using chicken anti-human C4b. In both the experiments, C4b deposition could not be detected on any of the pneumococcal strains. Mannan and E. coli were used as positive controls. The results are means ± the standard errors of the mean (SEM) of three different experiments. The differences between the controls (mannan and E. coli) and the S. pneumoniae strains were significant (P < 0.001); there were no differences among the S. pneumoniae strains (two-way analysis of variance [ANOVA] with Dunnett’s correction for multiple comparisons).
Inactivation of PspA and/or PspC reduces the degradation of C4b on S. pneumoniae.
We opsonized formalin-fixed wild-type S. pneumoniae D39, along with PspA–, PspC–, and PspA–/C– knockout strains of D39, with serial dilutions of normal human serum and assayed for C4b and C3dg using a microtiter plate assay (Fig. 2). C4b could not be detected on the surface of the wild-type D39 strain but increased when PspA or PspC was knocked out and was highest in the absence of both PspA and PspC (Fig. 2A). The results for C4dg were the opposite; high levels of C4dg were observed on the wild-type D39 strain, reduced in the PspA and PspC knockout strains, and almost completely absent in the PspA–/C– double-knockout strain (Fig. 2B). These results suggested a synergistic role for PspA and PspC in the rapid degradation of C4b to C4dg on the pneumococcal surface.
FIG 2.
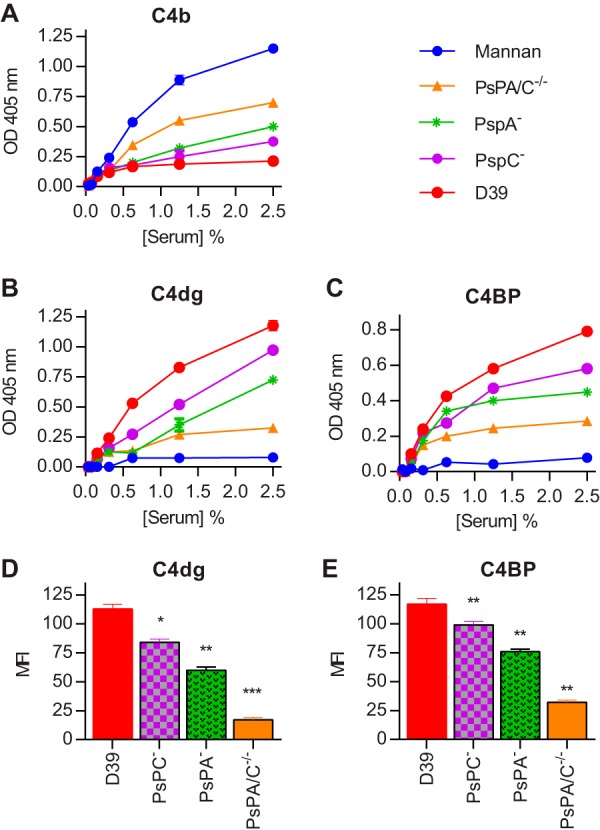
Deposition of C4b, C4dg and C4BP on wild-type S. pneumoniae D39 and its PspA–, PspC–, and PspA–/C– mutant strains. Microtiter plates were coated with formalin-fixed bacteria and incubated with serial dilutions of human serum. C4b (A), C4dg (B), and C4BP (C) deposition was assayed using specific antibodies. C4b is not detected on wild-type D39 but appears on the pneumococcal surface in the absence of PspA and/or PspC, with the highest level detected on the PspA–/C– double-mutant strain. In contrast, C4dg (the final product of C4 degradation) and C4BP are abundant on wild-type D39, less so on the PspA- and PspC-deficient strains, and almost completely absent on the PspA–/C– double-knockout strain. Neither C4dg nor C4BP was detected on mannan. When live bacteria were opsonized with NHS and then analyzed for C4dg (D) and C4BP (E) by FACS, the results were consistent with those obtained using immobilized formalin-fixed bacteria. The results are means ± the SEM of three different experiments. In panels A, B, and C, results were analyzed by two-way ANOVA with Dunnett’s correction for multiple comparisons. The P value was ≤0.001 for every comparison between the Psp mutants and D39. Statistical significance is indicated by asterisks in panels D and E (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [two-tailed t test]).
C4 degradation is associated with high levels of surface bound C4bp.
Next, we compared the deposition of C4dg and C4BP on the surface of formalin-fixed bacteria using microtiter plate-based assays (Fig. 2B and C) and on live bacteria by fluorescence-activated cell sorting (FACS) (Fig. 2D and E). C4dg deposition was decreased in the absence of PspA or PspC, while deficiency of both PspA and PspC resulted in negligible levels of C4dg being detected on the surface of pneumococci. Results for C4BP deposition were directly comparable; the highest levels were observed on the wild-type strain, with significantly lower levels on the single PspA and PspC knockout strains and very low levels of C4BP on the double-knockout strain (Fig. 2C and E). Taken together, these results provide strong evidence for the hypothesis that the pneumococcal Psp proteins cooperate to sequester host C4BP and so inactivate C4b on the bacterial surface. Mannan, which was used as a control in these experiments, showed high C4b and C4c depositions (Fig. 2), no C4dg deposition, and no C4BP binding, suggesting that the ability to bind C4BP is crucial for the conversion of C4b and C4c into C4dg.
Recombinant PspA and PspC bind to serum C4bp.
Microtiter plates coated with recombinant PspA or PspC were used in a pulldown assay to capture C4BP from human serum. Serum C4BP bound to both proteins in a concentration-dependent and saturable manner (Fig. 3). The apparent 50% effective concentration for the binding to PspC was approximately one-quarter that for PspA, indicating that the affinity of PspC for C4BP is ∼4-fold greater than that of PspA. No binding was seen with wells coated with bovine serum albumin (BSA), which were used as a negative control. In another experiment, we preincubated 1% NHS with serial dilutions of the recombinant proteins before using the mixture to opsonize formalin-fixed bacteria on microtiter plates. Both proteins increased C4b deposition and inhibited C4dg deposition on the bacteria to approximately the same extent (Fig. 3B and C).
FIG 3.
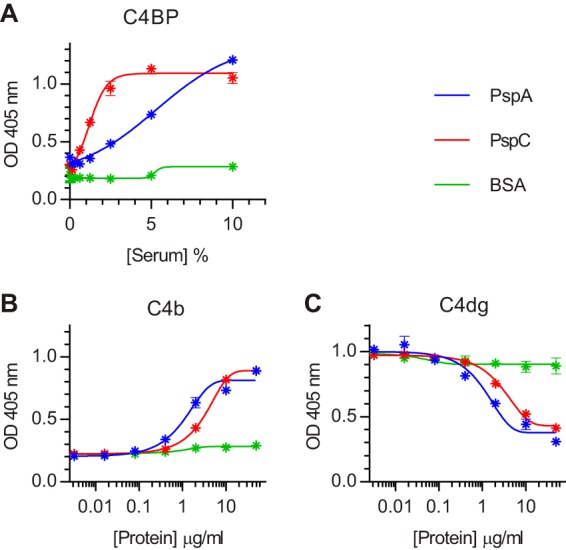
Recombinant PspA and PspC bind serum C4BP, inhibit C4dg deposition, and augment C4b deposition on S. pneumoniae D39. (A) Microtiter plates were coated with recombinant PspA or PspC (0.5 µg/well) and blocked with BSA. Serial dilutions of NHS were incubated on the plates, and bound C4BP was detected with anti-human C4BP. Wells just blocked with BSA were used as a negative control. (B and C) Microtiter plates were coated with formalin-fixed D39 and incubated with 1% NHS supplemented with the indicated concentrations of recombinant PspA, PspC, or BSA (for controls). Mixing serum with either PspA or PspC led to increased C4b deposition (B) and reduced C4dg deposition (C). The results are means ± the SEM of duplicates and are representative of two independent experiments.
C3b deposition on the surface of wild-type and mutant strains of S. pneumoniae D39.
C3 deposition was assessed on the surface of wild-type D39 and its PspA– and PspC– strains to test whether the lack of C4b fragments would affect downstream complement activation. C3b deposition was not completely absent on D39, even at serum concentrations too low to be permissive for effective alternative pathway activation. However, a deficiency of either PspA or PspC significantly increased the level of C3 deposition, and the highest level of C3b deposition was observed on the PspA–/C– double mutant (Fig. 4). This result suggests that effective C4b deposition helps but is not an absolute requirement for C3b opsonization of pneumococci, a finding that is consistent with our previous observation that C3b can be deposited via the lectin pathway, even in the complete absence of C4 (7).
FIG 4.
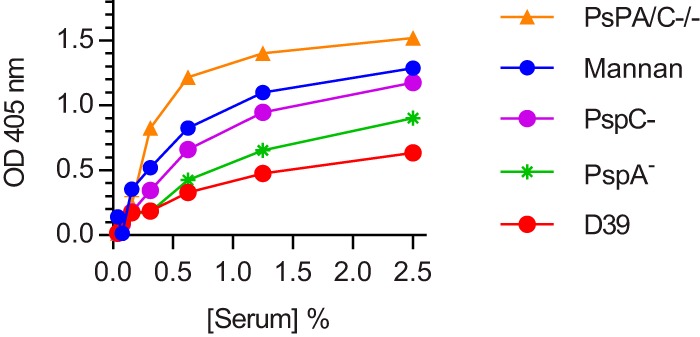
The deficiency of either PspA or PspC enhances C3b deposition on the surface of S. pneumoniae. C3b deposition was detected by anti-human C3c antibody. The absence of C4b deposition on D39 does not lead to completely defective C3 opsonization of the pneumococcal surface. However, higher levels of C3b deposition were detected on PspA–, PspC–, and PspA–/C– strains, where C4b can also be detected. The results are expressed as means ± the SEM, representing three different experiments. P < 0.001 for all Psp mutant versus D39 comparisons (two-way ANOVA with Dunnett’s correction for multiple comparisons; results obtained with mannan were excluded from the analysis).
DISCUSSION
It has previously been reported that the complement activation product C4b is undetectable on the surface of S. pneumoniae after exposure to human and mouse serum. Instead, the pneumococcal surface is liberally decorated with the final breakdown product of C4, C4dg, suggesting that the bacteria do not avoid C4 deposition altogether but possess a mechanism to inactivate the larger opsonic fragments (C4b and iC4b). The details of this mechanism have not been completely elucidated (42).
Li et al. (40) showed that the absence of PspA resulted in increased C1q, C4, and C3 deposition on the pneumococcal surface and increased immune adherence to erythrocytes. In a PspA- and PspC-deficient strain, the same effect was observed, but C3 opsonization was further increased. Enhanced C3b deposition on PspA and PspC doubly deficient bacteria was not seen when factor B-deficient mouse serum was used, implying that the high level of C3b deposition in the double-knockout PspA- and PspC-deficient strain is alternative pathway dependent. A deficiency of PspC alone had no effect on the alternative pathway or immune adherence. Together, these results suggest that PspA alone increases classical pathway activation and then acts together with PspC to further increase C3 opsonization and immune adherence, although it should be noted that Li et al. did not give full consideration to the contribution of the lectin pathway, which might account for all of the effects described, except the enhanced C1q binding. The reported susceptibility of PspC-deficient S. pneumoniae to alternative pathway-driven C3 opsonization might be explained by the fact that PspC can sequester factor H from host serum to the bacterial surface (43), where it prevents the formation of alternative pathway C3 convertases by accelerating the decay of C3bB and C3bBb complexes and by acting as a cofactor in the factor I-mediated conversion of C3b to smaller cleavage products.
A recent paper by Dieudonne-Vatran et al. reported the attachment of host C4BP to multiple clinical isolates of S. pneumoniae (41). C4BP bound to the bacteria retained its biological function, i.e., it was able to direct the factor I-mediated degradation of C4. C4BP binding was particularly high in several serotype 14 strains. Using isogenic variants of TIGR4 expressing different capsule types, as well as isogenic capsule deletion mutants (Δcps) of serotype 14, these authors showed that C4BP attachment was not directly related to capsular serotype. C4BP binding was, however, shown to be a function of the PspC allotype. It is particularly high in strains expressing PspC allotype 4, which includes most serotype 14 strains, and is significantly reduced, but not abolished, in ΔpspC strains (41).
Using mutant strains of S. pneumoniae D39 with targeted deletions of PspA, PspC, or both, we investigated whether only PspC, or also PspA, can sequester host C4BP to the bacterial surface and so promote the decay of C4b and iC4b to C4dg (neither of which can form the C3 convertase, C4b2a). Bacteria with single absences of either PspC or PspA showed increased levels of C4b (and iC4b) deposition. C4b deposition was maximized in the absence of both PspA and PspC. There was a negative correlation between the levels of C4b deposition on the one hand and the levels of C4BP and C4dg on the other (Fig. 2). This implies that both PspA and PspC can bind serum C4BP and so accelerate the conversion of C4b to C4dg.
Crucially, we were able to demonstrate a direct interaction between C4b and recombinant PspA and PspC. Using a pulldown assay, we were able to show that, when recombinant Psp proteins were immobilized on a microtiter plate, they captured C4BP from human serum (Fig. 3). Likewise, PspA and PspC both acted as competitive inhibitors of pneumococcal PspA and C, augmenting C4b deposition on D39 and, at the same time, reducing C4dg deposition (Fig. 3B and C). In a direct binding assay, it appeared that PspC bound C4 about four times as strongly as PspA; in the inhibition assays, the 50% inhibitory concentrations of the proteins were similar.
We demonstrated that the factors that determine resistance to C4b deposition on S. pneumoniae are conserved across 11 different encapsulated strains, covering 6 serotypes and 8 nonencapsulated strains (Fig. 1). Thus, resistance to C4b deposition is a function of the cell wall and is independent of capsule. Dieudonne-Vatran et al. (41) noted that encapsulation significantly reduces C4BP binding; our results indicate that even in the presence of capsule sufficient C4BP binds to the pneumococci to ensure the complete inactivation of C4b.
The results reported here show that both PspA and PspC can capture and bind C4BP from serum, which in turn drastically restricts the half-life of C4b deposited on the bacterial surface through its rapid conversion to C4dg via factor I-mediated and C4BP cofactor-catalyzed cleavage.
We recently described a mechanism by which the lectin pathway can activate C3 in the absence of C4, i.e., MASP-2 can directly cleave C3 to C3b and iC3b (44, 45). In pneumococcal infection, this partially compensates for the loss of C4 (and the resulting loss of the classical and lectin pathway C3 convertase, C4b2a) and explains why MASP-2-deficient mice are more susceptible to infection (7). It also offers an explanation for the residual C3b deposition seen on S. pneumoniae opsonized with serum at concentrations too low to be permissive for alternative pathway activation (Fig. 4).
In conclusion, we describe the mechanism that S. pneumoniae uses to evade complement C4 deposition on its surface, where C4b is not inhibited on the surface of the pneumococcus, but it is rapidly degraded into the smaller hemolytically inactive product C4dg through sequestration of C4BP by the noncapsular virulence factors PspA and PspC in close proximity to C4b. This finding explains how the deficiency of either PspA or PspC significantly increases the level of C3b deposition on the surface of the bacteria, enhances opsonophagocytosis, and hence minimizes the virulence of S. pneumoniae in a mouse model of S. pneumoniae infection.
MATERIALS AND METHODS
Bacterial strains.
S. pneumoniae serotype 2 strain D39 (NCTC 7466) was obtained from the National Collection of Type Cultures, London, UK. Clinical isolates of serotypes 3, 18C, and 6B were provided by Hermínia de Lencastre, ITQB Nova, Oeiris, Portugal. Mutant strains used in this study were all derived from serotype 2 strain D39. The PspC– strain was provided by Aras Kadioglu, University of Liverpool, Liverpool, UK. Other strains were from the collection of Peter Andrew.
Construction of pspA and pspA pspC mutant strains.
In vitro mariner mutagenesis was used to construct the pspA mutant (46). The genetic region containing SPD_0126 was amplified using SPD0126F (CAAGTCTAGCCAGCGTCGCT) and SPD0126R (CAGAATCAGCCCCTCCAAG) primers. For this, 200 ng of PCR fragment was mixed with 200 to 400 ng of donor mariner plasmid pR412, conferring resistance to spectinomycin. The mixture was incubated in the presence of Himar1 transposase, as described previously (46, 47). Gaps in transposition products were repaired with T4 DNA polymerase (New England BioLabs, Hitchin, UK) and subsequently by E. coli ligase (New England Biolabs). Using a synthetic competence-inducing peptide, repaired transposition products were transformed into S. pneumoniae D39. Transformants were selected with 100 µg/ml spectinomycin. Later, the insertion of the resistance cassette was confirmed by PCR using the transposon-specific primer MP127 (CCGGGGACTTATCAGCCAACC) or MP128 (TACTAGCGACGCCATCTATGTG) with appropriate chromosomal primers. Moreover, further confirmation of mutations was done by sequencing (48). To construct the pspA pspC mutant strain, the mutated region was amplified from the pspA mutant using SPD0126F and SPD0126R and transformed into the pspC mutant as described above.
Heterologous expression and purification of PspC and PspA.
PspC (SH13) and PspA (QP2) were purified by using Ni2+-affinity chromatography from E. coli BL21 as described earlier (49–51). Briefly, E. coli strains were grown in Luria-Bertani medium (Roth), and protein expression was induced (at an optical density at 600 nm [OD600] of 0.8) with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h. His6-tagged proteins from cell lysates were purified using an ÄKTA purifier system (GE Healthcare) and 1-ml HisTrap FF crude columns (GE Healthcare). Finally, the proteins were dialyzed against phosphate-buffered saline (PBS; pH 7.4) and analyzed by SDS-PAGE and Coomassie brilliant blue R-250 staining.
Collection of sera.
Blood was collected from healthy individuals after receipt of written consent, as required by the local ethics committee. Next, 10 ml of blood was drawn by venipuncture and immediately transferred onto ice to avoid the activation of complement and left for 3 h. Blood was then centrifuged at 7,000 × g for 7 min. Serum was separated and stored at –80°C until further use.
Preparation of bacteria.
S. pneumoniae was inoculated into brain heart infusion broth (Oxoid), incubated at 37°C overnight, and centrifuged the next day at 3,000 × g for 10 min. The bacterial pellet was washed three times in PBS and resuspended in 0.5% (vol/vol) formalin in PBS for 3 h at room temperature. Bacteria were centrifuged after fixation, washed with PBS again, and resuspended in carbonate coating buffer (35 mM NaHCO3, 15 mM Na2CO3 [pH 9.6]).
Complement activation assays.
Portions (100 μl) of formalin-fixed S. pneumoniae D39 or E. coli (OD550 of 0.6), suspended in coating buffer, were added to each well of Nunc Maxisorb microtiter enzyme-linked immunosorbent assay plates, followed by incubation overnight at 4°C. Then, 100 μl of mannan (10 μg/ml) was added to the plates as a positive control. To block the residual protein binding sites, 250 μl of 1% (wt/vol) BSA in TBS buffer (10 mM Tris, 140 mM NaCl [pH 7.4]) was added to each well, and the plates were left at room temperature for 2 to 3 h. The plates were then washed three times with wash buffer (TBS with 0.05% Tween 20). Serum was diluted 2-fold in barbital buffered saline (4 mM barbital, 1 mM MgCl2, 2 mM CaCl2, 145 mM NaCl [pH 7.4]). Serum was not added to each well of the last row; these wells were used as negative controls. After the serum was added, the plates were incubated at 37°C for 90 min and washed three times.
To investigate whether C4 is completely inhibited or deposited and rapidly degraded on the bacterial surface, C4b and C4dg were assayed using chicken anti-human C4, which reacts with human C4b and iC4b (Agrisera), and mouse anti-human C4dg (Quidel), which recognizes only C4dg. After incubation for 90 min at room temperature, the plates were washed again, and appropriate alkaline phosphatase (AP)-conjugated secondary antibodies, diluted in TBS, were added to the plates. The plates were then left for a further 90 min at room temperature. Finally, 100 μl of the colorimetric p–nitrophenyl phosphate substrate was added to all wells to determine the extent of deposition of different C4 fragments. The absorbance at 405 nm was then measured using a microtiter plate reader (Bio-Rad).
Similarly, C3b deposition and C4BP binding were also determined by using specific rabbit anti-human C3 (Dako) and mouse anti-human C4BP (Quidel) as primary antibodies and their respective AP-conjugated anti-rabbit and anti-mouse as secondary antibodies.
Flow cytometry.
Bacteria from stock culture were thawed, washed three times with wash buffer (TBS with 0.05% Tween 20), and then suspended in barbital buffered saline to a final concentration of 106 CFU/ml. A 10-μl portion of human serum was added to 200 μl of bacterial suspension, followed by incubation at 37°C for 1 h. The bacteria were washed again and then resuspended in wash buffer containing primary antibodies targeted against different complement fragments. These antibodies included mouse anti-human C4dg (Quidel) and mouse anti-human C4BP (Quidel). After incubation for 90 min at room temperature, the bacteria were washed and resuspended in wash buffer containing fluorescein isothiocyanate-conjugated anti-mouse IgG (Dako). After further incubation, the bacteria were washed again and fixed using 1% (wt/vol) paraformaldehyde. The fluorescence intensity was measured by FACS (FACSCalibur; Becton Dickinson).
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00742-18.
REFERENCES
- 1.Weiser JN. 2010. The pneumococcus: why a commensal misbehaves. J Mol Med 88:97–102. doi: 10.1007/s00109-009-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Musher DM. 2003. How contagious are common respiratory tract infections? N Engl J Med 348:1256–1266. doi: 10.1056/NEJMra021771. [DOI] [PubMed] [Google Scholar]
- 3.Scott JA. 2008. The global epidemiology of childhood pneumonia 20 years on. Bull World Health Organ 86:494–496. doi: 10.2471/BLT.08.052753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams BG, Gouws E, Boschi-Pinto C, Bryce J, Dye C. 2002. Estimates of world-wide distribution of child deaths from acute respiratory infections. Lancet Infect Dis 2:25–32. doi: 10.1016/S1473-3099(01)00170-0. [DOI] [PubMed] [Google Scholar]
- 5.Lim WS, Macfarlane JT, Boswell TC, Harrison TG, Rose D, Leinonen M, Saikku P. 2001. Study of community acquired pneumonia aetiology (SCAPA) in adults admitted to hospital: implications for management guidelines. Thorax 56:296–301. doi: 10.1136/thorax.56.4.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brow JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, Walport MJ, Botto M. 2002. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc Natl Acad Sci U S A 99:16969–16974. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, Holmskov U, Takahashi K, Stahl GL, Dudler T, Girija UV, Wallis R, Kadioglu A, Stover CM, Andrew PW, Schwaeble WJ. 2012. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog 8:e1002793. doi: 10.1371/journal.ppat.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ali YM, Hayat A, Saeed BM, Haleem KS, Alshamrani S, Kenawy HI, Ferreira VP, Saggu G, Buchberger A, Lachmann PJ, Sim RB, Goundis D, Andrew PW, Lynch NJ, Schwaeble WJ. 2014. Low-dose recombinant properdin provides substantial protection against Streptococcus pneumoniae and Neisseria meningitidis infection. Proc Natl Acad Sci U S A 111:5301–5306. doi: 10.1073/pnas.1401011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zipfel PF, Skerka C. 2009. Complement regulators and inhibitory proteins. Nat Rev Immunol 9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 10.Wallis R, Mitchell DA, Schmid R, Schwaeble WJ, Keeble AH. 2010. Paths reunited: initiation of the classical and lectin pathways of complement activation. Immunobiology 215:1–11. doi: 10.1016/j.imbio.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genster N, Ostrup O, Schjalm C, Eirik MT, Cowland JB, Garred P. 2017. Ficolins do not alter host immune responses to lipopolysaccharide-induced inflammation in vivo. Sci Rep 7:3852. doi: 10.1038/s41598-017-04121-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujita T, Matsushita M, Endo Y. 2004. The lectin-complement pathway—its role in innate immunity and evolution. Immunol Rev 198:185–202. doi: 10.1111/j.0105-2896.2004.0123.x. [DOI] [PubMed] [Google Scholar]
- 13.Zipfel PF, Mihlan M, Skerka C. 2007. The alternative pathway of complement: a pattern recognition system. Adv Exp Med Biol 598:80–92. doi: 10.1007/978-0-387-71767-8_7. [DOI] [PubMed] [Google Scholar]
- 14.Thurman JM, Holers VM. 2006. The central role of the alternative complement pathway in human disease. J Immunol 176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 15.Walport MJ. 2001. Complement. First of two parts. N Engl J Med 344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 16.Fujita T. 2002. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol 2:346–353. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 17.Carroll MV, Sim RB. 2011. Complement in health and disease. Adv Drug Deliv Rev 63:965–975. doi: 10.1016/j.addr.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Nagasawa S, Ichihara C, Stroud RM. 1980. Cleavage of C4b by C3b inactivator: production of a nicked form of C4b, C4b′, as an intermediate cleavage product of C4b by C3b inactivator. J Immunol 125:578–582. [PubMed] [Google Scholar]
- 19.Scharfstein J, Ferreira A, Gigli I, Nussenzweig V. 1978. Human C4-binding protein. I. Isolation and characterization. J Exp Med 148:207–222. doi: 10.1084/jem.148.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blom AM, Villoutreix BO, Dahlback B. 2004. Complement inhibitor C4b-binding protein—friend or foe in the innate immune system? Mol Immunol 40:1333–1346. doi: 10.1016/j.molimm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Carlsson F, Berggård K, Stålhammar-Carlemalm M, Lindahl G. 2003. Evasion of phagocytosis through cooperation between two ligand-binding regions in Streptococcus pyogenes M protein. J Exp Med 198:1057–1068. doi: 10.1084/jem.20030543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ram S, Cullinane M, Blom AM, Gulati S, McQuillen DP, Monks BG, O’Connell C, Boden R, Elkins C, Pangburn MK, Dahlback B, Rice PA. 2001. Binding of C4b-binding protein to porin: a molecular mechanism of serum resistance of Neisseria gonorrhoeae. J Exp Med 193:281–295. doi: 10.1084/jem.193.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngampasutadol J, Ram S, Blom AM, Jarva H, Jerse AE, Lien E, Goguen J, Gulati S, Rice PA. 2005. Human C4b-binding protein selectively interacts with Neisseria gonorrhoeae and results in species-specific infection. Proc Natl Acad Sci U S A 102:17142–17147. doi: 10.1073/pnas.0506471102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blom AM, Berggard K, Webb JH, Lindahl G, Villoutreix BO, Dahlback B. 2000. Human C4b-binding protein has overlapping, but not identical, binding sites for C4b and streptococcal M proteins. J Immunol 164:5328–5336. doi: 10.4049/jimmunol.164.10.5328. [DOI] [PubMed] [Google Scholar]
- 25.Meri T, Blom AM, Hartmann A, Lenk D, Meri S, Zipfel PF. 2004. The hyphal and yeast forms of Candida albicans bind the complement regulator C4b-binding protein. Infect Immun 72:6633–6641. doi: 10.1128/IAI.72.11.6633-6641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hallstrom T, Jarva H, Riesbeck K, Blom AM. 2007. Interaction with C4b-binding protein contributes to nontypeable Haemophilus influenzae serum resistance. J Immunol 178:6359–6366. doi: 10.4049/jimmunol.178.10.6359. [DOI] [PubMed] [Google Scholar]
- 27.Agarwal V, Hammerschmidt S, Malm S, Bergmann S, Riesbeck K, Blom AM. 2012. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J Immunol 189:3575–3584. doi: 10.4049/jimmunol.1102934. [DOI] [PubMed] [Google Scholar]
- 28.Bruyn GA, Zegers BJ, van Furth R. 1992. Mechanisms of host defense against infection with Streptococcus pneumoniae. Clin Infect Dis 14:251–262. doi: 10.1093/clinids/14.1.251. [DOI] [PubMed] [Google Scholar]
- 29.Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. 2010. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun 78:704–715. doi: 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melin M, Trzcinski K, Antonio M, Meri S, Adegbola R, Kaijalainen T, Kayhty H, Vakevainen M. 2010. Serotype-related variation in susceptibility to complement deposition and opsonophagocytosis among clinical isolates of Streptococcus pneumoniae. Infect Immun 78:5252–5261. doi: 10.1128/IAI.00739-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuste J, Botto M, Paton JC, Holden DW, Brown JS. 2005. Additive inhibition of complement deposition by pneumolysin and PspA facilitates Streptococcus pneumoniae septicemia. J Immunol 175:1813–1819. doi: 10.4049/jimmunol.175.3.1813. [DOI] [PubMed] [Google Scholar]
- 32.Paton JC. 1996. The contribution of pneumolysin to the pathogenicity of Streptococcus pneumoniae. Trends Microbiol 4:103–106. doi: 10.1016/0966-842X(96)81526-5. [DOI] [PubMed] [Google Scholar]
- 33.Dave S, Brooks-Walter A, Pangburn MK, McDaniel LS. 2001. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun 69:3435–3437. doi: 10.1128/IAI.69.5.3435-3437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jarva H, Janulczyk R, Hellwage J, Zipfel PF, Bjorck L, Meri S. 2002. Streptococcus pneumoniae evades complement attack and opsonophagocytosis by expressing the pspC locus-encoded Hic protein that binds to short consensus repeats 8-11 of factor H. J Immunol 168:1886–1894. doi: 10.4049/jimmunol.168.4.1886. [DOI] [PubMed] [Google Scholar]
- 35.Yother J, Briles DE. 1992. Structural properties and evolutionary relationships of PspA, a surface protein of Streptococcus pneumoniae, as revealed by sequence analysis. J Bacteriol 174:601–609. doi: 10.1128/jb.174.2.601-609.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu AH, Fulgham RL, McCrory MA, Briles DE, Szalai AJ. 1999. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect Immun 67:4720–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukerji R, Mirza S, Roche AM, Widener RW, Croney CM, Rhee DK, Weiser JN, Szalai AJ, Briles DE. 2012. Pneumococcal surface protein A inhibits complement deposition on the pneumococcal surface by competing with the binding of C-reactive protein to cell surface phosphocholine. J Immunol 189:5327–5335. doi: 10.4049/jimmunol.1201967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren B, Szalai AJ, Hollingshead SK, Briles DE. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect Immun 72:114–122. doi: 10.1128/IAI.72.1.114-122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quin LR, Moore QC, McDaniel LS. 2007. Pneumolysin, PspA, and PspC contribute to pneumococcal evasion of early innate immune responses during bacteremia in mice. Infect Immun 75:2067–2070. doi: 10.1128/IAI.01727-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Glover DT, Szalai AJ, Hollingshead SK, Briles DE. 2007. PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect Immun 75:5877–5885. doi: 10.1128/IAI.00839-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dieudonne-Vatran A, Krentz S, Blom AM, Meri S, Henriques-Normark B, Riesbeck K, Albiger B. 2009. Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J Immunol 182:7865–7877. doi: 10.4049/jimmunol.0802376. [DOI] [PubMed] [Google Scholar]
- 42.Krarup A, Sorensen UB, Matsushita M, Jensenius JC, Thiel S. 2005. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, L-ficolin, and H-ficolin. Infect Immun 73:1052–1060. doi: 10.1128/IAI.73.2.1052-1060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herbert AP, Makou E, Chen ZA, Kerr H, Richards A, Rappsilber J, Barlow PN. 2015. Complement evasion mediated by enhancement of captured factor H: implications for protection of self-surfaces from complement. J Immunol 195:4986–4998. doi: 10.4049/jimmunol.1501388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yaseen S, Demopulos G, Dudler T, Yabuki M, Wood CL, Cummings WJ, Tjoelker LW, Fujita T, Sacks S, Garred P, Andrew P, Sim RB, Lachmann PJ, Wallis R, Lynch N, Schwaeble WJ. 2017. Lectin pathway effector enzyme mannan-binding lectin-associated serine protease-2 can activate native complement C3 in absence of C4 and/or C2. FASEB J 31:2210–2219. doi: 10.1096/fj.201601306R. [DOI] [PubMed] [Google Scholar]
- 45.Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, Dudler T, Parent B, Lhotta K, Wallis R, Farrar CA, Sacks S, Lee H, Zhang M, Iwaki D, Takahashi M, Fujita T, Tedford CE, Stover CM. 2011. Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A 108:7523–7528. doi: 10.1073/pnas.1101748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terra VS, Zhi X, Kahya HF, Andrew PW, Yesilkaya H. 2015. Pneumococcal 6-phospho-β-glucosidase (BglA3) is involved in virulence and nutrient metabolism. Infect Immun 84:286–292. doi: 10.1128/IAI.01108-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaspar P, Al-Bayati FA, Andrew PW, Neves AR, Yesilkaya H. 2014. Lactate dehydrogenase is the key enzyme for pneumococcal pyruvate metabolism and pneumococcal survival in blood. Infect Immun 82:5099–5109. doi: 10.1128/IAI.02005-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karlyshev AV, Pallen MJ, Wren BW. 2000. Single-primer PCR procedure for rapid identification of transposon insertion sites. Biotechniques 28:1078–1082. doi: 10.2144/00286bm05. [DOI] [PubMed] [Google Scholar]
- 49.Voss S, Hallström T, Saleh M, Burchhardt G, Pribyl T, Singh B, Riesbeck K, Zipfel PF, Hammerschmidt S. 2013. The choline-binding protein PspC of Streptococcus pneumoniae interacts with the C-terminal heparin-binding domain of vitronectin. J Biol Chem 288:15614–15627. doi: 10.1074/jbc.M112.443507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Binsker U, Kohler TO, Krauel K, Kohler S, Schwertz H, Hammerschmidt S. 2015. Pneumococcal adhesins PavB and PspC are important for the interplay with human thrombospondin. J Biol Chem 290:14542–14555. doi: 10.1074/jbc.M114.623876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammerschmidt S, Bethe G, Remane PH, Chhatwal GS. 1999. Identification of pneumococcal surface protein A as a lactoferrin-binding protein of Streptococcus pneumoniae. Infect Immun 67:1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.