

Summary
Autoimmune Addison’s disease (AAD), or primary adrenocortical insufficiency, is a classical organ‐specific autoimmune disease with 160 years of history. AAD is remarkably homogeneous with one major dominant self‐antigen, the cytochrome P450 21‐hydroxylase enzyme, which is targeted by both autoantibodies and autoreactive T cells. Like most autoimmune diseases, AAD is thought to be caused by an unfortunate combination of genetic and environmental factors. While the number of genetic associations with AAD is increasing, almost nothing is known about environmental factors. A major environmental factor commonly proposed for autoimmune diseases, based partly on experimental and clinical data and partly on shared pathways between anti‐viral immunity and autoimmunity, is viral infections. However, there are few reports associating viral infections to AAD, and it has proved difficult to establish which immunological processes that could link any viral infection with the initiation or progression of AAD. In this review, we will summarize the current knowledge on the underlying mechanisms of AAD and take a closer look on the potential involvement of viruses.
Keywords: Addison’s disease, autoimmunity, chemokines, virus infections
Introduction
Appropriate production of steroid hormones by the adrenal cortex is vital for human life. Mineralocorticoids such as aldosterone regulate the body’s salt and water balance, while glucocorticoids such as cortisol are important for many aspects of metabolism and for mobilization of energy during stressful conditions. In the adrenals of patients with autoimmune Addison’s disease (AAD), the steroid hormone‐producing cells of the adrenal cortex are targeted by an immune‐mediated attack resulting in organ failure and lack of steroid hormone production 1, 2. Patients with AAD therefore depend upon lifelong supplementation therapy which does not restore full health, but leaves patients exposed to increased morbidity, heightened mortality and reduced quality of life 3, 4, 5, 6. There is therefore a need to improve understanding of the underlying pathological mechanisms behind AAD in order to form the basis for rational design of molecular and cellular strategies that target the genesis of the disease. The development of AAD and most autoimmune diseases is probably a multi‐factorial process involving a combination of genetic and environmental factors and the failure to control autoreactive lymphocytes at multiple stages 7. Several genetic risk factors are known, in particular certain variants within the major histocompatibility complex (MHC) 6, 8, but almost nothing is known about environmental factors.
While AAD may occur in isolation, more than half the patients suffer from additional autoimmune diseases, frequently autoimmune thyroiditis (hypo‐ or hyperthyroidism) and type 1 diabetes 6, 9, 10. The co‐existence of two of these three is commonly referred to as autoimmune polyendocrine syndrome type 2 (APS‐2). AAD is also a major disease component of autoimmune polyendocrine syndrome type 1 (APS‐1), in which the presence of two of the following three manifestations is diagnostic: AAD, chronic mucocutaneous candidiasis and hypoparathyroidisim 11. While the two syndromes share several features such as the presence of serum adrenal autoantibodies and immune infiltration of the adrenal cortex 12, the genetic background of APS‐1 differs from APS‐2 as the former is caused by mutations in the AIRE (autoimmune regulator) gene, and APS‐2 (including isolated AAD) is a polygenic disorder. AIRE plays a critical role in the negative selection of immature T cells in the thymus, preventing the escape of autoreactive T cells and development of autoimmunity 13. Hence, mutations in AIRE lead to multi‐organ autoimmunity. The mechanisms by which AAD develops in APS‐1 and APS‐2 are therefore thought to differ. Thus, we will focus mainly on the latter form here.
A recent twin study from Sweden revealed that the heritability of AAD is extremely high, indicating a strong genetic influence on the occurrence of disease 14. However, although the concordance rate of monozygotic twins is high at 0·73, the involvement of additional variables such as environmental factors is very likely. This is supported further by an apparent increase in the incidence of AAD in Europe during recent decades 15, 16, 17, 18, 19. Knowledge of AAD is scarce in other parts of the world, but epidemiological data indicate that the prevalence is exceedingly far lower in the Far East and South Africa than in Europe 20, 21, 22. However, the same major histocompatibility complex (MHC) risk alleles are shared between European, North American and South African patients 23, indicating additional environmental factors. Of course, these may include protective as well as predisposing factors, including childhood infections that may actually protect against autoimmune diseases 24. Nevertheless, suspected environmental triggers in autoimmune diseases include infectious agents such as viruses, bacteria and other pathogens, and these will remain the focus of this current review 25. Unfortunately, unlike certain other autoimmune diseases such as type 1 diabetes, multiple sclerosis or autoimmune liver diseases 26, 27, 28, no specific candidate infectious agents have been identified for AAD. However, many infectious pathogens are known to infect the adrenal cortex and some of them may also cause adrenal insufficiency 29. In the following we will review some of these infectious agents, and highlight how they may take part in the pathogenesis of AAD.
Microbial causes of adrenal insufficiency
In earlier years, adrenal insufficiency was caused predominantly by infection with Mycobacterium tuberculosis, which spreads from the lungs to the adrenals, causing necrosis and calcification 30. However, this cause of adrenal insufficiency is now generally confined to regions where the bacterium is still endemic, and is not associated with the autoimmune form that dominates in developed countries today. When considering that an infection could serve as a trigger for adrenal autoimmunity, a variety of other microorganisms has also been shown to infect the adrenal gland and could be of interest 29. These include a diverse array of viruses, bacteria, fungi and parasites. However, we find viruses to be the most relevant, given the immune responses involved in AAD, the frequent links to other autoimmune endocrinopathies (such as type 1 diabetes), and the fact that many of the other microbes with a tropism for the adrenals, such as M. tuberculosis or Paracoccidioides brasiliensis, are not endemic in the western world 31, 32. The latter pathogens are also examples of agents capable of mediating direct adrenal destruction. Conversely, viruses are often capable of establishing chronic or latent infections in humans. Such chronic infections often have features typical for autoimmune diseases, such as autoantibodies and organ‐specific inflammation.
Many of the infectious agents known to infect the adrenal cortex and cause adrenal insufficiency are opportunistic infections, primarily affecting immunocompromised individuals. In particular, patients with acquired immunodeficiency syndrome (AIDS) caused by HIV are susceptible to adrenalitis. In a morphological evaluation of adrenal autopsies from 128 patients dying from AIDS, more than 99% had adrenalitis, with cytomegalovirus (CMV) being the cause in half the cases 33. Bacterial, fungal and protozoan infectious agents such as Mycobacterium sp., Histoplasma and Cryptococcus sp. and Tryponosoma and Toxoplasma sp. were also detected. Other studies have reported that the adrenal gland is the most frequently CMV‐affected organ in AIDS patients 34. CMV and adrenalitis have also been described in other acquired immunodeficient states, such as allogeneic haematopoetic stem‐cell transplantation, kidney transplantation and liver transplantation after chronic hepatitis C virus infection 35, 36, 37, 38. Finally, adrenal infections by CMV and also other members of the Herpesviridae family, such as herpes simplex virus types 1 and 2 (HSV‐1/HSV‐2) and human herpesvirus 6 (HHV‐6), have been reported in neonates and infants 39, 40, 41, 42. Although the children described with these infections were seemingly immunocompetent (e.g. no signs of concomitant HIV infection or genetic causes of severe immunodeficiency), the immune systems of neonates and infants are immature with suboptimal responses to infections and vaccines 43, 44. Viral adrenalitis in primary immunodeficiencies have also been described, including adrenal insufficiency caused by Epstein–Barr virus (EBV) infection in an adolescent with Wiscott–Aldrich syndrome and subclinical adrenal CMV infection discovered at autopsy in children with severe combined immunodeficiency 45, 46. However, some of the viruses described above, including HSV‐1 and CMV, have also been associated with adrenalitis in apparently immunocompetent adults 29, 47, 48. Hepatitis B virus (HBV) and hepatitis C virus (HCV) infections have also been reported in connection with adrenal insufficiency 49, 50. Interestingly, in one patient hepatitis B surface antigen was detected in autopsy material from remaining adrenal tissue, indicating that HBV can have a tropism for the adrenal cortex 49.
The susceptibility of the adrenals to viral infections in immunosuppressed or immunodeficient individuals is mirrored by reports suggesting impaired immunity and increased susceptibility to infections in patients with AAD. Recently it was found that AAD patients have impaired natural killer (NK) cell functions, potentially compromising their early recognition and elimination of virus‐infected cells 51. Furthermore, it has been demonstrated that peripheral blood cells from AAD patients respond poorly to in‐vitro stimulation with interferons (IFNs), which substantiates the notion of impaired early anti‐viral immune responses 52. Epidemiological investigations have also suggested that AAD patients have more infections, and are prescribed with more anti‐microbial agents (including anti‐virals), than the general population 15, 53. However, the interpretation of these data is complicated by the fact that AAD patients are medicated with exogenous glucocorticoids that have many immunomodulatory effects 54. Although AAD patients have little to no endogenous glucocorticoid production and replacement doses are attempted to be kept within physiological borders, it is recognized that excessive use of glucocorticoids increases the risk of infectious complications 55. It is therefore unclear whether the increased risk of infections in AAD patients is related to glucocorticoid replacement therapy or to a partial immune defect. Importantly, however, the increased susceptibility to infections in AAD patients does not show a clear relationship with glucocorticoid dosage, and is present already in incident patients prior to any glucocorticoid treatment 53. In a Danish nationwide study investigating more than 4·5 million people born between 1945 and 2000, an association between infection‐related hospital admissions and subsequent diagnoses of 29 different autoimmune diseases was found 56. AAD was among the diseases with the strongest association to hospitalization for serious infections prior to diagnosis. Intriguingly, for AAD in particular, an increase in the number of infections increased the risk for autoimmune disease in a dose‐dependent manner with patients having five or more infections. However, a word of caution is needed when interpreting these data. Serious infections (e.g. involving sepsis) require rapid activation of adrenocortical glucocorticoid production as a fundamental part of the stress response 57. As AAD can have a long subclinical phase with adrenal impairment, infections requiring rapid glucocorticoid production may easily precipitate clinically overt adrenocortical failure 12. It is therefore possible that the increased number of infections in AAD patients prior to diagnosis is merely reflecting the increased severity of infections in individuals with impaired adrenal function, and that these infections are unmasking the adrenocortical insufficiency rather than causing it. Indirect support for a role for infections early in life does, however, exist. A recent study from the United Kingdom and Poland demonstrated that month of birth exerts an effect on the risk of developing AAD, with peaks in December and January and troughs in May and July, suggesting increased exposure to seasonal viral infections in the perinatal period in individuals who develop AAD later in life 58.
Some studies have also suggested that the adrenal cortex is a natural reservoir for several viruses that are able to establish latent infections in humans, and that excessive glucocorticoid production could reactivate these viruses 59, 60. Viral agents detected in both normal adrenals and adrenocortical tumours (both benign and malignant) included most members of the Herpesviridae family and the polyomaviruses SV40 and BK virus. Primary infections with herpesviruses and polyomaviruses usually occur early in childhood, and thereafter these viruses establish latent or persistent infections that may be reactivated by immunosuppression. Interestingly, experimental infections with HSV‐1 in mice have revealed that the adrenal cortex is among the first organs to be infected before spreading of the virus to the central nervous system 61. In mice surviving primary HSV‐1 infection, subcapsular lesions developed in the adrenal cortex and persisted for up to 1 year after inoculation 62. These findings suggest that infections early in life, such as primary HSV‐1 infections, may leave lesions and scarring behind in the adrenal tissue, perhaps rendering individuals more susceptible to develop AAD later in life.
Gene–environment interactions
Several genetic factors that increase the risk of developing AAD have been identified, most of which play important roles in the interactions between antigen‐presenting cells (including B cells) and T cells 63. This is very typical for organ‐specific autoimmune diseases and many genetic risk factors for AAD are, in fact, shared with related diseases 64. A common assumption is that autoimmunity risk gene products are involved in the activation and/or regulation of pathogenic or self‐specific lymphocytes 65. However, it is highly likely that these genetic variants also influence the immune responses against microbial agents. A selection of genes associated with AAD that may also play an important role in anti‐viral immune responses is shown in Fig. 1. Here we describe how variants in these genes may interact with immune responses against viruses.
Figure 1.
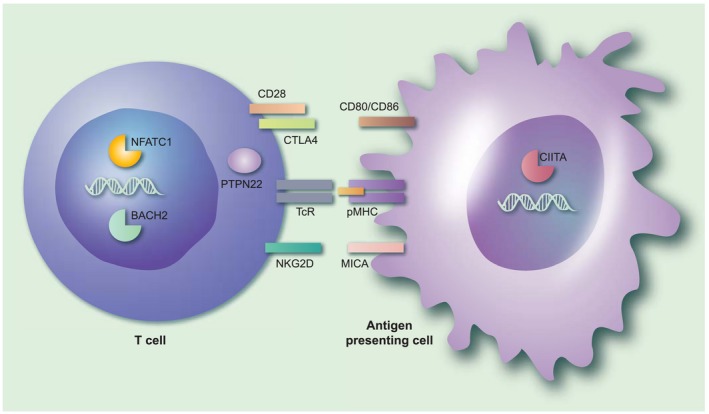
The immunological context of genetic susceptibility factors for autoimmune Addison’s disease that may interact with infectious agents. The figure gives an overview of the immunological processes implicated by the genetic susceptibility factors associated with autoimmune Addison’s disease (AAD). Note that both the products encoded by the risk genes themselves (denoted in bold type below) and their interaction partners are shown in the figure. Professional antigen‐presenting cells, such as dendritic cells or activated B cells, present antigenic peptides (from self or from microorganisms) on their major histocompatibility complex/human leucocyte antigen (MHC/HLA) molecules (pMHC in the figure) that may be recognized by T cells through their T cell receptor (TCR). An additional co‐stimulatory signal, usually mediated by CD80/CD86, is required for T cell activation. On the T cell, CD28 acts as a positive co‐stimulator, while cytotoxic T lymphocyte antigen‐4 (CTLA‐4) transmits inhibitory (negative) signals upon CD80/CD86 ligation. Polymorphisms in CTLA‐4 that may predispose to AAD may also lead to excessive immune responses in response to certain viruses. Protein tyrosine phosphatase, non‐receptor type 22 (PTPN22) serves critical roles in the regulation of T cell activation signalling events downstream of the TCR. A specific genetic variant of PTPN22 may predispose to the activation of self‐specific T cells, but also may lead to suboptimal immune responses against influenza. MHC class I polypeptide‐related sequence A (MICA), a ligand for the co‐stimulatory molecule natural killer group 2 member D (NKGD2) on CD8+ T cells, is up‐regulated by viral infection. Genetic variants in the transcription factors broad complex‐tramtrack‐bric a brac and cap’n’collar homology 2 (BACH2), nuclear factor of activated T cells (NFATC1) and class II major histocompatibility complex transactivator (CIITA) have all been associated with AAD. Mutations in BACH2 may give rise to an immunodeficiency syndrome with recurrent viral pulmonary infections; NFATC1 plays a role in the process of T cell exhaustion during viral infections, while CIITA is a well‐known target for immune evasive strategies for several viruses. [Colour figure can be viewed at wileyonlinelibrary.com]
AAD is a classic example of a human leucocyte antigen (HLA)‐associated organ‐specific autoimmune disease 6, 8, 66. Similar to type 1 diabetes and coeliac disease, the strongest associations are with the HLA‐DR3‐DQ2 and HLA‐DR4‐DQ8 haplotypes 11. In particular, these two haplotypes in heterozygous combination confer a considerable stronger risk of developing AAD 6. The high incidence of type 1 diabetes and coeliac disease, and relatively speaking also AAD, in Scandinavia may be the result of the HLA haplotypes HLA‐DR3‐DQ2 and HLA‐DR4‐DQ8 being common, because they protected people from succumbing to common infections 67. Indeed, heritability of susceptibility to infectious diseases is an important functional aspect of HLA class II alleles 68. For several millennia, in the absence of vaccinations, people survived on the sheer ability to mount protective immune responses to common virus infections, particularly those affecting children 67. Therefore, the better HLA class II presentation of viral peptides to T cells, and subsequent T cell and antibody responses to measles, rubella, mumps, etc., the greater the chance of survival. Unfortunately, the same HLA class II molecules that have been selected evolutionarily based on their protection against common pathogens might also be particularly suited to present peptides from self‐antigens and thereby participate in autoimmunity 69. The AAD‐associated HLA class II subtype DR4 may serve as an example, as this particular allele is associated with clearance of HBV and HCV viral infections 70, 71. It has also been suggested that the ability of CD4+ T cells to produce certain cytokines in response to infections, such as interferon (IFN)‐γ and interleukin (IL)‐17, is connected to distinct HLA haplotypes 69. These particular proinflammatory cytokines may be important in clearing infections, but at the same time act in a detrimental way to the infected host tissue and induce significant immunopathology.
Within the HLA locus, a connection to AAD has also been made with the MHC‐class I chain‐related gene A (MICA) 8, 72, which encodes a ligand for the activating natural killer (NK) and T cell co‐receptor natural killer group 2 (NKG2D). MICA can be induced on many cell types in response to cellular stress, such as during a virus infection 73. The binding of MICA to NKG2D triggers cytotoxicity and cytokine production in NK cells and γδT cells 74, but act as a co‐stimulator for activation of conventional CD8+ αβT cells 75. Ligation of NKG2D on an autoreactive CD8+ T cell by MICA on a virus‐infected cell in the setting of self‐peptide presentation could therefore lead to excessive activation and expansion of pathogenic CD8+ T cells.
A gene commonly associated with autoimmune diseases is the cytotoxic T lymphocyte antigen 4 (CTLA‐4), which acts as an inhibitory immunological checkpoint on activated T cells 76. Specifically, CTLA‐4 is expressed upon T cell activation and opposes continuous co‐stimulation of T cells through CD28, as it binds to CD80/CD86 on antigen‐presenting cells (APCs) with superior affinity 77. CTLA‐4 therefore accumulates at the immunological synapse between T cells and APCs upon T cell activation, and eventually blocks further co‐stimulation and abrogates the T cell response. A recent meta‐analysis of European AAD cohorts strengthened the link to CTLA‐4 single nucleotide polymorphisms (SNPs) 78, hypothesizing that the associated genetic variants may impair CTLA‐4 function and thereby lower the threshold for T cell activation, as suggested in autoimmune thyroid diseases 79. However, these CTLA‐4 polymorphisms may also influence anti‐viral immune responses and are associated with certain treatment outcomes in viral infections, e.g. clearance of hepatitis C virus (HCV) after treatment with IFN‐α and ribavirin 80. Another SNP associated with AAD is the 1858T allele of the protein tyrosine phosphatase, non‐receptor type 22 (PTPN22) 81. PTPN22 has both enzymatic and adaptive functions, and modulates signalling through both antigen receptors (T and B cells) and innate immune receptors 82. The SNP associated with AAD and many other autoimmune diseases may be of particular importance to the regulation of T cell responses to weak self‐peptide agonists and promotion of type I IFN production in myeloid cells following Toll‐like receptor (TLR) stimulation 83, 84. Interestingly, carriers of the 1858T allele also show an increased susceptibility to infections, including influenza 85.
Polymorphisms in the genes encoding transcription factors broad complex‐tramtrack‐bric a brac and cap’n’collar homology 2 (BACH2), nuclear factor of activated T cells, cytoplasmic 1 (NFATC1) and the class II, major histocompatibility complex, transactivator (CIITA) have been associated with increased risk of developing AAD 66, 86, 87, 88, 89. These transcription factors all play crucial roles in immunity. BACH2 is essential for B cell differentiation into plasma cells and for the generation of regulatory T cells 90. Mutations in BACH2 lead to an immunodeficiency syndrome with autoimmune components and recurrent pulmonary infections with both virus and bacteria 91. NFATC1 is involved in gene transcription in activated T cells and is a major target of the immunosuppressive drug cyclosporin 92. NFATC1 also takes part in T cell exhaustion during chronic viral infections and promotes the expression of inhibitory receptors such as programmed cell death protein 1 (PD‐1) 93. CIITA is considered a global regulator for the expression of MHC class II proteins and related molecules (including MHC class I) 93, and repression of CIITA is part of the immune evasion strategy of several viruses such as EBV and varicella zoster virus 94, 95.
Taken together, most of the AAD risk genes regulate or participate in anti‐viral immune responses, and the sum of these genetic risk variants could therefore modulate the course of a virus infection. Very little is currently known about such actual gene–environment interactions in the pathogenesis of AAD.
Pathogenesis of autoimmune Addison’s disease and implications for viral agents
Although the mechanisms by which the cells of the adrenal cortex are killed by the immune system are starting to unravel, the triggering and perpetuating factors of AAD are still almost completely unknown. The histopathological picture in AAD is characterized by lymphocytic infiltration of the adrenal cortex, while the adrenal medulla remains intact 96, 97, 98. During the course of the disease, the integrity of the cortex is gradually lost as steroidogenic cells are progressively destroyed and replaced by fibrous tissue 12, 99. Despite continuous loss of adrenocortical cells, AAD may remain subclinical for long periods before overt disease develops 12. In fact, adrenocortical failure may not manifest itself until 90% of the cells are destroyed 100. Unfortunately, histochemical studies have been restricted to post‐mortem analyses due to practical and ethical reasons, and a detailed analysis of the mononuclear infiltrate in AAD is lacking. Conversely, considerable progress on the disease‐specific immunological processes at play in AAD has been made in last decade. Autoimmunity‐mediated adrenocortical destruction is a gradual process developing over months and even years involving both T cells and B cells. This process is accompanied by 21‐hydroxylase (21OH)‐autoantibodies that can be detected in patient serum, constituting an early biomarker of this otherwise silent autoimmune process 6, 101. Although 21OH autoantibodies correlate with the degree of adrenal dysfunction in the preclinical phase of AAD, they do not seem to inhibit 21OH activity in vivo 102, 103. Instead, autoreactive T cells specific for 21OH peptides are considered to drive the immunopathology of AAD and indeed, both CD4+ and CD8+ T cells that recognize 21OH‐peptides and secrete IFN‐γ have been identified 104, 105, 106, 107. 21OH‐specific CD8+ T cells from AAD have also been shown to be able to kill adrenocortical cells in vitro in an antigen‐specific and HLA‐restricted manner through perforin and granzyme B‐mediated cytolysis 105.
A process in which three major cell types, CD4+/CD8+ T cells and B cells, with specificities for the same antigen, are mobilized and recruited to the adrenal cortex has clear parallels to immunity against infections. It may be assumed that the development of AAD is the result of misdirected processes originally meant to be protective. Given the strong genetic associations to HLA class II haplotypes, the highly co‐ordinated specific immune responses against 21OH by both CD4+ and CD8+ T cells and B cells probably depend upon the actions of 21OH‐specific CD4+ T helper cells. In this context, many functional aspects of CD8+ cytotoxic T cells against viruses have been shown to depend upon the actions of virus‐specific CD4+ T cells, including the primary effector response, the generation of memory and the recruitment to sites of infection 108, 109. This process is known as ‘licensing’, referring to the ability of CD4+ T cells to license cognate effector CD8+ T cell responses. In AAD, 21OH‐specific CD4+ T cells may ‘license’ 21OH‐specific CD8+ T cells to become activated and recruited to the adrenal cortex, leading to adrenalitis and adrenal insufficiency. This process may share mechanisms with the processes that have been described to co‐ordinate CD4+ and effector T cell responses to viruses.
Hypothetically speaking, certain viruses could play a role as major environmental factors in AAD, either by infecting steroid hormone‐producing cells of the adrenal cortex or by affecting the balance of the immune system. Given the highly co‐ordinated immunological attack on 21OH in AAD, and the parallels of these immune responses to those against viruses, we currently believe more in the former. Viruses could spread to the adrenal cortex and induce a strong inflammatory response. In individuals who cannot eradicate the virus efficiently it will remain in the adrenal cortex in a slowly replicating form, continuously producing viral nucleic acids and proteins activating the innate and the adaptive immune system and thus driving chronic inflammation and eventually autoimmunity (probably depending on the genetic background of the host). In addition to the autoimmune reaction against cells of the adrenal cortex, anti‐viral immune responses could also participate in the immunopathology. Furthermore, the virus itself may cause cellular damage and stress, and may directly interfere with adrenocortical function (e.g. glucocorticoid production) 110. Other mechanisms may also be involved, including immunological cross‐reactions between viral and host proteins, known as molecular mimicry 111. We are currently investigating this phenomenon in AAD, as the dominating CD4+ restricted T cell epitopes of 21OH have been identified 105. Interestingly, some of these epitopes seem to be shared between different HLA class II molecules which could, indeed, implicate molecular mimicry. T cell responses against 21OH epitopes are detectable for decades after disease onset, which could indicate continuous stimulation from cross‐reactive viral or microbial peptides. However, remaining or even regenerating adrenal tissue expressing 21OH could also be responsible for the consistent restimulation of 21OH‐specific T cells. Fig. 2 describes one possible scenario in which viral infections could play a role in the pathogenesis of AAD, based on current knowledge of disease‐specific immunological mechanisms. The figure is not meant to be extensive, and for more comprehensive overviews of the pathogenesis underlying AAD we refer to other reviews written by us and others 2, 63. The IFN‐induced chemokine C‐X‐C motif chemokine 10 (CXCL10) has been shown to be elevated in the serum of patients with AAD 52, 112, 113, 114, 115. This elevated CXCL10 production is not due to the activation of peripheral leucocytes, as peripheral blood mononuclear cells (PBMCs) from AAD patients actually show diminished production of CXCL10 upon stimulation with IFNs, compared to healthy blood donors 52. Instead, primary adrenocortical cells as well as adrenocortical cell‐lines of different origins have been shown to be able to produce large amounts of CXCL10 upon stimulation with proinflammatory cytokines such as IFN‐γ and TNF‐α 112. Adrenocortical cells are also able to express the TLR for double‐stranded RNA (dsRNA), TLR‐3 115. Stimulation with the synthetic double‐stranded nucleic acid polyinosinic:polycytidylic acid [poly (I:C)], a dsRNA and a ligand for TLR‐3, can also induce production of CXCL10, and along with IFN‐γ and to a certain degree also TNF‐α poly (I:C) is able to drastically increase the CXCL10 production in a synergistic manner. The CXCL10 produced by the adrenal cortex acts as a chemotactic signal for IFN‐γ‐producing CD4+ and CD8+ T cells bearing the chemokine receptor CXCR3. In concordance with this we have demonstrated that 21OH‐specific T cells from AAD patients express high levels of CXCR3 and are able to migrate towards CXCL10‐containing cell culture supernatants from IFN‐γ/poly (I:C)‐stimulated adrenocortical cells 104. In a genetically predisposed individual infected by a virus able to infect and replicate in the adrenal cortex, TLR‐3 may sense the infection through dsRNA. TLR‐3 activation could, in turn, lead to type I IFN production and activation of IFN‐stimulated genes (ISGs) such as CXCL10. Alternatively, as the adrenal cortex seems to be a poor producer of type I IFNs 115, 116, dendritic cells or macrophages (which are abundantly present in the adrenal cortex) may sense the virus and produce type I IFN. The presence of type I IFNs along with the stimulation of TLR‐3 in the adrenal cortex may initiate production of CXCL10, which recruits CXCR3‐bearing 21OH‐specific T cells from the nearest lymph node. These T cells could, in principle, have arisen years ahead of the ongoing infection, but are only now migrating to the tissue in response to CXCL10 production. Once autoreactive CD4+ and CD8+ T cells arrive in the adrenal cortex, they may become activated through the recognition of cognate HLA‐presented 21OH peptides and secrete large amounts of IFN‐γ and TNF‐α. This could initiate a self‐perpetuating inflammatory loop where the adrenal cortex is stimulated to boost the CXCL10 production even further, increasing the influx of proinflammatory CXCR3‐positive lymphocytes. The destructive process will continue until the antigen, in this case 21OH, is cleared, which eventually means that the adrenal cortex will be completely atrophied.
Figure 2.
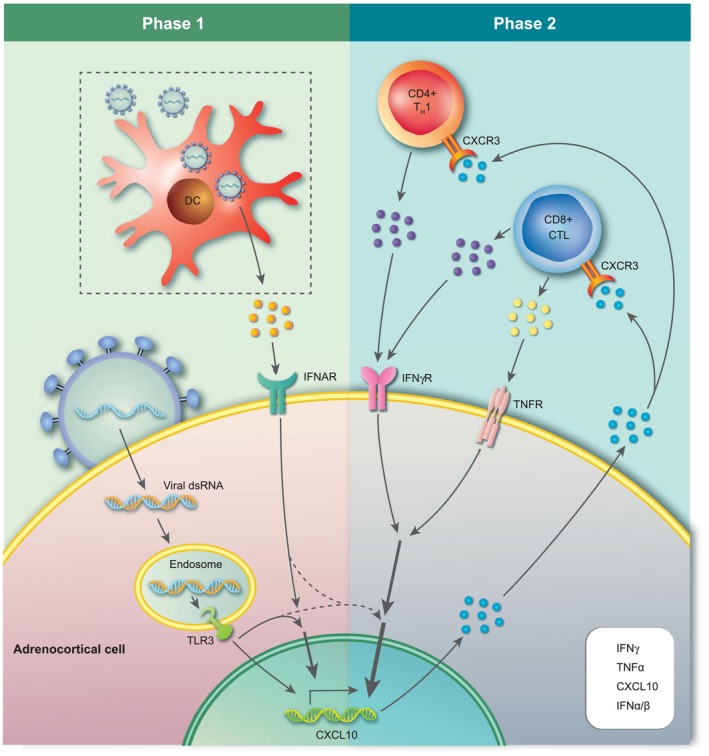
Hypothetical recruitment of autoreactive T cells to the adrenal cortex triggered by viral infection. Phase 1: a viral infection of the adrenal cortex may serve as the initial trigger of adrenalitis in AAD. Recognition of viral dsRNA by endosomal Toll‐like receptor 3 (TLR‐3) along with the action of locally produced type I interferons (IFNs) [here by virus‐infected dendritic cells (DCs)], stimulates the production of C‐X‐C motif chemokine 10 (CXCL10) by adrenocortical cells. Phase 2: CXCL10 promotes the recruitment of CXCR3‐expressing activated autoreactive CD4+ and CD8+ T cells (e.g. 21OH‐specific) that produce proinflammatory cytokines. These T cells are activated and expanded in a local draining lymph node or in secondary lymphoid organs elsewhere in the body (not shown in the figure). This event could be related to the virus infection in phase 1, with DCs and other antigen‐presenting cells (APCs) carrying and presenting adrenal components (including 21OH peptides) under proinflammatory conditions, or it could be a result of an earlier infection at a different location than the adrenal cortex (e.g. through molecular mimicry). T cell‐derived IFN‐γ and tumour necrosis factor (TNF)‐α intensify CXCL10 production by adrenocortical cells, leading to a vicious cycle that could strongly expand the mononuclear infiltrate and sustain inflammation of the adrenal cortex. For illustrative reasons, CXCL9 is not included in the figure, but may play an accompanying role in the recruitment of autoaggressive T cells in phase 2. [Colour figure can be viewed at wileyonlinelibrary.com]
Concluding remarks
Unlike other organ‐specific autoimmune diseases such as type 1 diabetes, which has been linked to enteroviruses, there are no single obvious viral candidates suggested to take active part in the pathogenesis of AAD. However, the highly targeted and co‐ordinated attack by the immune system on the steroidogenic cells of the adrenal cortex, driven primarily by one dominant self‐antigen (21OH), is highly reminiscent of that against viruses: CD4+ and CD8+ T cells, as well as antibodies specific for an intracellular antigen. This view is strengthened further by the genetic risk factors associated with AAD, as most of these encode proteins involved in normal anti‐viral immune responses. Epidemiological data also point towards a role for infections in the development of AAD. However, the long subclinical phase of AAD makes it difficult to identify any potential viruses that could play a role in the early phases of the pathogenesis. Prospective studies where children are followed from birth to disease onset, such as the TEDDY study for type 1 diabetes117, are not realistic with a rare disease such as AAD, and as AAD most commonly manifests itself in adults between 20 and 40 years of age. However, if common viruses such as CMV, EBV or HSV‐1 are involved in the pathogenesis, several years of prospective studies are a necessity. A possible strategy in the future could be to take advantage of the extremely high heritability of AAD and collect a large cohort of families with aggregation of AAD 14, 63. All family members could then be followed prospectively and monitored for their exposure to infectious agents or other environmental agents, and for any signs of functional adrenal impairment.
Disclosures
The authors declare that they have no conflicts of interest to disclose.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Pathogen infection and autoimmune disease. Clinical and Experimental Immunology 2019, 195:10–14.
Enterovirus infection and type 1 diabetes: unraveling the crime scene. Clinical and Experimental Immunology 2019, 195:15–24.
Pathogen infections and primary biliary cholangitis. Clinical and Experimental Immunology 2019, 195:25–34.
Pathogens and autoimmune hepatitis. Clinical and Experimental Immunology 2019, 195:35–51.
Mechanisms of lymphatic system‐specific viral replication and its potential role in autoimmune disease. Clinical and Experimental Immunology 2019, 195:64–73.
The microbiome in autoimmune diseases. Clinical and Experimental Immunology 2019, 195:74–85.
References
- 1. Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med 2009; 360:2328–39. [DOI] [PubMed] [Google Scholar]
- 2. Bratland E, Husebye ES. Cellular immunity and immunopathology in autoimmune Addison’s disease. Mol Cell Endocrinol 2011; 336:180–90. [DOI] [PubMed] [Google Scholar]
- 3. Bensing S, Brandt L, Tabaroj F et al Increased death risk and altered cancer incidence pattern in patients with isolated or combined autoimmune primary adrenocortical insufficiency. Clin Endocrinol (Oxf) 2008; 69:697–704. [DOI] [PubMed] [Google Scholar]
- 4. Bensing S, Hulting AL, Husebye ES, Kampe O, Lovas K. Management of endocrine disease: epidemiology, quality of life and complications of primary adrenal insufficiency: a review. Eur J Endocrinol 2016; 175:R107–16. [DOI] [PubMed] [Google Scholar]
- 5. Lovas K, Loge JH, Husebye ES. Subjective health status in Norwegian patients with Addison’s disease. Clin Endocrinol (Oxf) 2002; 56:581–8. [DOI] [PubMed] [Google Scholar]
- 6. Erichsen MM, Lovas K, Skinningsrud B et al Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry. J Clin Endocrinol Metab 2009; 94:4882–90. [DOI] [PubMed] [Google Scholar]
- 7. Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell 2007; 130:25–35. [DOI] [PubMed] [Google Scholar]
- 8. Skinningsrud B, Lie BA, Lavant E et al Multiple loci in the HLA complex are associated with Addison's disease. J Clin Endocrinol Metab 2011; 96:E1703–8. [DOI] [PubMed] [Google Scholar]
- 9. Dalin F, Nordling Eriksson G, Dahlqvist P et al Clinical and immunological characteristics of autoimmune addison's disease: a nationwide Swedish multicenter study. J Clin Endocrinol Metab 2017; 102:379–89. [DOI] [PubMed] [Google Scholar]
- 10. Fichna M, Fichna P, Gryczynska M, Walkowiak J, Zurawek M, Sowinski J. Screening for associated autoimmune disorders in Polish patients with Addison’s disease. Endocrine 2010; 37:349–60. [DOI] [PubMed] [Google Scholar]
- 11. Husebye ES, Anderson MS, Kampe O. Autoimmune polyendocrine syndromes. N Engl J Med 2018; 378:1132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 2002; 23:327–64. [DOI] [PubMed] [Google Scholar]
- 13. Mathis D, Benoist C. Aire. Annu Rev Immunol 2009; 27:287–312. [DOI] [PubMed] [Google Scholar]
- 14. Skov J, Hoijer J, Magnusson PKE, Ludvigsson JF, Kampe O, Bensing S. Heritability of Addison’s disease and prevalence of associated autoimmunity in a cohort of 112,100 Swedish twins. Endocrine 2017; 58:521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bjornsdottir S, Sundstrom A, Ludvigsson JF, Blomqvist P, Kampe O, Bensing S. Drug prescription patterns in patients with Addison’s disease: a Swedish population‐based cohort study. J Clin Endocrinol Metab 2013; 98:2009–18. [DOI] [PubMed] [Google Scholar]
- 16. Lovas K, Husebye ES. High prevalence and increasing incidence of Addison’s disease in western Norway. Clin Endocrinol (Oxf) 2002; 56:787–91. [DOI] [PubMed] [Google Scholar]
- 17. Meyer G, Neumann K, Badenhoop K, Linder R. Increasing prevalence of Addison’s disease in German females: health insurance data 2008–2012. Eur J Endocrinol 2014; 170:367–73. [DOI] [PubMed] [Google Scholar]
- 18. Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003; 361:1881–93. [DOI] [PubMed] [Google Scholar]
- 19. Mason AS, Meade TW, Lee JA, Morris JN. Epidemiological and clinical picture of Addison’s disease. Lancet 1968; 2:744–7. [DOI] [PubMed] [Google Scholar]
- 20. Nomura K, Demura H, Saruta T. Addison’s disease in Japan: characteristics and changes revealed in a nationwide survey. Intern Med 1994; 33:602–6. [DOI] [PubMed] [Google Scholar]
- 21. Hong AR, Ryu OH, Kim SY, Kim SW, Korean Adrenal Gland and Endocrine Hypertension Study Group, Korean Endocrine Society . Characteristics of Korean patients with primary adrenal insufficiency: a registry‐based nationwide survey in Korea. Endocrinol Metab (Seoul) 2017; 32:466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ross IL, Levitt NS. Addison’s disease symptoms – a cross sectional study in urban South Africa. PLOS ONE 2013; 8:e53526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ross IL, Babu S, Armstrong T et al HLA similarities indicate shared genetic risk in 21‐hydroxylase autoantibody positive South African and United States Addison’s disease. Tissue Antigens 2014; 84:361–9. [DOI] [PubMed] [Google Scholar]
- 24. Bach JF. Infections and autoimmune diseases. J Autoimmun 2005; 25 (Suppl):74–80. [DOI] [PubMed] [Google Scholar]
- 25. Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol 2009; 155:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christen U, Hintermann E. Autoantibodies in autoimmune hepatitis: can epitopes tell us about the etiology of the disease? Front Immunol 2018; 9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM, NeuroproMiSe EBV Working Group . Epstein–Barr virus in the multiple sclerosis brain: a controversial issue – report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain 2011; 134:2772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hyoty H. Viruses in type 1 diabetes. Pediatr Diabetes 2016; 17 (Suppl 22):56–64. [DOI] [PubMed] [Google Scholar]
- 29. Paolo WF Jr, Nosanchuk JD. Adrenal infections. Int J Infect Dis 2006; 10:343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dunlop D. Eighty‐six cases of Addison’s disease. BMJ 1963; 2:887–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Glaziou P, Sismanidis C, Floyd K, Raviglione M. Global epidemiology of tuberculosis. Cold Spring Harb Perspect Med 2014; 5:a017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bocca AL, Amaral AC, Teixeira MM, Sato PK, Shikanai‐Yasuda MA, Soares Felipe MS. Paracoccidioidomycosis: eco‐epidemiology, taxonomy and clinical and therapeutic issues. Future Microbiol 2013; 8:1177–91. [DOI] [PubMed] [Google Scholar]
- 33. Rodrigues D, Reis M, Teixeira V et al Pathologic findings in the adrenal glands of autopsied patients with acquired immunodeficiency syndrome. Pathol Res Pract 2002; 198:25–30. [DOI] [PubMed] [Google Scholar]
- 34. Pulakhandam U, Dincsoy HP. Cytomegaloviral adrenalitis and adrenal insufficiency in AIDS. Am J Clin Pathol 1990; 93:651–6. [DOI] [PubMed] [Google Scholar]
- 35. Matsumura T, Narimatsu H, Kami M et al Cytomegalovirus infections following umbilical cord blood transplantation using reduced intensity conditioning regimens for adult patients. Biol Blood Marrow Transplant 2007; 13:577–83. [DOI] [PubMed] [Google Scholar]
- 36. Ardalan M, Shoja MM. Cytomegalovirus‐induced adrenal insufficiency in a renal transplant recipient. Transplant Proc 2009; 41:2915–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tejura N, Sonyey A. CMV‐associated adrenal insufficiency in a renal transplant recipient. IDCases 2018; 11:44–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh N, Zeevi A, Gayowski T, Marino IR. Late onset cytomegalovirus disease in liver transplant recipients: de novo reactivation in recurrent hepatitis C virus hepatitis. Transpl Int 1998; 11:308–11. [DOI] [PubMed] [Google Scholar]
- 39. Schmitt K, Deutsch J, Tulzer G, Meindi R, Aberle S. Autoimmune hepatitis and adrenal insufficiency in an infant with human herpesvirus‐6 infection. Lancet 1996; 348:966. [DOI] [PubMed] [Google Scholar]
- 40. Dinleyici EC, Dogruel N, Dinleyici M, Us T. Adrenal insufficiency associated with cytomegalovirus infection in two infants. Int J Infect Dis 2009; 13:e181–4. [DOI] [PubMed] [Google Scholar]
- 41. Levy NL, Notkins AL. Viral infections and diseases of the endocrine system. J Infect Dis 1971; 124:94–103. [DOI] [PubMed] [Google Scholar]
- 42. Nakamura Y, Yamamoto S, Tanaka S et al Herpes simplex viral infection in human neonates: an immunohistochemical and electron microscopic study. Hum Pathol 1985; 16:1091–7. [DOI] [PubMed] [Google Scholar]
- 43. Burchett SK, Corey L, Mohan KM, Westall J, Ashley R, Wilson CB. Diminished interferon‐gamma and lymphocyte proliferation in neonatal and postpartum primary herpes simplex virus infection. J Infect Dis 1992; 165:813–8. [DOI] [PubMed] [Google Scholar]
- 44. Vekemans J, Ota MO, Wang EC et al T cell responses to vaccines in infants: defective IFNgamma production after oral polio vaccination. Clin Exp Immunol 2002; 127:495–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hertel NT, Jacobsen BB, Pedersen FK, Heilmann C. Adrenocortical insufficiency associated with Epstein–Barr virus infection in a patient with the Wiskott–Aldrich syndrome. Eur J Pediatr 1987; 146:603–4. [DOI] [PubMed] [Google Scholar]
- 46. Gupta K, Rawat A, Agrawal P et al Infectious and non‐infectious complications in primary immunodeficiency disorders: an autopsy study from North India. J Clin Pathol 2018; 71:425–35. [DOI] [PubMed] [Google Scholar]
- 47. Miyazaki Y, Akizuki S, Sakaoka H, Yamamoto S, Terao H. Disseminated infection of herpes simplex virus with fulminant hepatitis in a healthy adult. A case report. APMIS 1991; 99:1001–7. [DOI] [PubMed] [Google Scholar]
- 48. Arend SM, Kroes AC. Look and ye shall find… cytomegalovirus infection in immunocompetent patients. Clin Infect Dis 2003; 37:1607–8. [DOI] [PubMed] [Google Scholar]
- 49. Somlo F, Berry GR. Extrahepatic manifestations of hepatitis B virus infection: Addison’s disease and myelofibrosis in a patient with persistent hepatitis B surface antigenemia. Can J Infect Dis 1993; 4:139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tran HA, Song S, Lojewski RJ, Reeves GE. Exacerbation of hepatitis C induced subclinical hypoadrenalism by Interferon‐alpha2beta: a case report. Cases J 2008; 1:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bancos I, Hazeldine J, Chortis V et al Primary adrenal insufficiency is associated with impaired natural killer cell function: a potential link to increased mortality. Eur J Endocrinol 2017; 176:471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Edvardsen K, Bjanesoy T, Hellesen A et al Peripheral blood cells from patients with autoimmune Addison’s disease poorly respond to interferons in vitro, despite elevated serum levels of interferon‐inducible chemokines. J Interferon Cytokine Res 2015; 35:759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smans LC, Souverein PC, Leufkens HG, Hoepelman AI, Zelissen PM. Increased use of antimicrobial agents and hospital admission for infections in patients with primary adrenal insufficiency: a cohort study. Eur J Endocrinol 2013; 168:609–14. [DOI] [PubMed] [Google Scholar]
- 54. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med 2005; 353:1711–23. [DOI] [PubMed] [Google Scholar]
- 55. Stuck AE, Minder CE, Frey FJ. Risk of infectious complications in patients taking glucocorticosteroids. Rev Infect Dis 1989; 11:954–63. [DOI] [PubMed] [Google Scholar]
- 56. Nielsen PR, Kragstrup TW, Deleuran BW, Benros ME. Infections as risk factor for autoimmune diseases – a nationwide study. J Autoimmun 2016; 74:176–81. [DOI] [PubMed] [Google Scholar]
- 57. Kanczkowski W, Sue M, Zacharowski K, Reincke M, Bornstein SR. The role of adrenal gland microenvironment in the HPA axis function and dysfunction during sepsis. Mol Cell Endocrinol 2015; 408:241–8. [DOI] [PubMed] [Google Scholar]
- 58. Pazderska A, Fichna M, Mitchell AL et al Impact of month of birth on the risk of development of autoimmune Addison’s disease. J Clin Endocrinol Metab 2016; 101:4214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barzon L, Trevisan M, Masi G et al Detection of polyomaviruses and herpesviruses in human adrenal tumors. Oncogene 2008; 27:857–64. [DOI] [PubMed] [Google Scholar]
- 60. Pomara G, Cappello F, Barzon L et al Cytomegalovirus and BK‐virus co‐infection of a clinically non‐functioning adrenal adenoma: innocent bystanders or new pathogenetic agents? Eur J Histochem 2006; 50:131–2. [PubMed] [Google Scholar]
- 61. Hill T, Yirrell D, Blyth W. Infection of the adrenal gland as a route to the central nervous system after viraemia with herpes simplex virus in the mouse. J Gen Virol 1986; 67:309–20. [DOI] [PubMed] [Google Scholar]
- 62. Nachtigal M, Caulfield J. Early and late pathologic changes in the adrenal glands of mice after infection with herpes simplex virus type 1. Am J Pathol 1984; 115:175. [PMC free article] [PubMed] [Google Scholar]
- 63. Mitchell AL, Pearce SH. Autoimmune addison disease: pathophysiology and genetic complexity. Nat Rev Endocrinol 2012; 8:306–16. [DOI] [PubMed] [Google Scholar]
- 64. Zenewicz LA, Abraham C, Flavell RA, Cho JH. Unraveling the genetics of autoimmunity. Cell 2010; 140:791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marson A, Housley WJ, Hafler DA. Genetic basis of autoimmunity. J Clin Invest 2015; 125:2234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eriksson D, Bianchi M, Landegren N et al Extended exome sequencing identifies BACH2 as a novel major risk locus for Addison’s disease. J Intern Med 2016; 280:595–608. [DOI] [PubMed] [Google Scholar]
- 67. Lernmark A. Environmental factors in the etiology of type 1 diabetes, celiac disease, and narcolepsy. Pediatr Diabetes 2016; 17 (Suppl 22):65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cooke GS, Hill AV. Genetics of susceptibility to human infectious disease. Nat Rev Genet 2001; 2:967–77. [DOI] [PubMed] [Google Scholar]
- 69. Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol 2013; 190:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yan ZH, Fan Y, Wang XH, Mao Q, Deng GH, Wang YM. Relationship between HLA‐DR gene polymorphisms and outcomes of hepatitis B viral infections: a meta‐analysis. World J Gastroenterol 2012; 18:3119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McKiernan SM, Hagan R, Curry M et al Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology 2004; 40:108–14. [DOI] [PubMed] [Google Scholar]
- 72. Gambelunghe G, Falorni A, Ghaderi M et al Microsatellite polymorphism of the MHC class I chain‐related (MIC‐A and MIC‐B) genes marks the risk for autoimmune Addison’s disease. J Clin Endocrinol Metab 1999; 84:3701–7. [DOI] [PubMed] [Google Scholar]
- 73. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol 2013; 31:413–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shafi S, Vantourout P, Wallace G et al An NKG2D‐mediated human lymphoid stress surveillance response with high interindividual variation. Sci Transl Med 2011; 3:113ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Groh V, Rhinehart R, Randolph‐Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus‐infected cells. Nat Immunol 2001; 2:255–60. [DOI] [PubMed] [Google Scholar]
- 76. Brand O, Gough S, Heward J. HLA, CTLA‐4 and PTPN22: the shared genetic master‐key to autoimmunity? Expert Rev Mol Med 2005; 7:1–15. [DOI] [PubMed] [Google Scholar]
- 77. Krummel MF, Allison JP. CD28 and CTLA‐4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995; 182:459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wolff AS, Mitchell AL, Cordell HJ et al CTLA‐4 as a genetic determinant in autoimmune Addison’s disease. Genes Immun 2015; 16:430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ban Y, Davies TF, Greenberg DA et al Analysis of the CTLA‐4, CD28, and inducible costimulator (ICOS) genes in autoimmune thyroid disease. Genes Immun 2003; 4:586–93. [DOI] [PubMed] [Google Scholar]
- 80. Yee LJ, Perez KA, Tang J, van Leeuwen DJ, Kaslow RA. Association of CTLA4 polymorphisms with sustained response to interferon and ribavirin therapy for chronic hepatitis C virus infection. J Infect Dis 2003; 187:1264–71. [DOI] [PubMed] [Google Scholar]
- 81. Skinningsrud B, Husebye ES, Gervin K et al Mutation screening of PTPN22: association of the 1858T‐allele with Addison’s disease. Eur J Hum Genet 2008; 16:977–82. [DOI] [PubMed] [Google Scholar]
- 82. Stanford SM, Bottini N. PTPN22: the archetypal non‐HLA autoimmunity gene. Nat Rev Rheumatol 2014; 10:602–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Salmond RJ, Brownlie RJ, Morrison VL, Zamoyska R. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol 2014; 15:875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang Y, Shaked I, Stanford SM et al The autoimmunity‐associated gene PTPN22 potentiates Toll‐like receptor‐driven, type 1 interferon‐dependent immunity. Immunity 2013; 39:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Crabtree JN, He W, Guan W, Flage M, Miller MS, Peterson EJ. Autoimmune variant PTPN22 C1858T Is associated with impaired responses to influenza vaccination. J Infect Dis 2016; 214:248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pazderska A, Oftedal BE, Napier CM et al A variant in the BACH2 gene is associated with susceptibility to autoimmune Addison’s disease in humans. J Clin Endocrinol Metab 2016; 101:3865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mitchell AL, Boe Wolff A, MacArthur K et al Linkage analysis in autoimmune Addison’s disease: NFATC1 as a potential novel susceptibility locus. PLOS ONE 2015; 10:e0123550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ghaderi M, Gambelunghe G, Tortoioli C et al MHC2TA single nucleotide polymorphism and genetic risk for autoimmune adrenal insufficiency. J Clin Endocrinol Metab 2006; 91:4107–11. [DOI] [PubMed] [Google Scholar]
- 89. Skinningsrud B, Husebye ES, Pearce SH et al Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab 2008; 93:3310–7. [DOI] [PubMed] [Google Scholar]
- 90. Roychoudhuri R, Hirahara K, Mousavi K et al BACH2 represses effector programs to stabilize T(reg)‐mediated immune homeostasis. Nature 2013; 498:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Afzali B, Gronholm J, Vandrovcova J et al BACH2 immunodeficiency illustrates an association between super‐enhancers and haploinsufficiency. Nat Immunol 2017; 18:813–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Northrop JP, Ho SN, Chen L et al NF‐AT components define a family of transcription factors targeted in T‐cell activation. Nature 1994; 369:497–502. [DOI] [PubMed] [Google Scholar]
- 93. Man K, Gabriel SS, Liao Y et al Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory‐like T cells during chronic infection. Immunity 2017; 47:1129–41.e5. [DOI] [PubMed] [Google Scholar]
- 94. Lin JH, Lin JY, Chou YC et al Epstein–Barr virus LMP2A suppresses MHC class II expression by regulating the B‐cell transcription factors E47 and PU.1. Blood 2015; 125:2228–38. [DOI] [PubMed] [Google Scholar]
- 95. Abendroth A, Kinchington PR, Slobedman B. Varicella zoster virus immune evasion strategies. Curr Top Microbiol Immunol 2010; 342:155–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Al Sabri AM, Smith N, Busuttil A. Sudden death due to auto‐immune Addison’s disease in a 12‐year‐old girl. Int J Legal Med 1997; 110:278–80. [DOI] [PubMed] [Google Scholar]
- 97. Drury MI, Keelan DM, Timoney FJ, Irvine WJ. Juvenile familial endocrinopathy. Clin Exp Immunol 1970; 7:125–32. [PMC free article] [PubMed] [Google Scholar]
- 98. Irvine WJ, Stewart AG, Scarth L. A clinical and immunological study of adrenocortical insufficiency (Addison’s disease). Clin Exp Immunol 1967; 2:31–70. [PMC free article] [PubMed] [Google Scholar]
- 99. Imam K, Abdullah M, Felicetta JV. Alopecia universalis as a feature of polyglandular autoimmunity type I. West J Med 1988; 149:338–41. [PMC free article] [PubMed] [Google Scholar]
- 100. Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. BMJ 1978; 1:1591–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Winqvist O, Karlsson FA, Kampe O. 21‐Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet 1992; 339:1559–62. [DOI] [PubMed] [Google Scholar]
- 102. Laureti S, De Bellis A, Muccitelli VI et al Levels of adrenocortical autoantibodies correlate with the degree of adrenal dysfunction in subjects with preclinical Addison's disease. J Clin Endocrinol Metab 1998; 83:3507–11. [DOI] [PubMed] [Google Scholar]
- 103. Boscaro M, Betterle C, Volpato M et al Hormonal responses during various phases of autoimmune adrenal failure: no evidence for 21‐hydroxylase enzyme activity inhibition in vivo . J Clin Endocrinol Metab 1996; 81:2801–4. [DOI] [PubMed] [Google Scholar]
- 104. Hellesen A, Edvardsen K, Breivik L, Husebye ES, Bratland E. The effect of types I and III interferons on adrenocortical cells and its possible implications for autoimmune Addison’s disease. Clin Exp Immunol 2014; 176:351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dawoodji A, Chen JL, Shepherd D et al High frequency of cytolytic 21‐hydroxylase‐specific CD8+ T cells in autoimmune Addison’s disease patients. J Immunol 2014; 193:2118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rottembourg D, Deal C, Lambert M et al 21‐Hydroxylase epitopes are targeted by CD8 T cells in autoimmune Addison’s disease. J Autoimmun 2010; 35:309–15. [DOI] [PubMed] [Google Scholar]
- 107. Bratland E, Skinningsrud B, Undlien DE, Mozes E, Husebye ES. T cell responses to steroid cytochrome P450 21‐hydroxylase in patients with autoimmune primary adrenal insufficiency. J Clin Endocrinol Metab 2009; 94:5117–24. [DOI] [PubMed] [Google Scholar]
- 108. Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8(+) T lymphocyte mobilization to virus‐infected tissue requires CD4(+) T‐cell help. Nature 2009; 462:510–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003; 300:337–9. [DOI] [PubMed] [Google Scholar]
- 110. Kino T, Chrousos GP. Virus‐mediated modulation of the host endocrine signaling systems: clinical implications. Trends Endocrinol Metab 2007; 18:159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Oldstone MB. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr Top Microbiol Immunol 2005; 296:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rotondi M, Falorni A, De Bellis A et al Elevated serum interferon‐gamma‐inducible chemokine‐10/CXC chemokine ligand‐10 in autoimmune primary adrenal insufficiency and in vitro expression in human adrenal cells primary cultures after stimulation with proinflammatory cytokines. J Clin Endocrinol Metab 2005; 90:2357–63. [DOI] [PubMed] [Google Scholar]
- 113. Ekman B, Alstrand N, Bachrach‐Lindstrom M, Jenmalm MC, Wahlberg J. Altered chemokine Th1/Th2 balance in Addison’s disease: relationship with hydrocortisone dosing and quality of life. Horm Metab Res 2014; 46:48–53. [DOI] [PubMed] [Google Scholar]
- 114. Kisand K, Link M, Wolff AS et al Interferon autoantibodies associated with AIRE deficiency decrease the expression of IFN‐stimulated genes. Blood 2008; 112:2657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bratland E, Hellesen A, Husebye ES. Induction of CXCL10 chemokine in adrenocortical cells by stimulation through toll‐like receptor 3. Mol Cell Endocrinol 2013; 365:75–83. [DOI] [PubMed] [Google Scholar]
- 116. Matkovic U, Pacenti M, Trevisan M, Palu G, Barzon L. Investigation on human adrenocortical cell response to adenovirus and adenoviral vector infection. J Cell Physiol 2009; 220:45–57. [DOI] [PubMed] [Google Scholar]
- 117. Hagopian WA, Lernmark A, Rewers MJ et al TEDDY – the environmental determinants of diabetes in the young: an observational clinical trial. Ann NY Acad Sci 2006; 1079:320–6. [DOI] [PubMed] [Google Scholar]