

Summary
Enteroviruses (EV) have been historically associated to type 1 diabetes. Definitive proof for their implication in disease development is lacking, but growing evidence suggests that they could be involved in beta cell destruction either directly by killing beta cells or indirectly by creating an exacerbated inflammatory response in the islets, capable of attracting autoreactive T cells to the ‘scene of the crime’. Epidemiological and serological studies have been associated with the appearance of islet autoimmunity and EV RNA has been detected in prospective studies. In addition, the EV capsid protein has been detected in the islets of recent‐onset type 1 diabetic donors, suggesting the existence of a low‐grade EV infection that could become persistent. Increasing evidence in the field shows that a ‘viral signature’ exists in type 1 diabetes and involves interferon responses that could be sustained during prolonged periods. These include the up‐regulation of markers such as protein kinase R (PKR), melanoma differentiation‐associated protein 5 (MDA5), retinoic acid inducible gene I (RIG‐I), myxovirus resistance protein (MxA) and human leukocyte antigen‐I (HLA‐I) and the release of chemokines able to attract immune cells to the islets leading to insulitis. In this scenario, the hyperexpression of HLA‐I molecules would promote antigen presentation to autoreactive T cells, favoring beta cell recognition and, ultimately, destruction. In this review, an overview is provided of the standing evidence that implicates EVs in beta cell ‘murder’, the time‐line of events is investigated from EV entry in the cell to beta cell death and possible accomplices are highlighted that might be involved in beta cell demise.
Keywords: beta cell destruction, interferon response, type 1 diabetes, virus infection
1. Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by the loss of insulin‐producing beta cells in the pancreas. The trigger(s) is still unknown. Genetic predisposition and environmental factors play an important role, acting as candidate triggers of islet autoimmunity. Viruses, and especially enteroviruses (EV), have been examined for their potential implication in T1D pathogenesis. The association between EVs and T1D has, on many occasions, been a matter of debate for the scientific community. This division between supporters and opponents has pushed the field forward, and interesting studies looking at the possible presence of EVs in the pancreas have been published during recent years. EVs are known to infect beta cells, which situates them as ‘possible suspects in beta cell murder’. However, other factors, such as the host‐immune response to the infection, could play an important role in beta cell demise through the stimulation of immune autoreactivity, first triggered and then maintained by potential persistent infections in the pancreas 1. Thus, EVs might not act alone and the establishment of an inflammatory environment could contribute to beta cell destruction. A careful examination of the ‘scene of the crime’ might therefore be necessary to uncover hidden evidence. Despite the cumulative proof of an association of EVs with T1D, causal evidence is still lacking and remains a challenge for the field. Access to human samples at different stages of the disease as well as the use of new technologies to study them will be fundamental in order to find it. Co‐ordinated efforts carried out by specialized working groups such as the Network for Pancreatic Organ Donors with Diabetes (nPOD)‐Virus group or the Persistent Virus Infection in Diabetes Network (PEVNET) are currently investigating the possible presence or absence of EVs in human samples as well as the host‐immune response, and are already performing groundbreaking research in the field. In this review we will focus on the most recent findings pertaining EV infections, the host response to a potential infection and possible interventions aimed to find definitive proof implicating these pathogens in T1D development.
1.1. There has been a murder: can we find the weapon?
Enteroviruses are non‐enveloped single‐stranded, positive sense RNA viruses that belong to the Picornaviridae family. Their genome has a 5′‐untranslated region (5′UTR), a coding region that is translated into a single polyprotein, and a 3′UTR with a polyadenylated tail (3′UTR) 2 (Fig. 1). The coding region is translated into 11 proteins comprising the structural proteins (VP1‐VP4, for capsid formation), the polymerase (for replication) and proteases (for polyprotein cleavage) (Fig. 1) 3, 4. These viruses are transmitted by the fecal–oral and respiratory routes, with an initial replication phase in the gastrointestinal or the upper respiratory tract, respectively 4. After this initial replication, and if the appropriate cellular receptors are present, they can spread to other organs. EVs enter the cell through to the decay accelerating factor (DAF) 5 and the Coxsackie‐adenovirus receptor (CAR) 6. The former has not been detected in the pancreas, but CAR is expressed in islet cells 7, representing a potential entrance for EVs to beta cells. Recent studies suggest that both alpha and beta cells express CAR but that EV replication is less efficient in alpha cells, due to a more efficient blocking of protein translation 8, which stops viral production. In addition, interferon (IFN) and signal transducer and activator of transcription 1 (STAT‐1)‐dependent genes are expressed at a higher level in alpha cells compared to beta cells, allowing them to rapidly eliminate the virus (Fig. 2). Conversely, beta cells present a more sustained EV protein and IFN‐induced markers expression, reflecting a more chronic and non‐cytolytic infection 8. These differences in viral clearance between alpha and beta cells correlate well with the absence of VP1 positivity in alpha cells in the pancreas of patients with T1D 9. In an analysis of the distribution of VP1‐positive cells between insulin‐deficient islets (IDIs) and insulin‐containing islets (ICIs) in 10 patients with frequent VP1‐positive endocrine cells, Richardson and colleagues reported that 78·7% of the ICIs were found to be positive for VP1. By contrast, only 2·6% of the IDIs showed evidence of VP1 10, suggesting that beta cells might fail to control viral replication and could harbor virus particles for a long time. Additionally, beta cells can harbor a persistent infection for up to 30 days post‐infection in intact human islets 11 and non‐cytopathic slow replicating viruses harboring a deletion at the 5′ terminus region have been detected in persistently infected non‐obese diabetic (NOD) mice 12. Therefore, after an initial replication phase, a non‐cytopathic, persistent infection could be established in some beta cells.
Figure 1.
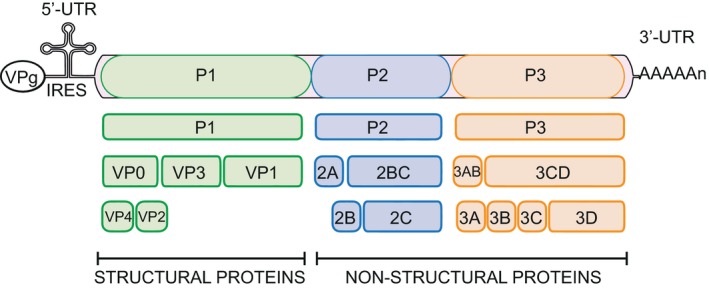
Schematic representation of the enterovirus genome. At the 5′ end the RNA contains an internal ribosomal entry site for cap‐dependent translation (IRES) and is covalently linked to the tyrosine‐3 residue of a small virus‐encoded peptide (VPg), which is used as a primer for RNA replication. The 3′ end has a polyadenylated tail [poly(A)] and contains an untranslated region (UTR). The genome encodes a single polyprotein that is cleaved into P1, P2 and P3. From the precursor P1, the capsid proteins VP0, VP3 and VP1 are generated. The non‐structural proteins 2A (protease) and 2ABC are formed from P2. Finally, P3 yields the production of 3AB and 3CD. The final processing steps lead to the production of VP4 and VP2 (capsid proteins), 2B (increases membrane permeability and inhibits cellular secretory pathways), 2C (vesicle formation), 3A (inhibits intracellular transport), 3B (VPg, primes RNA synthesis), 3C (protease) and 3D (polymerase).
Figure 2.
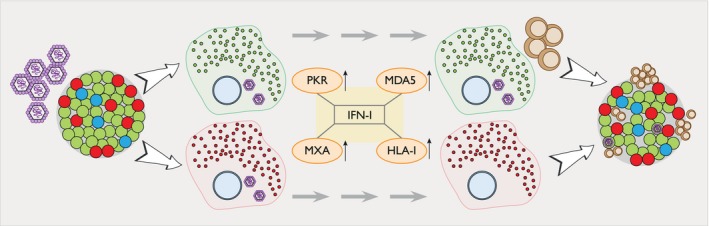
Beta cells might fail to control enterovirus (EV) replication. EVs are able to replicate in both alpha and beta cells. Once the virus has entered the cells through its interaction with Coxsackie‐adenovirus receptor (CAR, viral RNA is sensed by protein kinase R (PKR) and melanoma differentiation‐associated protein 5 (MDA5), which induce the production of interferon (IFN)‐I and the creation of an anti‐viral state in the cell. This includes the up‐regulation of myxovirus resistance protein (MxA) (increasing IFN production) and islet human leukocyte antigen‐I (HLA‐I) expression. Due to a more efficient blocking of protein translation 8, alpha cells are able to stop viral production. In addition, IFN and other IFN‐related molecules are expressed at a higher level in alpha cells compared to beta cells, allowing them to rapidly eliminate the virus. Conversely, beta cells present a more sustained EV protein and IFN‐induced markers expression, reflecting a more chronic and non‐cytolytic infection 8. Additionally, beta cells can harbor persistent infections 11 and non‐cytopathic slow replicating viruses have been detected in the pancreas of persistently infected NOD mice 12, suggesting that beta cells might fail to control viral replication. Lastly, a sustained IFN‐stimulated HLA‐I hyperexpression could enhance beta cell antigen presentation, potentially leading to beta cell destruction by autoreactive CD8 T cells. Alpha cells are shown in red, beta cells in green, delta cells in blue, EVs in purple and CD8 T cells in brown.
While the detection of VP1 in the islets of diabetic donors suggests that EV capsid antigens are present in positive cells, it does not provide information regarding the EV type that infected them. The widely used Dako 5D8/1 antibody, originally generated using CVB5 as immunogen, reacts with most of the enterovirus strains of the Coxsackie, echo and poliovirus groups 13, leaving a long list of potential suspects. Investigators are focusing on producing more suitable reagents to specifically detect EVs, which might have important consequences for vaccine development. A good example is the recent study by Laitinen et al. 14, in which new monoclonal antibodies to the CVB‐encoded proteases 2Apro and 3Cpro were generated. These antibodies were able to detect the vast majority of EV‐B species in EV‐A‐, B‐ and C‐infected cells. In addition, the authors reported their suitable use for Western blotting, immunocyto‐ and immunohistochemistry and flow cytometry. Although this is just a small step towards EV identification in human samples, ongoing efforts are generating new reagents with increased specificity. In addition, alternatives to the Dako 5D8/1 have been produced recently. The new 3A6 antibody is able to detect all CVBs but has no reactivity against EV‐A species, providing a new tool for EV identification 15. Moreover, this antibody can be used in Western blotting, peptide immunofluorescence, immunotransmission electron microscopy, immunohistochemistry and immunofluorescent assay, and constitutes a complementary option to the use of the commercial 5D8/1 or the previously reported 5D9 antibody for the detection of EVs in the pancreas of diabetic donors 13.
Despite these difficulties, several studies in the field have presented a large body of evidence that proves that an association between certain EVs serotypes and T1D exists. Epidemiological studies 16, detection of RNA 17, 18, 19 and serological analyses 20, 21 have confirmed a link between different CVB infections and T1D. In a recent report, stool samples were analyzed to evaluate whether the presence of EVs was associated with the appearance of islet autoimmunity in the Type 1 Diabetes Prediction and Prevention study (DIPP). Children with multiple autoantibodies had more EV infections than control children, and this enhanced level of infection occurred more than 1 year before the first detection of islet autoantibodies 19. Several serotypes have been identified as potential triggers of the disease 19. CVB1 infections have been associated with the first appearance of insulin autoantibodies 21. CVB5 tended to have a similar association, while none of the CVBs were associated with the appearance of GAD autoantibodies 22. In an interesting study, antibody responses to CVB capsid proteins were evaluated in sera and cord blood serum from 440 children or adolescents enrolled in the prospective German BABYBIAB, BABYDIET, DiMelly and the TEENDIAB studies 23. Children who developed early insulin autoimmunity had response profiles associated with weak protection against CVB infection, whereas competent responses were observed in children who developed autoimmunity against GAD 23. There was also a clear association between the production of VP1 antibodies and the induction of neutralizing antibodies, suggesting an important link between VP1 responses and protection 23. Similarly, children who had islet autoimmunity before 1 year of age or had multiple autoantibodies by 9 months had defective humoral responses after tetanus toxoid booster vaccination, while no differences were found for rubella between autoantibody‐positive and ‐negative children 24. These studies therefore suggest an impaired ability to mount effective humoral immune responses to exogenous antigens and raise the possibility that viral clearance may be impaired in children with early islet autoimmunity 23, 24.
Coxsackieviruses have also been detected in pancreatic biopsies obtained from six individuals with recent‐onset T1D, providing further evidence of the possible association of EV with T1D in individuals very close to disease onset 25. VP1 was detected in the islets of all patients and in two of nine controls. Enterovirus‐specific RNA sequences were detected in four of six patients and in none of the six controls. All patients and controls were polymerase chain reaction (PCR)‐negative for rhinovirus, norovirus, rotavirus and parechovirus. The CVBs isolated from these samples failed to efficiently amplify in vitro, suggesting a poor replicative capacity and pointing to a replication‐deficient virus that could have persistently infected the pancreas. Persistent infections can generate replication deficient viruses with naturally occurring 5′ terminus deletions 26. This has been proved in NOD murine pancreas, where the generation of these terminally deleted viruses was associated with persistence in the absence of cytopathic effect 12. This could be favored by a deficient humoral response in genetically predisposed individuals. As described above, children who develop early insulin autoimmunity had an incomplete antibody response to CBV 23.
1.2. The scene of the crime: what is the evidence telling us?
Increasing evidence in the field shows that a ‘viral signature’ exists in T1D, one that could be expected to appear after an EV infection. Viral sensors appear at the ‘scene of the crime’ when viruses manage to enter the cells and initiate their replication cycle. Protein kinase R (PKR) is a double stranded (dsRNA)‐dependent protein kinase that plays a critical role in anti‐viral defense 27. Its activation leads to the impairment of eukaryotic initiation factor 2 (eIF‐2a), which results in the inhibition of protein synthesis 28. It also has a role in cellular signal transduction and transcriptional control. As a consequence of dsRNA accumulation in infected cells, PKR is activated to inhibit the translation of viral mRNAs 27 in an attempt to stop viral replication. In the pancreas of diabetic patients, PKR has been detected in cells that also express VP1 29, providing additional proof of the existence of an EV infection in these individuals. The presence of long‐length dsRNA is sensed by the RNA‐helicase melanoma differentiation‐associated protein 5 (MDA5) 30. It is encoded by the IFN induced with helicase C domain 1 (IF1H1) gene, in which various single nucleotide polymorphisms (SNPs) have been found to confer a higher risk of T1D development, increasing inflammation and IFN responses 31. MDA5 is expressed in isolated human islets in response to EV infection 32 and IFN production 33. However, its pancreatic expression has not been fully investigated in prediabetic and diabetic donors. The myxovirus resistance protein (MxA) is a GTPase induced exclusively by IFN production that inhibits RNA virus replication. Studies on a very limited number of samples have shown that its expression is elevated in the islets of T1D and autoantibody‐positive donors 34. Lastly, retinoic acid inducible gene I (RIG‐I) is a cytosolic pattern recognition receptor (PRR) involved in sensing RNA, virus infection and in inducing IFN‐I production 35. Human islets infected with CVB3 express high levels of IFN‐I, IFN‐III, MDA5, RIG‐I and Toll‐like receptor (TLR)‐3, along with a variety of inflammatory cytokines 36. Studies in patients with fulminant T1D potentially infected with EVs reported a strong expression of MDA5, RIG‐I and VP1 in the islets compared to controls 37 and significantly increased numbers of alpha cells expressing RIG‐I and IRF3 38.
The interaction of these viral sensors with its ligand activates downstream signaling cascades leading to the production of IFNs, which results in the activation of an anti‐viral state and enables the detection of viral signatures 39. It has been proposed that EVs could be inducing interferonopathy‐like conditions within the islet microenvironment 40. In isolated islets and EndoC‐bH1 cells IFN‐α is capable of inducing ER stress, reducing insulin content, increasing the proinsulin to insulin ratio and decreasing the expression of the prohormone convertases PC1 and PC2, responsible for the processing of proinsulin to insulin 41. Accordingly, increases in the proinsulin to insulin ratio in the pancreas of prediabetic and recent‐onset donors have been reported 42. Thus, IFN‐α produced locally in the islets could alter the functional state of beta cells.
IFN production induces the expression of other gene products, such as STAT‐1, which binds to the IFN‐regulatory factor 9 (IRF9) and ultimately induces the transcription of expression of IFN‐stimulated genes (ISGs) aimed to inhibit viral transcription, translation and replication 43. STAT‐1 is present at low levels in the islets of non‐diabetic donors and in IDIs of T1D donors 44. However, its expression is elevated in ICIs and higher in beta cells. In addition, laser‐microdissected islets also present higher STAT‐1 mRNA levels, especially close to disease onset 44. Viral infections or exposure to IFN‐γ/IL‐1β or IFN‐α increase the islet expression of ISGs 45. Similarly, islets of recent‐onset T1D patients express high levels of ISGs 46. Quantitative mass spectrometry analysis on cultured primary human islets infected with CVB4 revealed a considerable decrease in insulin and an increase in proteins that participate in the host anti‐viral response, such as MxA, ISG15, IFN‐induced protein with tetratricopeptide repeats 1 (IFIT1), IFIT2, IFIT13, STAT‐1 and 2′‐5′‐oligoadenylate synthase 3 (OAS3) 47. IFN‐I signaling culminates in the induction of an anti‐viral response which aims to interfere at various steps with the viral cycle 45. These studies suggest that an anti‐viral signature exists in the islets of T1D patients and highlights the need to characterize these responses in the human pancreas, especially during the prediabetic phase and close to onset of disease.
IFN‐I can also induce HLA‐I hyperexpression, a hallmark of T1D 48, 49. Gian Franco Bottazzo and colleagues, 35 years ago, were the first to discuss the possibility that insults (like viruses) could induce the aberrant expression of human leukocyte antigen (HLA) molecules, leading to organ‐specific autoimmunity 50, 51. In 1985, the careful examination of the pancreas from a diabetic child who had died at onset of T1D revealed the hyperexpression of HLA‐I molecules in the islets 52. During recent decades, HLA‐I expression has become a hallmark of type 1 diabetes and multiple studies have reported its elevated expression in the islets of prediabetic and diabetic donors, especially in insulin‐containing islets (ICIs), at the protein and mRNA level 44, 53, 54, 55, 56, 57, 58. In addition, Eizirik and colleagues have recently shown that the hyperexpression of HLA‐I at the cell surface of beta cells remains even 7 days post‐IFN‐α stimulation, while other markers of inflammation and ER stress rapidly return to baseline 59. Moreover, continuous IFN‐α stimulation is not required for persistent HLA‐I hyperexpression at the cell surface, indicating that its prolonged expression could be due to the abundant presence of antigenic peptides generated in response to IFN‐α 59. Alternatively, the presence of a small amount of persistently infected cells in the islets could be sufficient to maintain a low‐level IFN response, which could maintain the hyperexpression of HLA‐I molecules years after onset, as suggested by studies of pancreas sections from long‐standing T1D donors (up to 12 years post‐onset) 44. As EVs can inhibit HLA‐I antigen presentation in infected cells through blockage of the secretory pathway in order to avoid recognition by the immune system 60, 61, IFN‐α‐stimulated HLA‐I hyperexpression could enhance beta cell antigen presentation early in the disease process and extend it for a long period of time after onset.
In the context of an enhanced antigen presentation, cytosolic peptides are transported into the ER lumen via the transporter associated with antigen processing (TAP) 62, which is part of the HLA‐I peptide loading complex. The ER aminopeptidase 1 (ERAP1) cuts these peptides into a suitable length for loading into the HLA‐I 63. Peptide–HLA complexes are transported to the cell surface for immune recognition. IFN‐I exposure induces the increase of TAP1, TAP2, TPBP, chaperones and the editing enzyme ERAP1, suggesting an increase in transport, stable processing and loading of peptides onto HLA‐I within the ER 45, 64. The resulting augmented autoantigen presentation could potentially lead to beta cell destruction by autoreactive CD8 T cells. TAP and ERAP1 have been shown to potentially contribute to the generation of preproinsulin (PPI) epitopes that target beta cells for killing by PPI‐specific cytotoxic CD8 T cells 65. Accordingly, TAP expression was elevated in ICIs of T1D donors compared to IDIs or islets from non‐diabetic donors. Similarly to HLA‐I expression, TAP was highly expressed in beta cells but was also present in other islet cells, while ERAP1 was not different in non‐diabetic and diabetic donors 65. This suggests that, in the context of an EV infection, IFN would induce the expression of HLA‐I and could create a fertile field for the presentation of beta cell antigens to autoreactive T cells, attracted to islets by the inflammatory environment.
1.3. The investigation: did it act alone?
Based on this evidence, one could envision a ‘crime scene’ or scenario in which viral infections might induce the production of IFN‐I, increase the expression of HLA‐I molecules and the presentation of antigens on the surface of beta cells. IFN‐I also influences the islet microenvironment and modulates both the innate and the adaptive immune responses by activating resident monocytes and plasmacytoid dendritic cells (pDCs), which can produce large amounts of IFNs themselves 66. This local signature creates a positive feedback loop of inflammation where cytokines and chemokines may recruit both anti‐viral and autoreactive T cells 40. Once T cells have reached the islet, IFN‐I provides the necessary signals to complete CD8 T cell expansion and activation, increasing their cytotoxic capacity 67. In addition, IFN‐I could also promote cross‐presentation of antigens from DCs directly to CD8 T cells 68. We have reported the presence of high numbers of CD11c+ cells in the pancreas of autoantibody‐positive and T1D donors 69. In addition, it has been shown that CVB3 infection drastically reduces the capacity of DCs to prime naive CD8 T cells in vitro and in vivo 70. This might have implications for the potential detection of anti‐viral T cell responses in individuals with T1D who have been exposed to EVs.
An important missing link between EV infections and T1D is the detection of circulating or infiltrating EV‐reactive T cells. Predicted CVB epitopes and in‐vitro peripheral blood mononuclear cell (PBMC) stimulation assays have tried to analyze T cell responses against EV 71. Peakman and colleagues have recently reported a novel strategy for the study of CVB epitopes by using in‐silico and ex‐vivo approaches 72. By combining conventional major histocompatibility complex (MHC)‐I‐binding algorithms with phylogenetic approaches, they identified regions of the CVB genome evolving under positive selection in order to identify epitopes from immunogenic regions. Interestingly, the 2A protein presented the highest variation within each serotype. The most conserved region between different CVB serotypes was the 3C protease. Conversely, the most variable regions were the capsid proteins, but they were also the most immunogenic across serotypes. PBMCs were stimulated ex vivo with pools of synthetic peptides based on the in‐silico predictions. Positive responses to CVB peptide pools were low, broad and not different between T1D and non‐diabetic patients 72. However, only adults and long‐standing T1D patients were included in this study. As EV infections are very common early in life and viral signatures have been found mainly in prediabetic and recent‐onset T1D donors, studies that target these populations are necessary to fully characterize T cell responses against EV and evaluate their possible role in disease pathogenesis.
The existence of molecular mimicry reflecting potential cross‐reactivity between EV protein epitopes and host islet proteins that are recognized by autoreactive T cells has been debated for a long time. Although many studies have attempted to prove this hypothesis, the evidence to support it is not strong. Technical challenges and restricted knowledge on the nature of the infiltrating cells have influenced the outcome of these studies. The majority reported possible cross‐reactivity between EV and GAD or IA‐2 epitopes 73, 74. A potential cross‐reactivity between the 2C of CVB4 and GAD247–266 at the TCR level in memory CD4 T cells has also been reported in one patient 23. This suggests that exposure to CVB4 could indeed activate GAD‐specific CD4 T cells via molecular mimicry. However, these findings will have to be confirmed in larger populations. An interesting hypothesis is that molecular mimicry does not act as a disease trigger and, rather, it contributes to disease pathogenesis 75. In this context, the expansion of previously primed autoreactive T cell populations via heterologous virus infections and molecular mimicry could lead to the acceleration of the disease in already prediabetic hosts 75. This brings to the table the concept that islet inflammation is a pre‐existing condition for the contribution of viral infections to molecular mimicry or to disease pathogenesis in general. For example, the presence of a persistent infection creates a chronic inflammatory environment in which autoreactive T cell populations could be activated and expanded 75. In this context, the careful examination of the clinical history of the patient becomes critical in order to identify if multiple infections could have contributed to disease development by increasing the inflammatory milieu and attracting immune cells, both anti‐viral and autoreactive, to the islets.
2. Innocent until proved guilty
With or without molecular mimicry, the reality is that there is a gap in our knowledge on the nature of the islet infiltrating antigen‐specific T cells (autoreactive and/or virus‐reactive). Only a small number of self‐reactive cells have been detected in the periphery or in the islets of T1D patients 53, 76 and the majority of infiltrating cells are considered of unknown reactivity or bystanders. Bystander activation can be caused by sensing of viral RNA and activation of the anti‐viral response through molecules such as PRK, MDA5, RIG‐I or Mx1, among others, with the subsequent production of IFN‐I. A virus infection that kills a limited amount of beta cells and generates the activation of an anti‐viral state could create a fertile field for autoreactive T cells. Interestingly, CVB4 infection accelerates diabetes only if a minimum number of autoreactive T cells are present 77. In addition, studies in mice have shown that the timing of the infection is also a critical factor for the development of T1D. Mice that were infected when low levels of insulitis (or autoreactive T cells) were present were protected from developing the disease through a non‐specific immunostimulatory mechanism 77. Studies in mice have shown that islets that have a high level of non‐islet specific T cells do not develop diabetes due to bystander suppression of antigen‐specific T cells. The authors hypothesized that the non‐specific T cells restrict the access of the antigen‐specific cells to their cognate antigen at the inflammatory site 78. Whether this scenario occurs in human T1D needs further investigation, but suggests that weak adaptive immune responses against viruses could also be detrimental for the host, if circulating autoreactive T cells are present and a predisposition to T1D exists.
3. Calling for reinforcements, co‐operation is the key
Many laboratories have investigated the role of EVs during the recent decades. The difficult timing and the heterogeneity of T1D has made data collection and interpretation extremely challenging. The nPOD‐Virus group was created a few years ago in an attempt to co‐ordinate studies regarding the role of EVs in disease pathogenesis in human samples, bringing together investigators with different expertise. This collaborative effort has a strong focus on EVs, with the aim to fill many of the critical knowledge gaps about their role in the disease. By gaining access to innovative and powerful technologies that had not been used previously, the group aims to find definitive proof for the association of EV and T1D. In addition, the ability to gain access and to analyze samples from the same donors in multiple laboratories by using different techniques provides an excellent opportunity to generate robust data. Ongoing analyses will provide strong evidence for the association of EV with T1D by not only identifying viral protein in the pancreas but also viral RNA and viral indicators of infection, such as the expression of HLA‐I, Mx1, PKR, MDA5 and dsRNA. In addition, the group is also looking at the presence of CD45, CD3 and CD8, as well as virus‐specific T cells, in an effort to provide strong evidence for the presence or absence of a viral signature in T1D. These efforts can lead to a better characterization of the virus–host interaction and the link between EV infections and pancreas pathology. If EVs play a role in T1D, the nPOD‐Virus group could provide enough evidence for the development of a vaccine or the use of anti‐virals that could prevent or delay the disease. An optimized and scalable protocol for the development of a CVB1 vaccine has been already tested in murine models 79. The vaccine was able to induce a strong, virus‐neutralizing antibody response, protected against a CVB1 challenge and did not accelerate diabetes in NOD mice. In a follow‐up study, the vaccine was tested on suppressor of cytokine signaling transgenic (SOCS1‐tg) mice, in which beta cells are unable to respond to IFNs and have an increased susceptibility to EV infection 80. Similarly to NOD mice, SOCS1‐tg mice developed a robust antibody response, were protected from a CVB1 challenge and from diabetes and had normal pancreas morphology 80.
The studies exposed above and the identification of different EVs serotypes with possible roles in T1D suggest that the development of an EVs vaccine for T1D could have great benefits, but could be challenging to produce. Prospective studies such as DIPP 81 and the Environmental Determinants of Diabetes in the Young (TEDDY) Study 82 could be very important for the identification of diabetogenic viruses and could provide a valuable platform to test the efficacy of vaccination strategies in preventing the disease. As several serotypes have been associated to T1D, a hexavalent vaccine including all six CVB serotypes seems to be the best approach 80. In addition, this vaccine would be able to prevent other diseases associated with CVB infection, such as myocarditis and septic meningitis 80. However, if EVs other than CVBs are implicated, the vaccine might not be able to halt the disease, and other non‐CVB EVs might have to be considered. Although the scientific community has recently increased efforts to identify specific EV species by virus amplification and sequencing techniques 83, the hostile environment of the pancreas is prone to RNA degradation 84, which does not facilitate such a quest. While this continues to limit the identification of EVs in the target organ, it is expected that new technologies and improved sensitivity and specificity of current techniques will overcome this challenge.
Based on the efficacy and safety observed in mice, investigators in Finland will begin clinical trials to study the use of a CVB vaccine in humans 85. In Phase I the vaccine will be tested for safety in adult healthy volunteers. In Phase II, it will be administered to children in order to test its safety and efficacy against EV infections. In Phase III the investigators will test if the vaccine is effective in preventing the onset of T1D 85. Parallel efforts by investigators in Norway will study the safety and efficacy of anti‐virals to successfully reduce virus amplification in recent onset T1D patients. This is also of interest in the context of persistent viral infections, as it could reduce pancreatic inflammation and recover insulin secretion.
4. Conclusions
In summary, a high number of studies have reported the association of EV infection with the development of islet autoimmunity and clinical onset, but have failed to find definitive proof for their implication as triggers of T1D. In humans, the heterogeneous disease development and the infection dynamics make EV detection very challenging. Longitudinal studies could provide a definitive answer but frequent sample collection and exhaustive follow‐ups seem necessary to catch EVs at the ‘scene of the crime’. In addition, an extensive characterization of the host response to infections prior to disease development and the access to human pancreata from individuals with two or more autoantibodies and at onset of disease will be crucial to investigate if an interferonopathy‐like condition exists in T1D. Whether beta cells are more susceptible to viral infections, EVs are directly responsible for beta cell death, or it is the host response which creates a fertile field for autoreactivity, a vaccine against EVs could provide definitive proof for the role of EV as triggers of T1D.
Disclosures
T. R.‐C. declares no conflicts of interest.
Acknowledgements
T. R.‐C. is supported by a research grant from Helmholtz Zentrum Muenchen and a JDRF research grant awarded to the nPOD‐V consortium (JDRF 25‐2012‐516).
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Pathogen infection and autoimmune disease. Clinical and Experimental Immunology 2019, 195:10–14.
Pathogen infections and primary biliary cholangitis. Clinical and Experimental Immunology 2019, 195:25–34.
Pathogens and autoimmune hepatitis. Clinical and Experimental Immunology 2019, 195:35–51.
The potential role for infections in the pathogenesis of autoimmune Addison’s disease. Clinical and Experimental Immunology 2019, 195:52–63.
Mechanisms of lymphatic system‐specific viral replication and its potential role in autoimmune disease. Clinical and Experimental Immunology 2019, 195:64–73.
The microbiome in autoimmune diseases. Clinical and Experimental Immunology 2019, 195:74–85.
References
- 1. Tang JW, Holmes CW. Acute and chronic disease caused by enteroviruses. Virulence 2017;8:1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zell R. Picornaviridae – the ever‐growing virus family. Arch Virol 2018;163:299–317. [DOI] [PubMed] [Google Scholar]
- 3. Racaniello VR. Picornaviridae: the viruses and their replicationIn Knipe DM, Howley PM, eds. Fields virology, vol. 1, 5th edn. Philadelphia, PA: Lippincott Williams & Wilkins; 2007, 795–838. [Google Scholar]
- 4. van der Linden L, Wolthers KC, van Kuppeveld FJ. Replication and inhibitors of enteroviruses and parechoviruses. Viruses 2015;7:4529–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J Virol 1995;69:3873–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bergelson JM, Cunningham JA, Droguett G et al Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997;275:1320–3. [DOI] [PubMed] [Google Scholar]
- 7. Hodik M, Anagandula M, Fuxe J et al Coxsackie‐adenovirus receptor expression is enhanced in pancreas from patients with type 1 diabetes. BMJ Open Diabetes Res Care 2016;4:e000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marroqui L, Lopes M, dos Santos RS et al Differential cell autonomous responses determine the outcome of Coxsackievirus infections in murine pancreatic alpha and beta cells. Elife 2015;4:e06990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dotta F, Censini S, van Halteren AG et al Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent‐onset type 1 diabetic patients. Proc Natl Acad Sci USA 2007;104:5115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009;52:1143–51. [DOI] [PubMed] [Google Scholar]
- 11. Chehadeh W, Kerr‐Conte J, Pattou F et al Persistent infection of human pancreatic islets by Coxsackievirus B is associated with alpha interferon synthesis in beta cells. J Virol 2000;74:10153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tracy S, Smithee S, Alhazmi A, Chapman N. Coxsackievirus can persist in murine pancreas by deletion of 5' terminal genomic sequences. J Med Virol 2015;87:240–7. [DOI] [PubMed] [Google Scholar]
- 13. Maccari G, Genoni A, Sansonno S, Toniolo A. Properties of two enterovirus antibodies that are utilized in diabetes research. Sci Rep 2016;6:24757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laitinen OH, Svedin E, Kapell S et al New Coxsackievirus 2A(pro) and 3C(pro) protease antibodies for virus detection and discovery of pathogenic mechanisms. J Virol Methods 2018;255:29–37. [DOI] [PubMed] [Google Scholar]
- 15. Saarinen NVV, Laiho JE, Richardson SJ et al A novel rat CVB1‐VP1 monoclonal antibody 3A6 detects a broad range of enteroviruses. Sci Rep 2018;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta‐analysis of observational molecular studies. BMJ 2011;342:d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schulte BM, Bakkers J, Lanke KHW et al Detection of enterovirus RNA in peripheral blood mononuclear cells of type 1 diabetic patients beyond the stage of acute infection. Viral Immunol 2010;23:99–104. [DOI] [PubMed] [Google Scholar]
- 18. Alidjinou EK, Sane F, Lefevre C et al Enteroviruses in blood of patients with type 1 diabetes detected by integrated cell culture and reverse transcription quantitative real‐time PCR. Acta Diabetol 2017;54:1025–9. [DOI] [PubMed] [Google Scholar]
- 19. Honkanen H, Oikarinen S, Nurminen N et al Detection of enteroviruses in stools precedes islet autoimmunity by several months: possible evidence for slowly operating mechanisms in virus‐induced autoimmunity. Diabetologia 2017;60:424–31. [DOI] [PubMed] [Google Scholar]
- 20. Oikarinen S, Tauriainen S, Hober D et al Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes 2014;63:655–62. [DOI] [PubMed] [Google Scholar]
- 21. Laitinen OH, Honkanen H, Pakkanen O et al Coxsackievirus B1 is associated with induction of beta‐cell autoimmunity that portends type 1 diabetes. Diabetes 2014;63:446–55. [DOI] [PubMed] [Google Scholar]
- 22. Sioofy‐Khojine AB, Lehtonen J, Nurminen N et al Coxsackievirus B1 infections are associated with the initiation of insulin‐driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018;61:1193–202. [DOI] [PubMed] [Google Scholar]
- 23. Ashton MP, Eugster A, Walther D et al Incomplete immune response to Coxsackie B viruses associates with early autoimmunity against insulin. Sci Rep 2016;6:32899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmid S, Molteni A, Füchtenbusch M et al Reduced IL‐4 associated antibody responses to vaccine in early pre‐diabetes. Diabetologia 2002;45:677–85. [DOI] [PubMed] [Google Scholar]
- 25. Krogvold L, Edwin B, Buanes T et al Detection of a low‐grade enteroviral infection in the islets of Langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015;64:1682–7. [DOI] [PubMed] [Google Scholar]
- 26. Bouin A, Nguyen Y, Wehbe M et al Major persistent 5' terminally deleted Coxsackievirus B3 populations in human endomyocardial tissues. Emerg Infect Dis 2016;22:1488–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia MA, Gil J, Ventoso I et al Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 2006;70:1032–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pindel A, Sadler A. The role of protein kinase R in the interferon response. J Interferon Cytokine Res 2011;31:59–70. [DOI] [PubMed] [Google Scholar]
- 29. Richardson SJ, Leete P, Bone AJ et al Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl‐1. Diabetologia 2013;56:185–93. [DOI] [PubMed] [Google Scholar]
- 30. Morse ZJ, Horwitz MS. Innate viral receptor signaling determines type 1 diabetes onset. Front Endocrinol (Lausanne) 2017;8:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Looney BM, Xia C‐Q, Concannon P et al Effects of type 1 diabetes‐associated IFIH1 polymorphisms on MDA5 function and expression. Curr Diab Rep 2015;15:96. [DOI] [PubMed] [Google Scholar]
- 32. Ylipaasto P, Kutlu B, Rasilainen S et al Global profiling of coxsackievirus‐ and cytokine‐induced gene expression in human pancreatic islets. Diabetologia 2005;48:1510–22. [DOI] [PubMed] [Google Scholar]
- 33. Hultcrantz M, Hühn MH, Wolf M et al Interferons induce an antiviral state in human pancreatic islet cells. Virology 2007;367:92–101. [DOI] [PubMed] [Google Scholar]
- 34. Chen J, Feigenbaum L, Awasthi P et al Insulin‐dependent diabetes induced by pancreatic beta cell expression of IL‐15 and IL‐15Ralpha. Proc Natl Acad Sci USA 2013;110:13534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu XX, Wan H, Nie L et al RIG‐I: a multifunctional protein beyond a pattern recognition receptor. Protein Cell 2018;9:246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Domsgen E, Lind K, Kong L et al An IFIH1 gene polymorphism associated with risk for autoimmunity regulates canonical antiviral defence pathways in Coxsackievirus infected human pancreatic islets. Sci Rep 2016;6:39378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aida K, Nishida Y, Tanaka S et al RIG‐I‐ and MDA5‐initiated innate immunity linked with adaptive immunity accelerates beta‐cell death in fulminant type 1 diabetes. Diabetes 2011;60:884–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoneda S, Imagawa A, Fukui K et al A histological study of fulminant type 1 diabetes mellitus related to human cytomegalovirus reactivation. J Clin Endocrinol Metab 2017;102:2394–400. [DOI] [PubMed] [Google Scholar]
- 39. Mostafavi S, Yoshida H, Moodley D et al Parsing the interferon transcriptional network and its disease associations. Cell 2016;164:564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jean‐Baptiste VSE, Xia C‐Q, Clare‐Salzler MJ, Horwitz MS. Type 1 diabetes and type 1 interferonopathies: localization of a type 1 common thread of virus infection in the pancreas. EBioMedicine 2017;22:10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lombardi A, Tomer Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J Autoimmun 2017;80:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodriguez‐Calvo T, Zapardiel‐Gonzalo J, Amirian N et al Increase in pancreatic proinsulin and preservation of beta‐cell mass in autoantibody‐positive donors prior to type 1 diabetes onset. Diabetes 2017;66:1334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Richardson SJ, Rodriguez‐Calvo T, Gerling IC et al Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 2016;59:2448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Newby BN, Mathews CE. Type I interferon is a catastrophic feature of the diabetic islet microenvironment. Front Endocrinol (Lausanne) 2017;8:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lundberg M, Krogvold L, Kuric E et al Expression of interferon‐stimulated genes in insulitic pancreatic islets of patients recently diagnosed with type 1 diabetes. Diabetes 2016;65:3104–10. [DOI] [PubMed] [Google Scholar]
- 47. Nyalwidhe JO, Gallagher GR, Glenn LM et al Coxsackievirus‐induced proteomic alterations in primary human islets provide insights for the etiology of diabetes. J Endocr Soc 2017;1:1272–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha‐interferon in insulin‐secreting beta cells in type 1 diabetes mellitus. Lancet 1987;2:1423–7. [DOI] [PubMed] [Google Scholar]
- 49. Huang X, Yuang J, Goddard A et al Interferon expression in the pancreases of patients with type I diabetes. Diabetes 1995;44:658–64. [DOI] [PubMed] [Google Scholar]
- 50. Bottazzo GF, Hanafusa T, Pujol‐Borrell R, Feldmann M. Role of aberrant HLA‐DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet 1983;2:1115–9. [DOI] [PubMed] [Google Scholar]
- 51. Bonifacio E, Bosi E, Leslie RD. Gian Franco Bottazzo, 1946–2017. Diabetologia 2018;61:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bottazzo GF, Dean BM, McNally JM et al In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med 1985;313:353–60. [DOI] [PubMed] [Google Scholar]
- 53. Coppieters KT, Dotta F, Amirian N et al Demonstration of islet‐autoreactive CD8 T cells in insulitic lesions from recent onset and long‐term type 1 diabetes patients. J Exp Med 2012;209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Foulis AK, Farquharson MA, Hardman R. Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin‐dependent) diabetes mellitus. Diabetologia 1987;30:333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Richardson SJ, Willcox A, Bone AJ et al Immunopathology of the human pancreas in type‐I diabetes. Semin Immunopathol 2011;33:9–21. [DOI] [PubMed] [Google Scholar]
- 56. Foulis AK. The pathology of the endocrine pancreas in type 1 (insulin‐dependent) diabetes mellitus. APMIS 1996;104:161–7. [DOI] [PubMed] [Google Scholar]
- 57. Rodriguez‐Calvo T, Suwandi JS, Amirian N et al Heterogeneity and lobularity of pancreatic pathology in type 1 diabetes during the prediabetic phase. J Histochem Cytochem 2015;63:626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Somoza N, Vargas F, Roura‐Mir C et al Pancreas in recent onset insulin‐dependent diabetes mellitus. Changes in HLA, adhesion molecules and autoantigens, restricted T cell receptor V beta usage, and cytokine profile. J Immunol 1994;153:1360–77. [PubMed] [Google Scholar]
- 59. Coomans de Brachene A, Dos Santos RS, Marroqui L et al IFN‐alpha induces a preferential long‐lasting expression of MHC class I in human pancreatic beta cells. Diabetologia 2018;61:636–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kemball CC, Harkins S, Whitmire JK et al Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLOS Pathog 2009;5:e1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Deitz SB, Dodd DA, Cooper S et al MHC I‐dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci USA 2000;97:13790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cresswell P. Intracellular surveillance: controlling the assembly of MHC class I‐peptide complexes. Traffic 2000;1:301–5. [DOI] [PubMed] [Google Scholar]
- 63. Saric T, Chang SC, Hattori A et al An IFN‐gamma‐induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I‐presented peptides. Nat Immunol 2002;3:1169–76. [DOI] [PubMed] [Google Scholar]
- 64. Neefjes J, Jongsma MLM, Paul P et al Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011;11:823–36. [DOI] [PubMed] [Google Scholar]
- 65. Kronenberg‐Versteeg D, Eichmann M, Russell MA et al Molecular pathways for immune recognition of preproinsulin signal peptide in type 1 diabetes. Diabetes 2018;67:687–96. [DOI] [PubMed] [Google Scholar]
- 66. Sozzani S, Del Prete A, Bosisio D. Dendritic cell recruitment and activation in autoimmunity. J Autoimmun 2017;85:126–40. [DOI] [PubMed] [Google Scholar]
- 67. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 2005;174:4465–9. [DOI] [PubMed] [Google Scholar]
- 68. Le Bon A, Etchart N, Rossmann C et al Cross‐priming of CD8+ T cells stimulated by virus‐induced type I interferon. Nat Immunol 2003;4:1009–15. [DOI] [PubMed] [Google Scholar]
- 69. Rodriguez‐Calvo T, Ekwall O, Amirian N et al Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes 2014;63:3880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kemball CC, Flynn CT, Hosking MP et al Wild‐type coxsackievirus infection dramatically alters the abundance, heterogeneity, and immunostimulatory capacity of conventional dendritic cells in vivo. Virology 2012;429:74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weinzierl AO, Rudolf D, Maurer D et al Identification of HLA‐A*01‐ and HLA‐A*02‐restricted CD8+ T‐cell epitopes shared among group B enteroviruses. J Gen Virol 2008;89:2090–7. [DOI] [PubMed] [Google Scholar]
- 72. Kundu R, Knight R, Dunga M, Peakman M. In silico and ex vivo approaches indicate immune pressure on capsid and non‐capsid regions of Coxsackie B viruses in the human system. PLOS ONE 2018;13:e0199323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Atkinson MA, Bowman MA, Campbell L, Darrow Bl, Kaufman Dl, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin‐dependent diabetes. J Clin Invest 1994;94:2125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Honeyman MC, Stone NL, Harrison LC. T‐cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA‐2: potential for mimicry with rotavirus and other environmental agents. Mol Med 1998;4:231–9. [PMC free article] [PubMed] [Google Scholar]
- 75. Christen U, Edelmann KH, McGavern DB et al A viral epitope that mimics a self antigen can accelerate but not initiate autoimmune diabetes. J Clin Invest 2004;114:1290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Babon JA, DeNicola ME, Blodgett DM et al Analysis of self‐antigen specificity of islet‐infiltrating T cells from human donors with type 1 diabetes. Nat Med 2016;22:1482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T‐cells in pancreatic islets. Diabetes 2000;49:708–11. [DOI] [PubMed] [Google Scholar]
- 78. Christoffersson G, Chodaczek G, Ratliff SS, Coppieters K, vonHerrath MG. Suppression of diabetes by accumulation of non‐islet‐specific CD8(+) effector T cells in pancreatic islets. Sci Immunol 2018;3:eaam6533. [DOI] [PubMed] [Google Scholar]
- 79. Hankaniemi MM, Laitinen OH, Stone VM et al Optimized production and purification of Coxsackievirus B1 vaccine and its preclinical evaluation in a mouse model. Vaccine 2017;35:3718–25. [DOI] [PubMed] [Google Scholar]
- 80. Stone VM, Hankaniemi MM, Svedin E et al A Coxsackievirus B vaccine protects against virus‐induced diabetes in an experimental mouse model of type 1 diabetes. Diabetologia 2018;61:476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hyoty H. Viruses in type 1 diabetes. Pediatr Diabetes 2016;17(Suppl 22):56–64. [DOI] [PubMed] [Google Scholar]
- 82. Lonnrot M, Lynch KF, Elding Larsson H et al Respiratory infections are temporally associated with initiation of type 1 diabetes autoimmunity: the TEDDY study. Diabetologia 2017;60:1931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Genoni A, Canducci F, Rossi A et al Revealing enterovirus infection in chronic human disorders: an integrated diagnostic approach. Sci Rep 2017;7:5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Philips T, Kusmartseva I, Gerling IC et al Factors that influence the quality of RNA from the pancreas of organ donors. Pancreas 2017;46:252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. JDRF . Clinical trials announced for preventative type 1 vaccine. 2017, JDRF: https://jdrf.org.uk/news/vaccine-type-1-clinical-trials/.