

Summary
Good’s syndrome (thymoma and hypogammaglobulinaemia) is a rare secondary immunodeficiency disease, previously reported in the published literature as mainly individual cases or small case series. We use the national UK‐Primary Immune Deficiency (UKPID) registry to identify a large cohort of patients in the UK with this PID to review its clinical course, natural history and prognosis. Clinical information, laboratory data, treatment and outcome were collated and analysed. Seventy‐eight patients with a median age of 64 years, 59% of whom were female, were reviewed. Median age of presentation was 54 years. Absolute B cell numbers and serum immunoglobulins were very low in all patients and all received immunoglobulin replacement therapy. All patients had undergone thymectomy and nine (12%) had thymic carcinoma (four locally invasive and five had disseminated disease) requiring adjuvant radiotherapy and/or chemotherapy. CD4 T cells were significantly lower in these patients with malignant thymoma. Seventy‐four (95%) presented with infections, 35 (45%) had bronchiectasis, seven (9%) chronic sinusitis, but only eight (10%) had serious invasive fungal or viral infections. Patients with AB‐type thymomas were more likely to have bronchiectasis. Twenty (26%) suffered from autoimmune diseases (pure red cell aplasia, hypothyroidism, arthritis, myasthenia gravis, systemic lupus erythematosus, Sjögren’s syndrome). There was no association between thymoma type and autoimmunity. Seven (9%) patients had died. Good’s syndrome is associated with significant morbidity relating to infectious and autoimmune complications. Prospective studies are required to understand why some patients with thymoma develop persistent hypogammaglobulinaemia.
Keywords: agammaglobulinaemia, autoimmunity, bronchiectasis, Good’s syndrome, thymoma
Introduction
Thymomas are rare tumours of thymic epithelium, originally derived from the endodermal lining of the third pharyngeal pouch. They have recently been linked to mutations in the thymoma‐specific transcription factor regulating growth factor signalling GTF2I and, to a lesser extent, HRAS, NRAS and TP53 1, 2. Thymomas typically present in adults aged 40–60 years, with only a few cases described in children 3. Approximately half the tumours are encapsulated, and only 10% relapse after surgery 4.
Thymomas commonly trigger paraneoplastic autoimmune complications affecting the nervous system and bone marrow precursors 4, 5. Disorders of the nervous system are secondary to autoantibodies disrupting neural transmission, e.g. anti‐acetylcholine receptor antibodies in myasthenia gravis (occurring in 17–40% of patients with thymoma); anti‐glutamic acid decarboxylase (GAD65) and anti‐glycine receptor antibodies in Stiff‐person syndrome; anti‐Kv1 Voltage‐Gated K+ Channels (VGKC) or anti‐Contactin‐associated protein‐like 2 (Caspr2) antibodies in acquired neuromyotonia and autoimmune encephalitis; and anti‐P/Q‐type Voltage‐Gated Calcium Channels (VGCC) antibodies in Lambert–Eaton syndrome and autoimmune encephalitis 6. The most common disorders affecting bone marrow precursors are pure red cell aplasia (PRCA) and ‘pure B cell aplasia’ (Good’s syndrome: thymoma and hypogammaglobulinaemia) (3–4% of patients). A few patients have also been described with agranulocytosis, myelodysplasia or paroxysmal nocturnal haemoglobinuria 7, 8, 9. The pathogenesis of these complications is thought to involve either the production of autoantibodies, e.g. anti‐erythropoetin antibodies in PRCA, or destruction of bone marrow precursors by CD8 T lymphocytes in PRCA and Good’s syndrome 10, 11, 12, 13, 14.
Since the original description by Dr Robert Good more than 60 years ago, most publications of Good’s syndrome are single case reports or small case series, the previous largest being a systematic review of 47 Chinese patients 15, and a compilation of 152 cases from cases published prior to 2010 10. The aim of our survey was to review the clinical and laboratory features of patients with Good’s syndrome identified through the UK‐PIN immunodeficiency registry and investigate what factors may be associated with specific complications and outcome.
Methods
Patient definition and identification
Patients with thymoma were identified from 14 clinical immunology referral centres across the United Kingdom contributing patients to the national UK‐Primary Immune Deficiency (UKPID) registry. All had associated hypogammaglobulinaemia consistent with a diagnosis of Good’s syndrome. This registry was established in 2009 and recruited patients prospectively after obtaining written consent. Permission from principal investigators of each centre is required before their data can be accessed and studied and those centres with registered cases of Good’s syndrome were approached. All responded and consented to take part in the study. The registry currently contains 4758 patients with primary immunodeficiency diseases, 78 (1.6%) of whom are classified as having thymoma and hypogammaglobulinaemia. As patients were added to the UKPID registry prospectively as part of regular patient care, mortality data are limited to those dying during the last 9 years.
The study had multi‐centre ethical approval (04/MRE07/68). After requesting permission from Centre Principle Investigators, data were collated into a single spss database (IBM SPSS Statistics 22.0) for analysis. Clinical information included demographics, past medical history of acute or recurrent respiratory tract infections and non‐infectious complications. UKPID registry entries are updated on an annual basis to ensure that subsequent morbidity and mortality, as well as latest laboratory parameters, are recorded. For the purposes of this survey, registry entries were rechecked with source data by M.Z., P.D.A. and centre investigators and where possible missing data points were filled. Clinical complications for which data were available included respiratory tract infections (RTI), myasthenia gravis, pure red cell aplasia and hypogammaglobulinaemia. Therapies, including surgery, radiotherapy and chemotherapy for the thymoma, and immunoglobulin replacement therapy for the antibody deficiency were also recorded.
Laboratory investigations
Laboratory data included routine haematology [haemoglobin, white blood count (WBC), absolute neutrophil and lymphocyte counts, and platelets] and immunology [lymphocyte subsets: absolute CD3, CD4, CD8, CD19, CD56 and serum immunoglobulin (Ig) concentrations] recorded at diagnosis and on immunoglobulin replacement therapy. Lymphocyte subsets were assessed using standard immunofluorescent staining and flow cytometry was performed in accredited regional clinical immunology laboratories. Proliferative T cell responses were not requested. Serum immunoglobulin concentrations were measured by nephelometry. Vaccine responses were not requested on the majority of patients, as serum immunoglobulin concentrations were so low. Thymomas were classified histologically according to the World Health Organization (WHO) classification of tumours of the thymus 16.
Statistical analysis
Analysis was conducted using the IBM spss Statistics version 22.0 program. Continuous variables are quoted as medians and interquartile ranges. Statistical differences between groups were determined by χ2, Kruskal–Wallis and Mann–Whitney U‐tests. Spearman’s rho test was used to assess the correlation between continuous variables. Differences were considered statistically significant with a P‐value < 0·05 unless otherwise stated.
Results
Demographics of the cohort
Data were collected from 78 patients with thymoma and hypogammaglobulinaemia (Good’s syndrome) with a median age at evaluation of 64 years (range = 24–90). There were 32 males (41%) and 46 females (59%), with no reported familial cases. Seventy‐six (97%) patients were white European. Fifty‐five (71%) were from England, 14 (18%) from Scotland, five (6%) from Northern Ireland and four (5%) from Wales.
Clinical features (Table 1)
Table 1.
Demographic and clinical parameters
| Parameter | |
|---|---|
| Age (years) | 64 (58–71) |
| Gender | 46 (59%) female |
| Ethnicity | 76 (97%) white European |
| Age of onset of symptoms (years) | 54 (48–60) |
| Age at diagnosis of hypogammaglobulinaemia (years) | 58 (51–62) |
| Age of diagnosis of thymoma (years) | 58 (51–62) |
| Clinical features | |
| Bronchiectasis (on chest CT scan) | 35 (45%) |
| Chronic sinusitis | 7 (9%) |
| Autoimmunity | 20 (26%) |
| Red cell aplasia | 8 (10%) |
| Thyroid | 6 (8%) |
| Arthritis | 4 (5%) |
| Myasthenia | 3 (4%) |
| SLE | 3 (4%) |
| Sjögren’s disease | 2 (3%) |
| Died | 7 (9%) |
Figures represent number (percentage) for discrete variables and median (interquartile range) for continuous variables. SLE = systemic lupus erythematosus; CT = computerized tomography.
The median age at which symptoms appeared was 54 (interquartile range = 48–60) years. Median age of diagnosis of hypogammaglobulinaemia [58 (51–62) years] and thymoma [58 (51–62) years] were the same and a median of 4 years later. In 58 (74%) patients, the diagnosis of hypogammaglobulinaemia and thymoma were concurrent. In nine (12%) patients, the diagnosis of hypogammaglobulinaemia was 1–8 years prior to finding the thymoma and in 11 (14%) patients hypogammaglobulinaemia was diagnosed 1–6 years after finding the thymoma. In 74 (94%) patients the initial symptoms were recurrent sinopulmonary infections, with 35 (45%) having radiological evidence of bronchiectasis. Seven patients (9%) had chronic sinusitis. Three patients suffered from candidiasis, three from retinal cytomegalovirus (CMV) and toxoplasmosis, one from persistent warts and one died of progressive multi‐focal encephalopathy. Only two patients had problems with diarrhoea, one of whom was diagnosed with ulcerative colitis.
Autoimmunity was documented in 20 patients (26%), with pure red cell aplasia in eight (10%), hypothyroidism in six (8%), inflammatory arthritis in four (5%), myasthenia gravis in three (4%), systemic lupus erythematosus (SLE) in three (4%) and Sjögren’s syndrome in two (3%). Patients with red cell aplasia were not more likely to have myasthenia gravis or hypothyroidism. All the patients with hypothyroidism were female, while for the other autoimmune disorders there was no significant propensity to the female gender. The age at diagnosis of the patients with bronchiectasis or autoimmunity were not significantly different from patients without these complications (Fig. 1).
Figure 1.
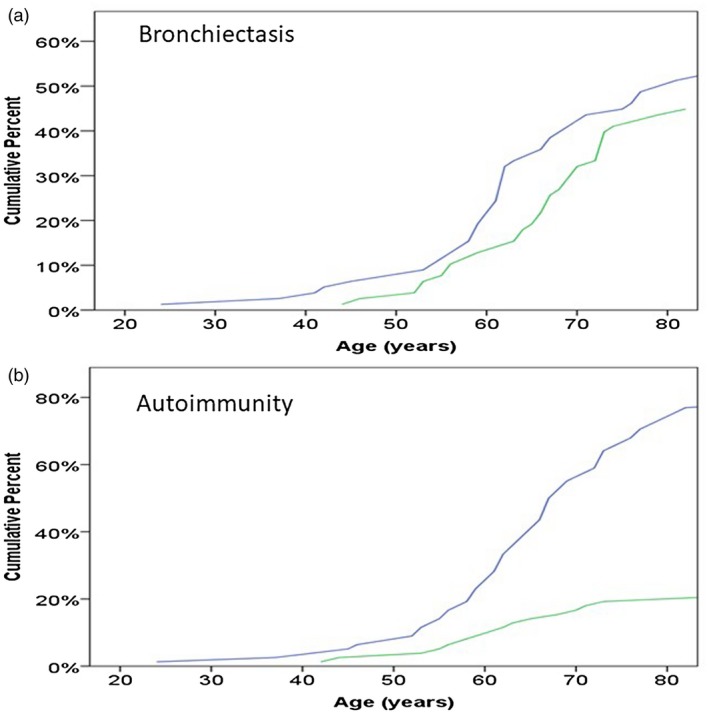
Age of diagnosis (years) in relation to whether or not the patients had (a) bronchiectasis or (b) autoimmune disease. Blue line = no disease; green line = disease.
Six patients (8%) had additional non‐thymic tumours (two basal cell carcinoma, one renal cell carcinoma, one breast carcinoma, one cervical carcinoma and one lung cancer). There was no significant association between thymic and extra‐thymic cancer. Seven (9%) patients had died, two of metastatic thymoma, two of bacterial sepsis, one of progressive multi‐focal encephalopathy and the causes of death in the other two (one of whom was aged 90 years old) were unclear.
Investigations
Chest computerized tomography (CT) scan was the preferred diagnostic procedure to identify both the thymoma and assess lung disease. Pathology and staging reports were available for 46 (59%) of the 78 patients (Table 2), as some patients had their surgery performed at a different hospital, and unfortunately pathology results were not relayed to the primary physician. Nine patients (12%) had evidence of thymic carcinoma, four locally invasive and five distally disseminated. Sixteen of 27 (59%) patients with stage AB disease had bronchiectasis compared with only three of 16 (19%) patients with other thymoma types (P = 0·02). There was no correlation between thymoma type and autoimmune complications.
Table 2.
Staging and pathology of thymoma
| Stage | ||
|---|---|---|
| I | Completely encapsulated | 32 (66%) |
| II | Invasion through thymic capsule | 8 (16%) |
| III | Local invasion into neighbouring organs | 4 (8%) |
| IV | Pleural/pericardial disseminationLymphogenous/hematogenous spread | 5 (10%) |
| Pathology | ||
| A | Medullary, spindle cell thymoma | 4 (9%) |
| AB | Mixed thymoma | 27 (59%) |
| B1–3 | Cortical thymoma (B3: well‐differentiated thymic carcinoma | 6 (13%) |
| C | Thymic carcinoma | 9 (19%) |
Pathology reports were available for 46 of the 78 patients. Based on Masaoka16.
Haematology and immunology results are shown in Table 3. Absolute B cell (CD19) numbers were less than 0·0 × 109/l, and serum immunoglobulin (Ig)M, IgG (prior to starting immunoglobulin replacement therapy) and IgA were well below the normal reference ranges. Patients with malignant thymomas (stage IV, classification C) had significantly lower absolute CD3+CD4+ counts compared with non‐malignant tumours (Table 4, Fig. 2). In fact, all patients with stage IV disease had absolute CD4 counts < 0·25 × 109/l compared with only 15% of patients with stages I–III. CD4/CD8 ratios did not vary significantly between the histological groups (P = 0·1). Neither stage nor absolute T lymphocyte count were significantly associated with autoimmunity.
Table 3.
Laboratory parameters
| Parameter | Result | Normal range |
|---|---|---|
| Haemoglobin (g/l) | 133 (125–145) | 115–165 |
| WBC (× 109/l) | 6·1 (4·3–8·6) | 4·0–11· 0 |
| Neutrophils (× 109/l) | 3·8 (2·4–6·0) | 1·80–7·50 |
| Lymphocytes (× 109/l) | 1·5 (0·9–2·5) | 1·00–4·00 |
| Platelets (× 109/l) | 250 (177–328) | 150–400 |
| CD3 (× 109/l) | 1·08 (0·53–2·05) | 0·62–2·40 |
| CD4 (× 109/l) | 0·53 (0·31–0·70) | 0·50–1·50 |
| CD8 (× 109/l) | 0·52 (0·20–1·06) | 0·19–0·90 |
| CD4/CD8 ratio | 0·93 (0·62–1·38) | 0·72–2·56 |
| CD19 (× 109/l) | 0·00 (0·00–0·00) | 0·12–0·64 |
| CD56 (× 109/l) | 0·11 (0·00–0·11) | 0·02–0·41 |
| IgM (g/l) | 0·09 (0·04–0·20) | 0·50–2·0 |
| IgG (g/l) | 2·92 (1·85–4·10) | 6·0–16·0 |
| IgA (g/l) | 0·20 (0·09–0·40) | 0·8–2·80 |
Numbers for continuous variables represent medians (interquartile ranges). Figures in red indicate that they are lower than the stated normal range. Immunoglobulin (Ig)G measurements were all before patients started on immunoglobulin replacement.
Table 4.
Clinical and laboratory parameters based on World Health Organization (WHO) histological type
| Parameter | A | AB | B | C | P‐value |
|---|---|---|---|---|---|
| Number | 4 | 27 | 6 | 9 | |
| Age (years) | 62 (62–69) | 64 (56–73) | 67 (55–78) | 59 (54–62) | 0·1 |
| Age onset | 54 (33–61) | 54 (44–60) | 60 (52–69) | 52 (40–56) | 0·1 |
| Females | 2 (50%) | 20 (74%) | 4 (67%) | 4 (44%) | 0·4 |
| Abs lymphs | 2·5 (1·9–3·2) | 1·6 (1·1–2·5) | 2·3 (1·0–2·6) | 0·8 (0·5–0·9) | 0·003 |
| Abs CD3 | 2·8 (2·3–4·1) | 1·1 (0·8–2·2) | 0·7 (0·4–1·7) | 0·7 (0·4–0·9) | 0·02 |
| Abs CD4 | 0·6 (0·3–1·0) | 0·6 (0·4–1·0) | 0·4 (0·2–) | 0·2 (0·1–0·3) | 0·02 |
| Abs CD8 | 1·4 (0·3–3·5) | 0·7 (0·2–1·5) | 0·2 (0·1–) | 0·3 (0·2–0·5) | 0·3 |
| Autoimmune | 1 (25%) | 6 (22%) | 1 (17%) | 4 (44%) | 0·6 |
| Myasthenia | 1 (25%) | 1 (4%) | 0 | 1 (11%) | 0·3 |
| RBC aplasia | 3 (9%) | 2 (7%) | 0 | 3 (33%) | 0·1 |
| Bronchiectasis | 1 (25%) | 16 (59%) | 0 | 2 (22%) | 0·02 |
Figures for continuous variables represent medians (interquartile ranges). Immunoglobulin (Ig)G measurements were all before patients started on immunoglobulin replacement. Statistical differences between groups determined by Kruskal–Wallis U‐test for continuous variables or χ2 test for discrete variables. Abs = absolute; RBC = red blood cells.
Figure 2.
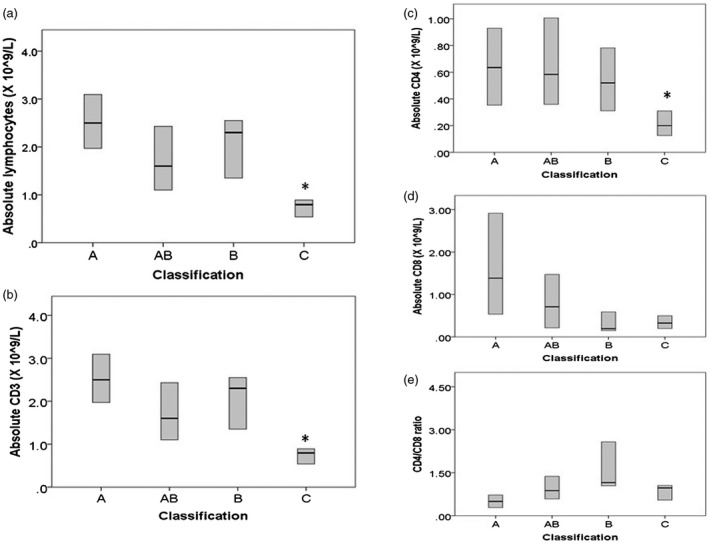
Association between World Health Organization (WHO) histology type and absolute blood (a) lymphocyte, (b) CD3 count, (c) CD4 count, (d) CD8 count, (e) CD4/CD8 ratio. Box plots represent median and interquartile ranges. *P < 0·05 by Kruskal–Wallis and Mann–Whitney U‐tests.
Management
All patients had undergone thymectomy and were on replacement immunoglobulin replacement therapy, administered either subcutaneously or intravenously. In addition to surgical resection, six patients were treated with radiotherapy, four with chemotherapy and three with both radiotherapy and chemotherapy.
Discussion
We present the largest primary case series of Good’s syndrome published to date. A review of 152 collated cases published in the literature prior to 2010 10 is the only larger series, collecting previously published cases. The age of onset of symptoms of the 78 UK patients ranged from 24 to 90 years and there were no children, in keeping with previous studies. Diagnosis of the thymoma and hypogammaglobulinaemia was concurrent in three‐quarters of the patients in this cohort. This might be expected, as all the patients were recruited to the UKPID registry by clinical immunologists caring for the patients’ hypogammaglobulinaemia. Chest symptoms are common in hypogammaglobulinaemia and chest radiographs would be part of the work‐up for these patients, leading to discovery of the mediastinal mass. Patients came from around the United Kingdom, with no specific ‘hot‐spots’. Most patients (94%) presented with infections and nearly half (45%) had radiological evidence of bronchiectasis. Although there were numerical reductions of T cells in a number of patients, infections usually associated with T cell deficiencies were not a prominent feature of this cohort, which is at odds with reported smaller studies 17, 18. Three patients had candidiasis, but antibiotics may have contributed to this problem. Two had problems with retinal CMV and toxoplasmosis and one died of progressive multi‐focal encephalopathy. Diarrhoea was not a prominent problem of this cohort, in contrast to another case series where it occurred in 38% 10. Twenty‐three per cent had evidence of other autoimmune disorders, the most commonly being PRCA (9%), with only 4% having myasthenia gravis, the predominant autoimmune disorder overall in patients with thymoma. Two patients (3%) had a significant neutropaenia with an absolute neutrophil counts less than 1·0 × 109/l. Eleven patients (15%) had a low platelet count of between 40 and 130 × 109/l. These findings might suggest that similar immune mechanisms underlie disorders of bone marrow precursors.
As with previous studies, the most common histological type was AB, with 58% having a moderate infiltrate of thymocytes 4, 10. The majority of tumours were benign, with only 19% spreading outside the capsule. This cohort seemed to have a more benign course than previously published series of thymoma patients not selected on the basis of hypogammaglobulinaemia, where more than half of patients’ tumours were invasive and 28% of patients had died 19, 20. In our cohort, only nine patients (11%) had thymic carcinoma and seven patients (9%) had died, two from their metastatic thymoma and three from infections. As with our previous study of patients with idiopathic hypogammaglobulinaemia, we found an association between a malignant phenotype and low T lymphocyte numbers, which might suggest a lack of immune surveillance in these patients 21.
A limitation of this study is that histology was only available for 46 of the 78 patients, as in a number of cases the thymectomy was carried out at a different centre and the histological report was not transferred to the immunologist looking after the patient.
In contrast to patients with common variable immunodeficiency (CVID), a prominent feature of patients with Good’s syndrome is the almost complete absence of B cells. Our understanding of the pathogenesis of Good’s syndrome is still very much in its infancy. Unlike patients with thymomas suffering from myasthenia gravis, where a third improve after thymectomy, we observed no improvement in the hypogammaglobulinaemia or B cell deficiency in our patients with Good’s syndrome after thymectomy 22. Research studies using bone marrow immunophenotyping are required to examine the stage at which B cell development is being disrupted 23. Prospectively, this could best be performed alongside thoracic surgery, reducing the need for additional hospital admissions and the discomfort of the procedure. In view of previous published findings which show over‐expression of specific CD8 clones by spectrotyping analysis, CD8 T cell phenotyping in bone marrow aspirates should also be performed to determine to what extent clonal proliferation of cytotoxic T cells was involved in the pathogenesis 11, 12. Additionally, serum should be investigated for evidence of autoimmunity as a cause of immunodeficiency. This may manifest, for instance, as anti‐cytokine antibodies or B cell precursor destruction.
A clearer understanding of the immune mechanisms underlying Good’s syndrome may not only suggest more focused therapies for this condition, but may also promote the development of novel drug targets in patients with antibody‐mediated autoimmune disorders and patients who have become resistant to biologicals because of neutralizing antibodies.
Disclosures
UKPIN receives sponsorship and financial support from Baxter, Biotest, BPL, CSL Behring, Grifols, Octapharma and Shire.
Author contributions
P.D.A. conceived of and led the study. Each regional centre lead was in charge of recruiting patients with idiopathic hypogammaglobulinaemia into the UKPID registry. All authors contributed to the writing of the manuscript and approved the final version.
Acknowledgements
This study was funded by a Rare Diseases Translational Research Collaboration Grant from the National Institute for Health Research.
References
- 1. Radovich M, Pickering CR, Felau I et al The integrated genomic landscape of thymic epithelial tumors. Cancer Cell 2018;33:244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrini I, Meltzer PS, Kim IK et al A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet 2014;46:844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yalçin B, Demir HA, Ciftçi AO et al Thymomas in childhood: 11 cases from a single institution. J Pediatr Hematol Oncol 2012;34:601–5. [DOI] [PubMed] [Google Scholar]
- 4. Maggi G, Casadio C, Cavallo A, Cianci R, Molinatti M, Ruffini E. Thymoma: results of 241 operated cases. Ann Thorac Surg 1991;51:152–6. [DOI] [PubMed] [Google Scholar]
- 5. Verstandig AG, Epstein DM, Miller WT Jr, Aronchik JA, Gefter WB, Miller WT. Thymoma – report of 71 cases and a review. Crit Rev Diagn Imaging 1992;33:201–30. [PubMed] [Google Scholar]
- 6. Evoli A, Lancaster E. Paraneoplastic disorders in thymoma patients. J Thorac Oncol 2014;9(Suppl 2):S143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen LP, Tsai JS, Lai WM, Yen LJ, Yu MS, Lin SJ. Myelodysplasia followed by Good’s syndrome: a unique manifestation associated with thymoma. Kaohsiung J Med Sci 2012;28:236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yip D, Rasko JE, Lee C, Kronenberg H, O’Neill B. Thymoma and agranulocytosis: two case reports and literature review. Br J Haematol 1996;95:52–6. [DOI] [PubMed] [Google Scholar]
- 9. Palmieri G, Selleri C, Montella L et al Thymoma followed by paroxysmal nocturnal hemoglobinuria: a unique clinical association in the context of multiorgan autoimmunity with a potential role for CD8+ T lymphocytes. Am J Hematol 2006;81:774–8. [DOI] [PubMed] [Google Scholar]
- 10. Kelesidis T, Yang O. Good’s syndrome remains a mystery after 55 years: A systematic review of the scientific evidence. Clin Immunol 2010;135:347–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masci AM, Palmieri G, Vitiello L et al Clonal expansion of CD8+ BV8 T lymphocytes in bone marrow characterizes thymoma‐associated B lymphopenia. Blood 2003;101:3106–8. [DOI] [PubMed] [Google Scholar]
- 12. Asherson GL, Johnson S, Platts‐Mills TA, Webster AD. Pathogenesis of hypogammaglobulinaemia with thymoma and late‐onset hypogammaglobulinaemia. J Clin Pathol Suppl (R Coll Pathol) 1979;13:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geary CG, Byron PR, Taylor G, MacIver JE, Zervas J. Thymoma associated with pure red cell aplasia, immunoglobulin deficiency and an inhibitor of antigen‐induced lymphocyte transformation. Br J Haematol 1975;29:479–85. [DOI] [PubMed] [Google Scholar]
- 14. Marmont A, Peschle C, Sanguineti M, Condorelli M. Pure red cell aplasia (PRCA): Response of three patients of cyclophosphamide and/or antilymphocyte globulin (ALG) and demonstration of two types of serum IgG inhibitors to erythropoiesis. Blood 1975;45:247–61. [PubMed] [Google Scholar]
- 15. Dong JP, Gao W, Teng GG, Tian Y, Wang HH. Characteristics of Good’s Syndrome in China: A Systematic Review. Chin Med J (Engl) 2017;130:1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marx A, Chan JK, Coindre JM et al The 2015 World Health Organization classification of tumors of the thymus: continuity and changes. J Thorac Oncol 2015;10:1383–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun X, Shi J, Wang M, Xu K, Xiao Y. Good’s syndrome patients hospitalized for infections: a single‐center retrospective study. Medicine (Balt) 2015;94:e2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malphettes M, Gérard L, Galicier L et al Good’s syndrome: an adult‐onset immunodeficiency remarkable for its high incidence of invasive infections and autoimmune complications. Clin Infect Dis 2015;61:e13–9. [DOI] [PubMed] [Google Scholar]
- 19. Weissferdt A, Kalhor N, Bishop JA et al Thymoma: a clinicopathological correlation of 1470 cases. Hum Pathol 2018;73:7–15. [DOI] [PubMed] [Google Scholar]
- 20. Yuan ZY, Gao SG, Mu JW et al Long‐term outcomes of 307 patients after complete thymoma resection. Chin J Cancer 2017;36:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brent J, Guzman D, Bangs C et al Clinical and laboratory correlates of lung disease and cancer in adults with idiopathic hypogammaglobulinaemia. Clin Exp Immunol 2016;184:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaufman AJ, Palatt J, Sivak M et al Thymectomy for myasthenia gravis: complete stable remission and associated prognostic factors in over 1000 cases. Semin Thorac Cardiovasc Surg 2016;28:561–68. [DOI] [PubMed] [Google Scholar]
- 23. Anzilotti C, Kienzler AK, Lopez‐Granados E et al Key stages of bone marrow B‐cell maturation are defective in patients with common variable immunodeficiency disorders. J Allergy Clin Immunol 2015;136:487–40. [DOI] [PubMed] [Google Scholar]