

Malaria is caused by the protozoan parasite Plasmodium, which undergoes a complex life cycle in a human host and a mosquito vector. The parasite’s cyclic GMP (cGMP)-dependent protein kinase (PKG) is essential at multiple steps of the life cycle.
KEYWORDS: Plasmodium falciparum, kinase, mechanism, proteasome, substrate
ABSTRACT
Malaria is caused by the protozoan parasite Plasmodium, which undergoes a complex life cycle in a human host and a mosquito vector. The parasite’s cyclic GMP (cGMP)-dependent protein kinase (PKG) is essential at multiple steps of the life cycle. Phosphoproteomic studies in Plasmodium falciparum erythrocytic stages and Plasmodium berghei ookinetes have identified proteolysis as a major biological pathway dependent on PKG activity. To further understand PKG’s mechanism of action, we screened a yeast two-hybrid library for P. falciparum proteins that interact with P. falciparum PKG (PfPKG) and tested peptide libraries to identify its phosphorylation site preferences. Our data suggest that PfPKG has a distinct phosphorylation site and that PfPKG directly phosphorylates parasite RPT1, one of six AAA+ ATPases present in the 19S regulatory particle of the proteasome. PfPKG and RPT1 interact in vitro, and the interacting fragment of RPT1 carries a PfPKG consensus phosphorylation site; a peptide carrying this consensus site competes with the RPT1 fragment for binding to PfPKG and is efficiently phosphorylated by PfPKG. These data suggest that PfPKG’s phosphorylation of RPT1 could contribute to its regulation of parasite proteolysis. We demonstrate that proteolysis plays an important role in a biological process known to require Plasmodium PKG: invasion by sporozoites of hepatocytes. A small-molecule inhibitor of proteasomal activity blocks sporozoite invasion in an additive manner when combined with a Plasmodium PKG-specific inhibitor. Mining the previously described parasite PKG-dependent phosphoproteomes using the consensus phosphorylation motif identified additional proteins that are likely to be direct substrates of the enzyme.
INTRODUCTION
Despite recent progress, malaria remains a devastating disease that kills over 400,000 people annually (1). Its causative agent, the protozoan parasite Plasmodium, undergoes a complex life cycle in the human host and the Anopheles mosquito vector (2). Plasmodium organisms are transmitted to humans by mosquitoes in the form of sporozoites. Sporozoites infect hepatocytes in the liver, within which they differentiate into exo-erythrocytic forms that give rise to hepatic merozoites. Hepatic merozoites invade erythrocytes and commence the symptomatic stage of infection. Some asexually replicating erythrocytic-stage parasites form dormant sexual stages, termed gametocytes. Gametocytes develop into gametes in the mosquito midgut when ingested in the blood meal. Zygotes, resulting from the fertilization of male and female gametes, differentiate into ookinetes. Ookinetes invade the midgut epithelium and give rise to oocysts within which the parasite differentiates to form sporozoites. Sporozoites invade salivary glands and are transmitted to a new host through the bite of the infected mosquito.
Second messenger signaling, especially through cyclic GMP (cGMP), plays a central role at several steps of this complex life cycle. cGMP signals are transduced primarily through the cGMP-dependent protein kinase (PKG) of the parasite. PKG is a serine/threonine kinase whose activity is regulated by cGMP. cGMP binding to the regulatory domain of the enzyme triggers an allosteric change (3) that activates the catalytic domain. Plasmodium PKG activity is essential for the parasite’s invasion of and egress from hepatocytes (4, 5) and erythrocytes (6, 7) and for its differentiation from gametocytes to gametes (8). It is also required for ookinete and sporozoite motility (5, 8, 9). Plasmodium PKG’s role in these different processes is likely explained by its regulation of phosphoinositide metabolism, which generates Ca2+ signals critical for merozoite egress, gametocyte activation, and ookinete motility (10, 11). In addition to phosphoinositide metabolism, protein proteolysis is a key PKG-dependent process, as revealed by PKG-dependent phosphoproteomes in both Plasmodium falciparum and Plasmodium berghei (10, 12). Despite the vital function of Plasmodium PKG in a variety of cellular processes, only a few direct substrates of the kinase have been identified so far (12). Some Plasmodium PKG-dependent protein sites have been inferred to be direct substrates of the kinase because they fall within the consensus sequence ascribed to mammalian PKG. However, it has never been demonstrated that parasite PKG has the same phosphorylation site preferences as its mammalian homologs, with which it shares only ∼43% sequence identity within the catalytic domain. Here, we define a consensus sequence for the P. falciparum PKG (PfPKG) preferred phosphorylation site and report PfPKG’s interaction with and potential phosphorylation of the regulatory subunit of the parasite proteasome.
RESULTS
Recombinant PfPKG interacts in vitro with the RPT1 subunit of the 19S proteasome and HSP90.
To better understand PKG’s essential role through the Plasmodium life cycle, it is critical to define its mechanism of action, for example, through identification of its downstream substrates. To identify potential substrates of P. falciparum PKG (PfPKG), we conducted yeast two-hybrid screens (data not shown) for P. falciparum proteins that interact with the PfPKG kinase domain (amino acids [aa] 409 to 853) using a previously described prey library encoding fragments of P. falciparum proteins (13). As bait, we utilized the PfPKG kinase domain (aa 409 to 761) modified to carry single amino acid substitutions that are predicted to increase the affinity of eukaryotic kinases for their substrates (PfPKG E706A and PfPKG R781A) (14). Yeast two-hybrid screens using PfPKG E706A and PfPKG R781A as baits identified 17 unique prey sequences as interacting with PfPKG. Of these, two prey sequences encoded fragments of annotated Plasmodium proteins: RPT1 (PF3D7_1311500) consisting of amino acids 114 to 420 (RPT1114–420), a subunit of the 19S regulatory particle of the proteasome, and heat shock protein 90 (PF3D7_0708400) consisting of amino acids 228 to 518 (HSP90228–518). RPT1114–420 and HSP90228–518 retested in a split-luciferase complementation assay, which tests the ability of two proteins to reconstitute active luciferase when fused to either the amino (N)- or carboxy (C)-terminal fragments of firefly luciferase (Nfluc and Cfluc, respectively) (15) (Fig. 1A).
FIG 1.

Recombinant PfPKG interacts with RPT1 and HSP90 in vitro. A split-luciferase protein complementation assay was used to test interaction between the PfPKG kinase domain (PfPKG E706A and PfPKG R781A) and a fragment of RPT1 (amino acids 114 to 420) or HSP90 (amino acids 228 to 518). The proteins were expressed in wheat germ extracts as fusions with the N-terminal domain (Nfluc) or the C-terminal domain (Cfluc) of luciferase. Reconstitution of functional luciferase was measured as average relative light units (RLU). (A) Western blotting confirmed expression of fusion proteins in in vitro-transcribed/translated wheat germ extracts. These fusion proteins were used in subsequent complementation assays. Fusions with Nfluc were detected using antiserum against the N-terminal domain of firefly luciferase. Fusions with Cfluc were detected using anti-FLAG antiserum. Smaller antibody-reactive bands are the likely translation products of incomplete transcripts. Expected molecular weights were as follows: Nfluc-PfPKG E706A, ∼100 kDa; Nfluc-HSP90228–518, ∼85 kDa; Nfluc-RPT1114–420, ∼85 kDa; Cfluc-PfPKG E706A, ∼85 kDa; Cfluc-PfPKG R781A, ∼85 kDa; Cfluc-HSP90228–518, ∼70 kDa; Cfluc-RPT1114–420, ∼60 kDa. (B) Incubation of Nfluc-PfPKG E706A with Cfluc-RPT1114–420, Cfluc-PfPKG E706A with Nfluc-RPT1114–420, or Cfluc-PfPKG R781A with Nfluc-RPT1114–420 significantly increased luciferase activity. In contrast, incubation of Nfluc-PfPKG E706A with Cfluc without a fusion partner [Cfluc(-)], Cfluc-PfPKG E706A with Nfluc without a fusion partner [Nfluc(-)], and Cfluc-RPT1114–420 with Nfluc(-) or Cfluc-PfPKG R781A with Nfluc(-) had a minimal effect on luciferase activity. Results shown are average of three replicates ± standard deviations. (C) Incubation of Nfluc-PfPKG E706A with Cfluc-HSP90228–518, Cfluc-PfPKG E706A with Nfluc-HSP90228–518, or Cfluc-PfPKG R781A with Nfluc-HSP90228–518 significantly increased luciferase activity. In contrast, incubation of Nfluc-PfPKGE706A with Cfluc(-), Cfluc-PfPKGE706A with Nfluc(-), Cfluc-RPT1114–420 with Nfluc(-), or Cfluc-PfPKG R781A with Nfluc(-) had a minimal effect on luciferase activity. Results shown are average of three replicates ± standard deviations. Data were analyzed using an unpaired t test (*, P < 0.05; **, P < 0.005).
The split-luciferase complementation assay confirmed interaction of PfPKG E706A and PfPKG R781A with RPT1114–420 and HSP90228–518 (Fig. 1B and C). Incubating a fusion of PfPKG E706A and the N-terminal fragment of luciferase (Nfluc-PfPKG E706A) with a fusion of RPT1114–420 and the C-terminal fragment of luciferase (Cfluc-RPT1114–420) significantly increased luciferase activity. Switching fusion partners, i.e., using Nfluc-RPT1114–420 and Cfluc-PfPKG E706A or Cfluc-PfPKG R781A, also resulted in a significant increase in luciferase activity. In contrast, there was minimal reconstitution of luciferase activity under the following conditions: (i) when Nfluc-PfPKG E706A was incubated with the C-terminal fragment of luciferase without a fusion partner [Cfluc(-)], (ii) when Cfluc-RPT1114–420 was incubated with the N-terminal fragment of luciferase without a fusion partner [Nfluc(-)], and (iii) when Cfluc-PfPKG E706A or Cfluc-PfPKG R781A was incubated with Nfluc(-). Fusions of HSP90 (amino acids 228 to 518) demonstrated similar specific interactions with fusions containing either PfPKG E706A or PfPKG E781A. These results demonstrated that PfPKG interacts in vitro with fragments of RPT1 and PfHSP90.
We reasoned that PKG’s interaction with RPT1 predicts that the proteasome participates in cellular processes that require parasite PKG, for example, host cell invasion (5). To examine the role of the parasite proteasome in this process, we tested the effect of MG132, a proteasomal inhibitor (16), on invasion of HepG2 cells by P. berghei sporozoites. MG132 causes a dose-dependent decrease in the fraction of sporozoites that become intracellular (Fig. 2A). These data demonstrate a role for the parasite proteasome in a cellular process that is PKG dependent. Viability of sporozoites after MG132 treatment, as measured by propidium iodide staining, was unaffected (see Fig. S1 in the supplemental material). Combinatorial treatment with MG132 and TSP, a trisubtituted pyrolle that is a specific inhibitor of Plasmodium PKG (5, 8, 17), has an additive effect on sporozoite invasion (Fig. 2B). Next, we tested the effect of proteasomal inhibition on P. berghei sporozoites that carry a hypomorphic allele of P. berghei PKG (PKG T619Q-HA, where HA is a hemagglutinin tag) with reduced PKG activity (5). Invasion by T619Q sporozoites is significantly more sensitive to MG132 than invasion by sporozoites expressing wild-type PKG (PKG-HA) (Fig. 2C). The additive effect of the MG132 and TSP combination and the sensitivity of PKG-hypomorph sporozoites to MG132 are consistent with P. berghei PKG (PbPKG) and the proteasome acting in the same pathway during sporozoite invasion.
FIG 2.
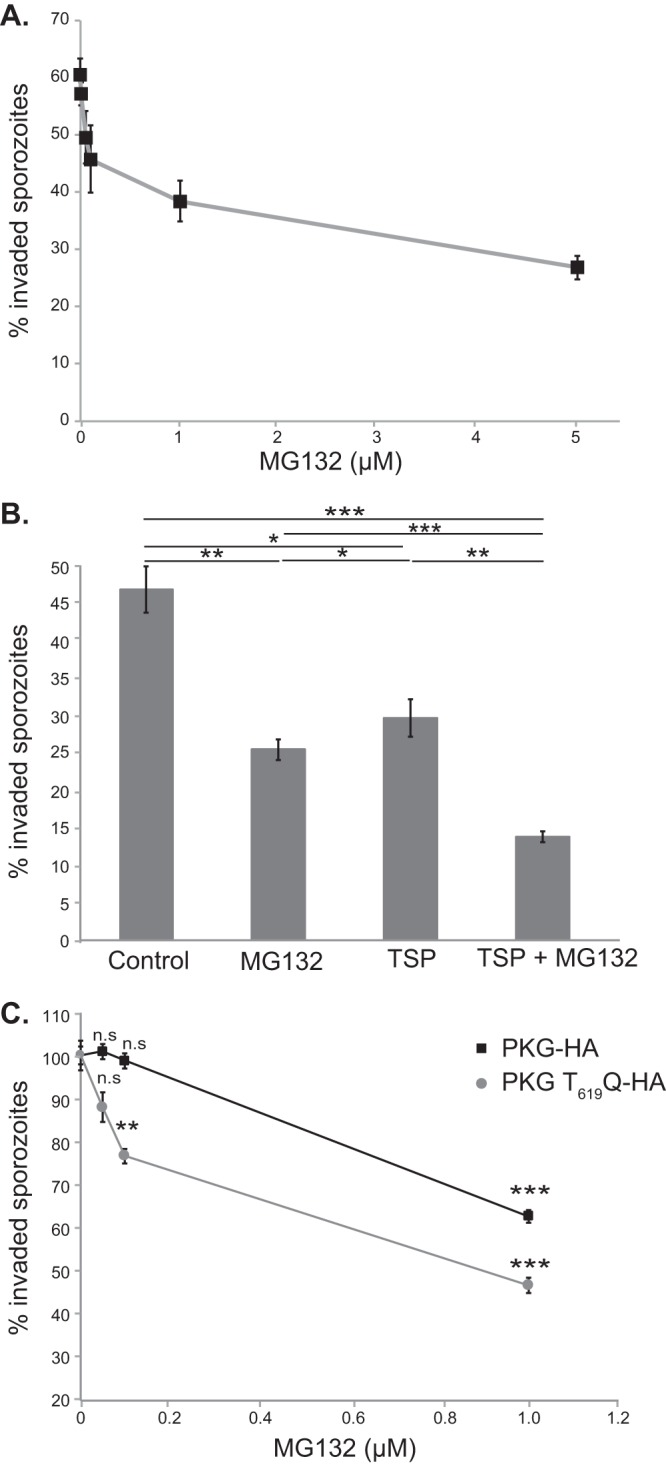
Sporozoite invasion requires the proteasome and PKG. (A) Treatment of sporozoites with MG132 leads to a dose-dependent decrease in sporozoite invasion. PbGFP-Luc sporozoites were treated with different doses of MG132 prior to addition to HepG2 cells. The fraction of sporozoites that are intracellular 90 min after addition to HepG2 cells was determined. Results shown are from a representative experiment (average of 4 replicates ± standard deviations). The experiment was performed twice. (B) Treatment of sporozoites with MG132 and TSP has an enhanced effect on sporozoite invasion compared to treatment with TSP or MG132 alone. PbGFP-Luc sporozoites were treated with either TSP (0.5 μM) alone or in combination with MG132 (5 μM) prior to addition to HepG2 cells. The fraction of sporozoites that are intracellular 90 min after addition to HepG2 cells was determined. Results shown are the average of three experiments, each done in triplicate, ± standard errors. (C) Invasion by sporozoites with reduced PKG activity (PKG T619Q-HA) is significantly more sensitive to MG132 inhibition than invasion by wild-type sporozoites (PKG-HA). Results shown are the average of four experiments, each done in triplicate or quadruplicate ± standard errors. Data were analyzed using an unpaired t test (*, P < 0.05; **, P < 0.005; ***, P < 0.0005; ns, not significant).
Next, we tested if inhibition of PfPKG has a global effect on protein degradation in the parasite. We quantified levels of ubiquitinated proteins in schizonts treated with a specific inhibitor of Plasmodium PKG (8) (TSP; termed compound 1 in reference 18). PfPKG inhibition through TSP treatment did not significantly alter the total level of ubiquitinated proteins in the parasite (Fig. S2). These data suggest that PKG-mediated phosphorylation of RPT1 is likely to affect specific, as yet unidentified, client proteins.
Analysis of P. falciparum PKG substrate specificity.
As a way to identify specific sites that are phosphorylated by PfPKG, we determined its phosphorylation site sequence specificity. We used a positional scanning peptide library approach in which the relative rate of phosphorylation is measured on a series of peptides in which each of the 20 amino acids is replaced at each of 9 positions surrounding the phosphorylation site (19) (Fig. 3A and Fig. S3A). This analysis revealed that PfPKG strongly prefers basic residues, in particular, Arg, at the −5 and −3 positions relative to the phosphorylation site. Basic residues (Arg, Lys, and His) were also preferentially phosphorylated at the −2 position. In addition, the kinase preferentially phosphorylated hydrophobic (Φ) residues at the +1 position. PfPKG appeared to generally deselect acidic residues at multiple positions both upstream and downstream of the phosphorylation site. Finally, PfPKG preferred Thr over Ser as the phosphoacceptor residue. The enhanced signal for peptides with fixed Thr residues at multiple positions is likely to be an artifact arising from those peptides having two potential sites of phosphorylation.
FIG 3.

PfPKG has a consensus phosphorylation site distinct from that of mammalian PKG. (A) A combinatorial peptide library consisting of 182 peptide mixtures with the general sequence shown at the top of the panel was used as the substrate in PfPKG enzymatic assays. The heat map shows the relative levels of phosphorylation of peptide mixtures having the indicated amino acid present at the indicated position. The sequence logo is scaled such that the height of the letter is proportional to the level of phosphorylation of the corresponding amino acid residue at the indicated position within the peptide. The logo was prepared using enoLOGOS (53). Results shown are the average from two separate determinations. (B) The X-ray crystal structure of PfPKG (PDB accession number 5DYK) was overlaid with that of an AKT-substrate peptide complex (PDB accession number 1O6K) using PyMOL. The substrate peptide (green) is modeled bound to PfPKG (cyan; the indicated interacting residues are shown in yellow). For clarity, AKT is not shown. (C) The consensus phosphorylation site of PfPKG was confirmed by determining the reaction rate of PfPKG using individual peptides as substrates. Peptides with amino acid replacements at key positions were used as substrates in PfPKG activity assays. The presumed phosphoacceptor site in each peptide is underlined. Reaction rates for each peptide were determined relative to the rate of the wild type (WT). PfPKG has a strong preference for amino acids at the −5 and +1 positions. PfPKG prefers Thr (AKTIDE-T versus AKTIDE-S) as the phosphoacceptor, a basic residue at the −5 position (BAD wild type versus BAD-5A), and a hydrophobic residue at the +1 position (BAD wild type versus BAD + 1A) relative to the phosphorylation site. It has weaker preference for peptides containing basic residues at the −2 position (BAD wild type versus BAD-2A). Results are the average of two experiments, each done in triplicate, ± standard deviations. (D) The domain of RPT1 that contacts PfPKG contains a phosphorylation site for PfPKG. Split-luciferase complementation assays between Nfluc-PfPKG E706A and Cfluc-RPT1114–420 were performed in the presence of peptides carrying either the wild-type (WT) or a mutant (T390A) consensus phosphorylation site from RPT1114–420. Wild-type peptide, but not the mutant, competed with RPT1 for interaction with PfPKG. Results are the average of three experiments, each performed in triplicate. (E) PfPKG efficiently phosphorylates peptides matching its consensus sequence present in two possible substrate proteins, RPT1 and PB_090940. Peptides with amino acid replacements at key positions were used as substrates in PfPKG activity assays. The presumed phosphoacceptor site in each peptide is underlined. Reaction rates for each peptide were determined relative to the rate of the wild type. Results are the average of three experiments, each performed using two or three replicates. Data were analyzed using an unpaired t test (**, P < 0.001; ***, P = 0.0001; ****, P < 0.0001; ns, not significant).
Prior analysis of the phosphorylation site specificity of mammalian PKG had revealed strong preferences for basic residues at both the −3 and the −2 positions, reminiscent of PKA (20–22). Our observation of a strong preference for Arg at the −5 position was therefore unanticipated for PfPKG. While not ascribed to PKG or PKA, selectivity for Arg at the −5 position is characteristic of a number of related mammalian kinases, most notably AKT isozymes (23). To rationalize the substrate specificity of PfPKG, we overlaid the X-ray crystal structure of an AKT-peptide complex (24) on the recently determined structure of PfPKG (Fig. 3B). In the resulting PfPKG-peptide model, Glu625 makes an ion pair with the −3 Arg residue in the substrate. In addition, three residues shared with AKT (Glu668, Tyr702, and Glu728) make polar contacts with the −5 Arg residue in the substrate. This model provides a likely explanation as to why PfPKG, like mammalian AKT, has a strong preference for Arg at the −5 position in its substrates.
To validate the results of the peptide library screen, we examined the phosphorylation rate of individual peptide substrates bearing point substitutions at residues strongly preferred in the peptide library experiments. A peptide corresponding to a phosphorylation site in the mouse protein BAD (Ser112) that incorporates multiple residues favored by PfPKG was an efficient substrate for the kinase (Fig. 3C and Fig. S3B). Furthermore, Ala replacement of the −3 Arg residue, the −2 His residue, or the +1 Tyr residue decreased the efficiency of phosphorylation. We tested the phosphoacceptor residue preference of PfPKG using versions of a peptide originally designed as an optimized substrate for the mammalian kinase, AKT (AKTIDE), in which the phosphorylation site residue was either Ser or Thr. The Thr peptide was phosphorylated about 4-fold faster than the equivalent Ser peptide, confirming that PfPKG prefers Thr over Ser as the phosphoacceptor residue. Phosphorylation of AKTIDE-Thr required cGMP and was inhibited by TSP, indicating that its phosphorylation was due to PfPKG itself and not to another kinase contaminating our preparations (Fig. 3C and Fig. S3B).
To determine if RPT1 or HSP90 could be a substrate of PKG, we searched the PKG-interacting domain of each for the presence of the minimal PfPKG phosphorylation site motif, (R/K)xx(S/T), and preferred amino acids at the −5, −2, 0 (phosphoacceptor), or +1 position relative to the sequence of the putative phosphoacceptor (Fig. 3A). RPT1114–420 contains six putative minimal sites for PfPKG phosphorylation (Table S1). Of these six sites, two contain an Arg or Lys at the −5 position, one contains Arg, Lys, or His at the −2 position, five contain a Thr at the 0 position, and three contain a hydrophobic residue at the +1 position. One of the six peptides contains preferred amino acids at all of the selected positions, i.e., RARRKT390I, suggesting that it could be phosphorylated by PfPKG.
Since interaction between the target amino sequence and a kinase is a prerequisite for substrate phosphorylation, we tested if the RARRKT390I sequence lies within the interaction interface of RPT1 and PfPKG. We measured the effect of adding a peptide containing RARRKT390I to the split-luciferase complementation assay that detects interaction between RPT1114–420 and the PfPKG kinase domain. Addition of the peptide inhibited luciferase activity that results when RPT1114–420 and the PfPKG kinase domain interact in a dose-dependent manner (Fig. 3D). A nearly identical peptide in which the putative phosphoacceptor T390 was replaced with Ala was significantly less effective in inhibiting luciferase reconstitution. These results strongly suggest that the PfPKG-contacting domain of RPT1 includes RARRKT390I and that T390 is likely to be the phosphoacceptor site, as predicted by the peptide library screen. To test if RARRKT390I can be phosphorylated by PfPKG, a peptide containing the sequence was used as the substrate in a PfPKG kinase reaction (Fig. 3E, left panel; Fig. S3C). As predicted by the positional scanning approach, PfPKG specifically phosphorylated the Thr residue (T390) that lies within the context of the consensus and not one that lies downstream (T392). Phosphorylation was dependent on Arg at the −5 (R385) and −3 (R387) positions and on a hydrophobic residue at +1 (I391). The phosphorylation of the peptide in vitro by PfPKG suggests that RPT1 could be a substrate for PKG. We tested if PfPKG phosphorylates full-length in vitro-transcribed/translated RPT1 but did not detect phosphorylation (data not shown). These data suggest that RPT1’s phosphorylation by PfPKG may require its assembly into the 19S proteasome or posttranslational modifications that are missing in the in vitro-transcribed/translated protein. We cannot exclude the possibility that RPT1 is not phosphorylated by PfPKG.
HSP90228–518 contains five putative minimal sites for PfPKG phosphorylation (Table S1). Of these five sites, one contains Arg or Lys at the −5 position, two contain Arg, Lys, or His at the −2 position, four contain a Thr at the 0 position, three contain a hydrophobic residue at the +1 position, and none contains preferred amino acids at all selected positions. These sites could potentially be phosphorylated by PfPKG.
To identify more potential direct targets of PKG, we examined the PKG-dependent phosphoproteomes of ookinetes (10) and schizonts (12) for phosphopeptides that contain the preferred amino acids at positions represented by the consensus site, (R/K)xR(H/K/R)x(S/T)Φ. Phosphopeptides that are phosphorylated in vivo in a PKG-dependent manner and carry the preferred amino acids at positions strongly selected by PfPKG in vitro are most likely to be direct targets.
Analysis of the PKG-dependent phosphoproteome of ookinetes (10) shows that 820 phosphopeptides undergo a significant decrease (P < 0.05) in phosphorylation when PKG activity is inhibited (Table S1). Of these, 165 contain the minimal motif, (R/K)xx(S/T). Of the 165 phosphosites, 18 contain an Arg or Lys at the −5 position, 28 contain a basic residue at the −2 position, 33 contain a Thr at the 0 position, and 67 contain a hydrophobic residue at the +1 position. These phosphosites are more likely to be direct targets of PKG.
A peptide from PBANKA_090940, a hypothetical protein, undergoes significant PKG-dependent phosphorylation and contains the PfPKG consensus RxR(H/K/R)x(S/T)Φ. We tested the PB_090940 peptide as a substrate in the PfPKG activity assay. The PB_090940 peptide was efficiently phosphorylated by PfPKG, and phosphorylation was dependent on the identified phosphoacceptor Ser (Ser450) (Fig. 3E, right panel, and Fig. S3C). These results are consistent with PB_090940 being a potential substrate for PbPKG.
The PKG-dependent phosphoproteome of schizonts contains 46 putative direct targets with the minimal (R/K)xx(S/T) motif (12) (Table S1). Of these, none contains an Arg or Lys at the −5 position, 10 contain a basic residue at the −2 position, 5 contain a Thr at the 0 position, and 21 contain a hydrophobic residue at the +1 position. Whether these sites are directly phosphorylated by PfPKG needs to be experimentally determined.
DISCUSSION
Since PKG serves multiple essential functions throughout the parasite life cycle, we attempted to better understand PKG’s mechanism of action by identifying PfPKG’s consensus phosphorylation site. PfPKG’s phosphorylation site motif is substantially different from the established mammalian PKG consensus sequence (20, 21), including that of human PKG1 determined previously using the same method (22). While PfPKG and mammalian PKG share a strong preference for Arg at the −3 position, mammalian PKG is nonselective at the −5 position, while PfPKG strongly prefers Arg at that position. At the −2 position, the mammalian kinase exclusively selected Lys and Arg residues, while PfPKG selected His as well. Finally, mammalian and parasite kinases have distinct phosphoacceptor residue preferences, with mammalian PKG preferring Ser and PfPKG preferring Thr. Among previously analyzed kinases, PfPKG appears most similar to mammalian ROCK1, which prefers Thr as the phosphoacceptor and has strong selectivity for Arg at the −5 and −3 positions (25, 26). We note that motifs determined on peptide substrates should not be taken as being strictly required for phosphorylation in vivo. Instead, true sites are expected to be highly enriched for selected residues, but no individual site will necessarily have all residues selected on peptides.
A relatively small fraction of kinase-dependent phosphorylation events found in proteomics approaches are direct substrates of the kinase. In different studies, direct substrates range from 5% to 20% of total sites identified (27, 28). Typically, sites identified in proteomics experiments that match the consensus sequence of a kinase and that decrease in abundance when the kinase is knocked out or inhibited are considered to be likely direct substrates. This criterion has been frequently used in phosphoproteomics studies to infer direct substrates of kinases (29–33).
From that standpoint, the fact that PfPKG has a novel consensus motif is significant. Our expectation is that the vast majority, if not all, of direct sites of PfPKG would have a Lys or Arg (His in rare cases) at the −3 position, with an overrepresentation of Arg or Lys at the −5 position, a basic residue at the −2 position, or a hydrophobic residue at the +1 position. Using these criteria to mine the PKG-dependent phosphoproteomes revealed that several proteins involved in gliding motility, such as GAP40 (34), coronin (35), MyoA (34, 36, 37), and ROM4 (38), are likely to be substrates of Plasmodium PKG. Phosphorylation of these proteins could be the mechanism through which PKG regulates motility, a prerequisite for invasion of host cells by different parasite stages. Future work will need to examine the biological significance of phosphorylation of these proteins by Plasmodium PKG.
Another parasite PKG-dependent cellular process identified by phosphoproteomics is proteolysis (12). In eukaryotic cells, regulated protein degradation by the proteasome is essential for protein homeostasis and influences processes such as the cell cycle, signal transduction, and stress response. Proteasomal function is essential in Plasmodium as well, as demonstrated by the potent parasiticidal activity of existing and novel proteasome inhibitors (39–42). The parasite proteasome plays an important role in many processes that also require PKG, such as erythrocytic-stage schizogony and the development of the liver stage (39, 42, 43), suggesting that PfPKG’s regulation of proteolysis is biologically significant. The importance of proteasomal function during sporozoite invasion, another process that requires Plasmodium PKG (5), has been investigated only once before (43). This study found that treatment with the proteasomal inhibitor lactacystin did not affect the invasion rate of sporozoites and concluded that proteasomal activity is not required for sporozoite invasion (43). Our data acquired using a different proteasomal inhibitor, MG132, demonstrate that sporozoite invasion requires proteolysis. One explanation for the differential effects of lactacystin and MG132 on sporozoite invasion could be that while the former inhibits the trypsin-like activity of the β2 subunit of the proteasome, the latter inhibits the caspase-like and chymotrypsin-like activities of the β1 and β5 subunits (44). For the first time, our data demonstrate that sporozoite invasion is sensitive to proteasomal inhibition, that this inhibition is additive with PKG inhibition, and that sporozoites with reduced PKG activity are more sensitive to proteasome inhibition than sporozoites with wild-type PKG. The regulation of cellular proteolysis by PKG may be evolutionarily conserved. Mammalian PKG regulates degradation of misfolded proteins in cardiomyocytes (45) via direct or indirect phosphorylation of the 19S regulatory particle of the proteasome (45, 46). Substrates of mammalian PKG in the proteasome have not been identified, but RPT6, a homolog of RPT1, is proposed as a potential target (45).
In this context, our findings that P. falciparum RPT1 physically interacts with the kinase domain of PfPKG and contains a consensus site for the kinase that is phosphorylated in vitro by PfPKG are intriguing. Characterization of the 26S proteasome in P. falciparum demonstrates stoichiometric incorporation of RPT1 (44), strongly suggesting that, as in higher eukaryotes, P. falciparum RPT1 is a critical component of the active proteasome. RPT1 is one of six AAA+ ATPases (RPT1 to RPT6) that are required for unfolding and translocation of the client proteins through the 19S regulatory particle prior to proteolysis by the catalytic 20S particle (47). The RPT1 homolog in Saccharomyces cerevisiae is phosphorylated at two Ser residues, but the responsible kinase(s) has not been identified (48, 49). Our data lead to a working hypothesis that PfPKG phosphorylation of RPT1 positively regulates proteasome function, possibly by promoting interactions between the 19S regulatory particle and client proteins. For example, phosphorylation could allosterically regulate RPT1 activity to promote substrate unfolding and translocation through the inner core of the proteasome, thereby regulating access of client proteins to the proteasome catalytic core. Alternatively, RPT1 phosphorylation could promote assembly of the 19S regulatory proteasome particle, as demonstrated for RPT6 in porcine cardiac tissue (50). Future work will focus on determining if RPT1 is a PKG substrate in vivo.
Our results also suggest interaction between PfPKG and PfHSP90. Interestingly, mammalian PKG regulates the activity of mammalian HSP90 (51). PfHSP90 contains several possible phosphorylation sites for PfPKG. While we did not detect phosphorylation of full-length protein in kinase assays, these data do not exclude phosphorylation of HSP90 by PfPKG in vivo, which may be preceded by additional posttranslational modifications absent in the in vitro-transcribed/translated protein. Alternatively, HSP90 could serve as a regulator of PfPKG activity. Indeed, mammalian HSP90 stabilizes and activates numerous client proteins that act in signal transduction pathways (51).
MATERIALS AND METHODS
Split luciferase assay.
PfPKGE706A (aa 409 to 853), PfPKGR781A (aa 409 to 853), PF3D7_1311500 (aa 114 to 420), and PF3D7_0708400 (aa 228 to 518), were fused to N- or C-terminal fragments of firefly luciferase using previously described Nfluc and Cfluc plasmids (15). Fusion proteins were expressed using a TNT SP6 High-Yield Wheat Germ Protein Expression System (Promega) according to the manufacturer’s protocol. Western blotting verified expression of Cfluc fusion proteins using anti-FLAG antibody (Sigma) and Nfluc fusion proteins using anti-N-terminal firefly luciferase (Santa Cruz Biotechnology) antibody. Equal volumes of Nfluc(-) and Cfluc(-) in vitro translation reactions were added to phosphate-buffered saline (PBS) supplemented with 1% bovine serum albumin (BSA) and protease inhibitors (Roche), incubated at 25°C for 2 h, and assayed for luciferase activity in duplicate or triplicate using a luminometer (BD Monolight 3010; BD Biosciences). To test the effect of peptides of luciferase reconstitution, peptides were added at 10-fold dilutions (0.71 to 714 μg/μl) to the incubation reaction mixture.
In vitro PKG activity assay.
PfPKG (aa 115 to 853) was amplified using the following primers: ATGACGATAAGGATCCAATGGG-AAAAGGTAGTTCTTTC and GCCAAGCTTCGAATTCTTAAAAATCTATGTCCCAGTTG. The PCR product was cloned (In-Fusion; Clontech) into pTrcHisB digested with BamHI and EcoRI. Recombinant protein was induced in BL21 cells at 18°C using 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and purified using Qiagen Ni-nitrilotriacetic acid (NTA) agarose according to the manufacturer’s protocol. Purified PfPKG was used in kinase reactions using 50 mM HEPES (pH 7.4), 10 mM MgCl2, 20 mM β-glycerophosphate, 2 mM dithiothreitol (DTT), 10 μM cGMP, 100 μM peptide, 0.1% BSA, 100 μM ATP, and 0.1 mM [32P]ATP (RPT1 and PB_090940 peptides, 3,000 Ci/mmol) or [33P]ATP (AKTIDE and BAD peptides, 0.3 μCi/μl) in the presence of absence of 50 nM TSP. The peptide concentration was 10 μM (AKTIDEs), 50 μM (BAD peptides), or 100 μM (RPT1 and PB_090940 peptides). Reaction mixtures were incubated at 30°C for 15 min. At 5-min intervals, 15-μl aliquots were removed and spotted onto P81 filter paper, which was washed, dried, and analyzed by scintillation counting as described previously (19).
Peptide library screening.
PfPKG peptide phosphorylation site specificity was determined using a peptide library (catalog no. 62017-1; Anaspec) consisting of 180 peptide mixtures with the general sequence YAXXXXXS/TXXXXAGKK-(biotin), in which 8 of the 9 X positions were a mixture of all 17 amino acids excluding Ser, Thr, and Cys (19). Within each mixture, a single X position was fixed as one of the 20 amino acids. In addition, three peptides were included in which all X positions were mixtures, but the central position was fixed as Ser, Thr, or Tyr. Peptide mixtures (50 μM) were arrayed in a 1,536-well plate in 2 μl of kinase reaction buffer (50 mM HEPES, pH 7.4, 10 mM MgCl2, 2 mM DTT, 20 mM β-glycerophosphate, 0.1% BSA, 0.1% Tween 20) per well. Reactions were initiated by adding PfPKG to each well in 200 nl of kinase reaction buffer containing 5 μM protein kinase inhibitor (PKI), 100 μM cGMP, and 0.55 mM [γ-33P]ATP (0.33 μCi/μl). Plates were incubated for 2 h at 30°C, after which 200-nl aliquots from each well were spotted onto streptavidin-coated membrane (Promega), which was washed as described, dried, and analyzed by phosphorimaging to measure the extent of radiolabel incorporation into each peptide.
Sporozoite invasion assay.
Anopheles stephensi mosquitoes were fed on infected Swiss-Webster mice. Mosquitoes infected with PbGFP-Luc, PKG-HA, and PKG T619Q-HA parasites were maintained at 20°C. Sporozoites were obtained at days 18 to 21 postfeeding through dissections of salivary glands.
HepG2 cells (obtained from ATCC) were cultured in Dulbecco’s modified Eagle medium (DMEM) (high glucose) supplemented with 10% fetal calf serum (FCS). Cells were seeded on collagen-coated multichambered slides for overnight growth at 37°C prior to addition of sporozoites. Invasion assays were performed using cells at 90% confluence, essentially as previously described (52). Sporozoites dissected in DMEM were pretreated with MG132, TSP, or vehicle for 30 min on ice in a volume of 20 μl. Sporozoites (4 × 104/well) were added to HepG2 cells in a final volume of 200 μl of DMEM supplemented with 10% FCS. Cells were fixed in 4% paraformaldehyde 1.5 h after addition of sporozoites. Cells were blocked in 1% BSA-PBS before incubation with anti-circumsporozoite (CS) antibody (3D11; 1 μg/ml) and anti-mouse Alexa 594. Cells were permeabilized with cold methanol, blocked, and incubated with 3D11 and anti-mouse Alexa 488. Extracellular sporozoites were quantified by determining the number of sporozoites that stained exclusively with anti-mouse Alexa 594. The total sporozoite number was quantified by determining the number of sporozoites immunostained with both Alexa 488 and Alexa 594. The effect of MG132 on sporozoite viability was determined by incubating MG132-treated sporozoites with propidium iodide (1 μg/ml) for 10 min. Viable sporozoites were identified as those excluding propidium iodide through microscopic examination.
Statistical analysis.
Statistical analysis was carried out using GraphPad Prism software, version 7.0.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge H. Li and M. Bogyo for helpful discussions.
This work was supported by grants from the National Science Foundation (IOS-1146221), National Institutes of Health (R21AI094167 and 5R01AI133633), and Department of Defense (W81XWH-13-1-0429) to P.B., the National Institutes of Health (R01 GM104047) to B.E.T., UGA startup funds to V.M., and NIH T32 AI060546 and an ARCS Foundation Award to H.M.K. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
P.B., B.E.T., and V.M. designed the experiments; P.B. and B.E.T. wrote the manuscript; K.G., R.K., M.S., H.J.L., and H.M.K. performed the experiments. P.D. conducted the bioinformatic analyses.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00523-18.
REFERENCES
- 1.WHO. 2016. World malaria report 2016. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Hill AV. 2011. Vaccines against malaria. Philos Trans R Soc Lond B Biol Sci 366:2806–2814. doi: 10.1098/rstb.2011.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim JJ, Flueck C, Franz E, Sanabria-Figueroa E, Thompson E, Lorenz R, Bertinetti D, Baker DA, Herberg FW, Kim C. 2015. Crystal structures of the carboxyl cGMP binding domain of the Plasmodium falciparum cGMP-dependent protein kinase reveal a novel capping triad crucial for merozoite egress. PLoS Pathog 11:e1004639. doi: 10.1371/journal.ppat.1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falae A, Combe A, Amaladoss A, Carvalho T, Menard R, Bhanot P. 2010. Role of Plasmodium berghei cGMP-dependent protein kinase in late liver stage development. J Biol Chem 285:3282–3288. doi: 10.1074/jbc.M109.070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Govindasamy K, Jebiwott S, Jaijyan DK, Davidow A, Ojo KK, Van Voorhis WC, Brochet M, Billker O, Bhanot P. 2016. Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol Microbiol 102:349–363. doi: 10.1111/mmi.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins CR, Hackett F, Strath M, Penzo M, Withers-Martinez C, Baker DA, Blackman MJ. 2013. Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog 9:e1003344. doi: 10.1371/journal.ppat.1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor HM, McRobert L, Grainger M, Sicard A, Dluzewski AR, Hopp CS, Holder AA, Baker DA. 2010. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot Cell 9:37–45. doi: 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McRobert L, Taylor CJ, Deng W, Fivelman QL, Cummings RM, Polley SD, Billker O, Baker DA. 2008. Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol 6:e139. doi: 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon RW, Taylor CJ, Bex C, Schepers R, Goulding D, Janse CJ, Waters AP, Baker DA, Billker O. 2009. A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS Pathog 5:e1000599. doi: 10.1371/journal.ppat.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, Schwach F, Chappell L, Gomes AR, Berriman M, Rayner JC, Baker DA, Choudhary J, Billker O. 2014. Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol 12:e1001806. doi: 10.1371/journal.pbio.1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brochet M, Billker O. 2016. Calcium signalling in malaria parasites. Mol Microbiol 100:397–408. doi: 10.1111/mmi.13324. [DOI] [PubMed] [Google Scholar]
- 12.Alam MM, Solyakov L, Bottrill AR, Flueck C, Siddiqui FA, Singh S, Mistry S, Viskaduraki M, Lee K, Hopp CS, Chitnis CE, Doerig C, Moon RW, Green JL, Holder AA, Baker DA, Tobin AB. 2015. Phosphoproteomics reveals malaria parasite protein kinase G as a signalling hub regulating egress and invasion. Nat Commun 6:7285. doi: 10.1038/ncomms8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE. 2005. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438:103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- 14.Deminoff SJ, Howard SC, Hester A, Warner S, Herman PK. 2006. Using substrate-binding variants of the cAMP-dependent protein kinase to identify novel targets and a kinase domain important for substrate interactions in Saccharomyces cerevisiae. Genetics 173:1909–1917. doi: 10.1534/genetics.106.059238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown HF, Wang L, Khadka S, Fields S, LaCount DJ. 2011. A densely overlapping gene fragmentation approach improves yeast two-hybrid screens for Plasmodium falciparum proteins. Mol Biochem Parasitol 178:56–59. doi: 10.1016/j.molbiopara.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldberg AL. 2012. Development of proteasome inhibitors as research tools and cancer drugs. J Cell Physiol 199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panchal D, Bhanot P. 2010. Activity of a trisubstituted pyrrole in inhibiting sporozoite invasion and blocking malaria infection. Antimicrob Agents Chemother 54:4269–4274. doi: 10.1128/AAC.00420-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurnett AM, Liberator PA, Dulski PM, Salowe SP, Donald RG, Anderson JW, Wiltsie J, Diaz CA, Harris G, Chang B, Darkin-Rattray SJ, Nare B, Crumley T, Blum PS, Misura AS, Tamas T, Sardana MK, Yuan J, Biftu T, Schmatz DM. 2002. Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites. A novel chemotherapeutic target. J Biol Chem 277:15913–15922. doi: 10.1074/jbc.M108393200. [DOI] [PubMed] [Google Scholar]
- 19.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma JL, Sheu YJ, Sassi HE, Sopko R, Chan CS, De Virgilio C, Hollingsworth NM, Lim WA, Stern DF, Stillman B, Andrews BJ, Gerstein MB, Snyder M, Turk BE. 2010. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal 3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell RD, Glass DB, Wong CW, Angelos KL, Walsh DA. 1995. Heat-stable inhibitor protein derived peptide substrate analogs: phosphorylation by cAMP-dependent and cGMP-dependent protein kinases. Biochemistry 34:528–534. doi: 10.1021/bi00002a018. [DOI] [PubMed] [Google Scholar]
- 21.Tegge W, Frank R, Hofmann F, Dostmann WR. 1995. Determination of cyclic nucleotide-dependent protein kinase substrate specificity by the use of peptide libraries on cellulose paper. Biochemistry 34:10569–10577. doi: 10.1021/bi00033a032. [DOI] [PubMed] [Google Scholar]
- 22.Wong A, Zhang YW, Jeschke GR, Turk BE, Rudnick G. 2012. Cyclic GMP-dependent stimulation of serotonin transport does not involve direct transporter phosphorylation by cGMP-dependent protein kinase. J Biol Chem 287:36051–36058. doi: 10.1074/jbc.M112.394726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. 1996. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399:333–338. doi: 10.1016/S0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. 2002. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol 9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 25.Belkina NV, Liu Y, Hao JJ, Karasuyama H, Shaw S. 2009. LOK is a major ERM kinase in resting lymphocytes and regulates cytoskeletal rearrangement through ERM phosphorylation. Proc Natl Acad Sci U S A 106:4707–4712. doi: 10.1073/pnas.0805963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C, Ha BH, Thevenin AF, Lou HJ, Zhang R, Yip KY, Peterson JR, Gerstein M, Kim PM, Filippakopoulos P, Knapp S, Boggon TJ, Turk BE. 2014. Identification of a major determinant for serine-threonine kinase phosphoacceptor specificity. Mol Cell 53:140–147. doi: 10.1016/j.molcel.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue L, Geahlen RL, Tao WA. 2013. Identification of direct tyrosine kinase substrates based on protein kinase assay-linked phosphoproteomics. Mol Cell Proteomics 12:2969–2980. doi: 10.1074/mcp.O113.027722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue L, Wang WH, Iliuk A, Hu L, Galan JA, Yu S, Hans M, Geahlen RL, Tao WA. 2012. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc Natl Acad Sci U S A 109:5615–5620. doi: 10.1073/pnas.1119418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. 2011. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braun KA, Vaga S, Dombek KM, Fang F, Palmisano S, Aebersold R, Young ET. 2014. Phosphoproteomic analysis identifies proteins involved in transcription-coupled mRNA decay as targets of Snf1 signaling. Sci Signal 7:ra64. doi: 10.1126/scisignal.2005000. [DOI] [PubMed] [Google Scholar]
- 32.Sacco F, Humphrey SJ, Cox J, Mischnik M, Schulte A, Klabunde T, Schafer M, Mann M. 2016. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat Commun 7:13250. doi: 10.1038/ncomms13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frohlich F, Olson DK, Christiano R, Farese RV Jr, Walther TC. 2016. Proteomic and phosphoproteomic analyses of yeast reveal the global cellular response to sphingolipid depletion. Proteomics 16:2759–2763. doi: 10.1002/pmic.201600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baum J, Richard D, Healer J, Rug M, Krnajski Z, Gilberger TW, Green JL, Holder AA, Cowman AF. 2006. A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J Biol Chem 281:5197–5208. doi: 10.1074/jbc.M509807200. [DOI] [PubMed] [Google Scholar]
- 35.Bane KS, Lepper S, Kehrer J, Sattler JM, Singer M, Reinig M, Klug D, Heiss K, Baum J, Mueller AK, Frischknecht F. 2016. The actin filament-binding protein coronin regulates motility in Plasmodium sporozoites. PLoS Pathog 12:e1005710. doi: 10.1371/journal.ppat.1005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones ML, Kitson EL, Rayner JC. 2006. Plasmodium falciparum erythrocyte invasion: a conserved myosin associated complex. Mol Biochem Parasitol 147:74–84. doi: 10.1016/j.molbiopara.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Siden-Kiamos I, Pinder JC, Louis C. 2006. Involvement of actin and myosins in Plasmodium berghei ookinete motility. Mol Biochem Parasitol 150:308–317. doi: 10.1016/j.molbiopara.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Baker RP, Wijetilaka R, Urban S. 2006. Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog 2:e113. doi: 10.1371/journal.ppat.0020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Ponder EL, Verdoes M, Asbjornsdottir KH, Deu E, Edgington LE, Lee JT, Kirk CJ, Demo SD, Williamson KC, Bogyo M. 2012. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem Biol 19:1535–1545. doi: 10.1016/j.chembiol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaMonte GM, Almaliti J, Bibo-Verdugo B, Keller L, Zou BY, Yang J, Antonova-Koch Y, Orjuela-Sanchez P, Boyle CA, Vigil E, Wang L, Goldgof GM, Gerwick L, O’Donoghue AJ, Winzeler EA, Gerwick WH, Ottilie S. 2017. Development of a potent inhibitor of the Plasmodium proteasome with reduced mammalian toxicity. J Med Chem 60:6721–6732. doi: 10.1021/acs.jmedchem.7b00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czesny B, Goshu S, Cook JL, Williamson KC. 2009. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob Agents Chemother 53:4080–4085. doi: 10.1128/AAC.00088-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, O’Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PCA, Bogyo M. 2016. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530:233–236. doi: 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gantt SM, Myung JM, Briones MR, Li WD, Corey EJ, Omura S, Nussenzweig V, Sinnis P. 1998. Proteasome inhibitors block development of Plasmodium spp. Antimicrob Agents Chemother 42:2731–2738. doi: 10.1128/AAC.42.10.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, Delahunty C, Fritz-Wolf K, Rahlfs S, Helena Prieto J, Yates JR, Becker K. 2015. Characterization of the 26S proteasome network in Plasmodium falciparum. Sci Rep 5:17818. doi: 10.1038/srep17818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. 2013. Protein kinase G positively regulates proteasome-mediated degradation of misfolded proteins. Circulation 128:365–376. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gillette TG, Hill JA. 2013. PKG primes the proteasome. Circulation 128:325–327. doi: 10.1161/CIRCULATIONAHA.113.003955. [DOI] [PubMed] [Google Scholar]
- 47.Collins GA, Goldberg AL. 2017. The logic of the 26S proteasome. Cell 169:792–806. doi: 10.1016/j.cell.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirano H, Kimura Y, Kimura A. 2016. Biological significance of co- and post-translational modifications of the yeast 26S proteasome. J Proteomics 134:37–46. doi: 10.1016/j.jprot.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Kikuchi J, Iwafune Y, Akiyama T, Okayama A, Nakamura H, Arakawa N, Kimura Y, Hirano H. 2010. Co- and post-translational modifications of the 26S proteasome in yeast. Proteomics 10:2769–2779. doi: 10.1002/pmic.200900283. [DOI] [PubMed] [Google Scholar]
- 50.Satoh K, Sasajima H, Nyoumura KI, Yokosawa H, Sawada H. 2001. Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 40:314–319. doi: 10.1021/bi001815n. [DOI] [PubMed] [Google Scholar]
- 51.Mollapour M, Neckers L. 2012. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta 1823:648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinnis P, De La Vega P, Coppi A, Krzych U, Mota MM. 2013. Quantification of sporozoite invasion, migration, and development by microscopy and flow cytometry. Methods Mol Biol 923:385–400. doi: 10.1007/978-1-62703-026-7_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. 2005. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res 33:W389–W392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.