

Summary
To investigate possible roles of T helper type 2 (Th2) cytokines in the anti‐arthritic effects of a blood fluke, Schistosoma mansoni (Sm), for mouse collagen‐induced arthritis (CIA), wild‐type (WT), signal transducer and activator of transcription 6 (STAT6) knock‐out (KO) and interleukin (IL)‐10 KO mice were infected with Sm. Three weeks after infection, the mice were immunized with bovine type II collagen (IIC). Arthritis severity was monitored by scoring, measurement of paw thickness and the presence of ankylosis. Serum anti‐IIC IgG levels, splenic cytokine production and cytokine gene expression in the popliteal lymph nodes (PLNs) were measured and compared among WT and gene‐KO mice. Consistent with our previous findings, Sm infection reduced the arthritis severity in WT mice. Splenic production of IL‐17A and tumor necrosis factor (TNF)‐α was reduced by the infection. In contrast, Sm infection markedly exacerbated CIA in STAT6 KO mice. In the KO mice, IL‐17A production was increased by the infection. Conversely, Sm infection did not affect the exacerbated arthritis in IL‐10 KO mice, although IL‐17A production was reduced by the helminth. Our results suggest that signaling via STAT6 (presumably IL‐4 and/or IL‐13) and IL‐10 is required for the suppression of CIA by Sm infection, but through different mechanisms. STAT6 was essential for helminth‐induced reduction of IL‐17A, whereas regulation of the basal arthritis severity by IL‐10 was needed in order for it to be sufficiently suppressed by the helminth.
Keywords: arthritis, collagen, helminth, IL‐4, IL‐10, IL‐17, Schistosoma, STAT6
Introduction
In recent decades, the incidence of autoimmune or allergic diseases has been increasing, whereas that of infectious diseases is decreasing worldwide 1. The prevalence of certain immune diseases is higher in developed countries than in developing countries 2. A recent explanation is the ‘hygiene hypothesis’; i.e. exposure to infectious agents, especially bacteria and parasites, prevents the onset of immune diseases. There are many reports to support this hypothesis, although there are controversial findings 3, 4.
Parasitic helminths have to survive by continuously struggling against or evading immune attacks by their hosts. Therefore, in general, they have immunomodulatory activity against the infected hosts. We have been studying the effects of parasitic helminths on experimental autoimmune or inflammatory diseases focusing on arthritis and diabetes 5, 6, 7, 8.
There are several reports on helminth infections in arthritis models, many of which demonstrated anti‐arthritic effects. In 1975, Pearson and Taylor found that a Syphacia oblevata‐infested rat colony was resistant to the development of adjuvant arthritis 9. Salinas‐Carmona et al. 10 found that other enteric nematode infections ameliorated arthritis in MRL/lpr mice. In 2009, we reported that a blood fluke distributed in Africa and South America (Schistosoma mansoni; Sm) reduced the severity of collagen‐induced arthritis (CIA), one of the most widely used rodent models of human rheumatoid arthritis (RA) 7. Subsequently, other researchers 11, 12 also reported anti‐arthritic effects of schistosome infection on CIA using an Asian blood fluke S. japonicum (Sj). In our study, Sm infection down‐regulated the splenic production of proarthritic cytokines [interleukin (IL)‐17A and tumor necrosis factor (TNF)‐α], accompanying the up‐regulation of anti‐arthritic cytokines (IL‐4 and IL‐10). Although such cytokine alterations [T helper type 2 (Th2) and/or regulatory T cell (Treg) polarization] are often observed in helminthic infections, the requirement of Th2 cytokines for disease prevention is not fully elucidated. Until now, varying results have been reported regarding the involvement of IL‐4 [or signal transducer and activator of transcription 6 (STAT6)] and IL‐10 in the anti‐autoimmune activity of helminths. For example, Sm ova lost their protective effects against experimental autoimmune encephalomyelitis (EAE) in the genetic absence of STAT6 13, whereas Fasciola hepatica (Fh) ES products protected mice from EAE in an IL‐4‐ and IL‐10‐independent manner 14. Several reports, including ours 5, 6, demonstrated that IL‐4 and IL‐10 were not essential for the anti‐diabetic effects of helminths 15, 16. Regarding arthritis, Shi et al. 17 reported that the anti‐arthritic effects of an intestinal tapeworm, Hymenolepis diminuta (Hd), on adjuvant‐induced monoarthiritis in mice (non‐permissive hosts) were lost under the genetic absence of IL‐4R or IL‐10. Recently, Chen et al. 18 found that the enteric nematode Nippostrogylus brasiliensis (Nb) protected mice against K/B×N serum‐induced arthritis (SIA) in an IL‐4/IL‐13‐dependent manner. Based on such findings, we hypothesized that Th2 cytokine signals are required for the anti‐arthritic effects of Sm. We evaluated the effects of Sm infection on CIA in STAT6 KO and IL‐10 KO mice, and discuss the possible roles of STAT6‐related cytokines (IL‐4, IL‐13) and IL‐10.
Materials and methods
Parasite
The Puerto Rican strain of Sm was maintained as described previously 5. Briefly, ICR mice were infected percutaneously with Sm by tail immersion in a cercarial suspension. Eight weeks or more after infection, portal perfusion was performed with 0·45% trisodium citrate containing physiological saline in order to harvest adult worms. Thereafter, the granulomatous livers of the mice were minced and digested with collagenase and actinase in 1·7% NaCl solution (2× saline) for 4–6 h at 37°C under continuous shaking. Eggs were purified by mesh filtration and then repeatedly washed by centrifugation. Miracidia were obtained by adding aged tap water onto the eggs, and used immediately for infection of the intermediate host snails (Puerto Rican strain of Biomphalaria glabrata).
Mice
STAT6 KO mice 19 were kindly provided by Dr Akira at Osaka University and IL‐10 KO mice 20 were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). As both mice strains were C57BL/6 background, we performed a 10‐generation back‐cross to the DBA1/J strain originated from SLC Japan (Hamamatsu, Japan). After back‐crossing, stable onset of CIA in both KO mice strains was confirmed by the same method used for wild‐type (WT) DBA/1J mice.
Infection of Sm and induction of CIA
Male STAT6 KO mice or IL‐10 KO mice (more than 6 weeks old at the time of infection) were used together with age‐matched WT DBA1/J mice. The mice were infected with 80 cercariae by tail immersion in aged tap water containing the cercariae for 30 min. Three weeks later, the mice were immunized intradermally (i.d.) with 200 μg of bovine type‐II collagen (IIC) (Cosmo Bio Co. Ltd, Tokyo, Japan) emulsified with Freund’s complete adjuvant (FCA) (Difco Laboratories, Detroit, MI, USA) at the dorsal skin near the base of the tail. From 3 weeks post‐immunization, the severity of arthritis was evaluated in each mouse every week, using both an arthritis score based on swelling (graded 0–3 for each paw; 0: normal, 1: slight to moderate swelling, 2: marked swelling, 3: maximal swelling) and the number of ‘arthritic’ paws, which had thickened by more than or equal to 2·2 mm. In addition to swelling, the presence of ankylosis was evaluated as positive or negative. At 6 weeks post‐immunization the mice were euthanized, and the paws, the popliteal lymph nodes (PLNs) of hind paws and spleens were sampled for histopathological examination, real‐time polymerase chain reaction (PCR) analysis and cytokine production assay, respectively. At the same time, adult worms were recovered from each infected mouse by portal perfusion and enumerated. All animal experiments were performed after approval by the Ethics Committee of Animal Care and Experimentation in accordance with the Guiding Principles for Animal Care Experimentation, The University of Occupational and Environmental Health, Japan and the Japanese Law for Animal Welfare and Care (no. 221).
Histopathology
At the end of the animal experiments, the paws of each mouse were removed and fixed in 10% neutral buffered formalin. The fixed paws were cut longitudinally with a razor, decalcified with formic acid and embedded in paraffin. The tissue sections were stained by hematoxylin and eosin (H&E).
Anti‐IIC antibody measurement
Antibodies against bovine IIC were measured by microplate enzyme‐linked immunosorbent assay (ELISA). The antigen (IIC) was diluted in 50 mM bicarbonate buffer (pH 9.6) to a final concentration of 1 μg/ml and used to coat wells of 96‐well microplates. Fifty microliters of the diluted antigen solution was added to each well. The plates were kept at 4°C overnight and briefly washed with washing buffer [phosphate‐buffered saline (PBS) containing 0·05% Tween 20]. The antigen‐coated plates were then blocked with 1% bovine serum albumin (BSA)/PBS containing 0·05% sodium azide for 2 h at room temperature. Serum samples were diluted 1 : 2000 with dilution buffer (1% BSA/PBS without sodium azide). Fifty microliters of diluted samples were added to the antigen‐coated plates and incubated at room temperature for 1 h. After washing three times with the washing buffer, appropriately diluted peroxidase‐conjugated anti‐mouse immunoglobulin (Ig)G (Sigma, St Louis, MO, USA), anti‐IgG1 or anti‐IgG2a (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) was added to the wells. After incubation at room temperature for 1 h, the plates were washed four times with the washing buffer. 2,2' azino‐bis[3‐ethilbenzthiazoline‐6‐sulfonic acid], diammonium salt (Moss, Inc., Pasadena, MD, USA) was used as a substrate. Optical density (OD) values were measured at 415 nm using a Model 550 microplate reader, and the results were analyzed using Microplate Manager III for Windows (Biorad Laboratories, Hercules, CA, USA).
Spleen cell culture and cytokine ELISA
At the end of the animal experiments, spleen cells were harvested and cultured in RPMI‐1640 medium supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin, at 6 million cells/ml on 24‐well culture plates. The splenocytes were stimulated with anti‐CD3 monoclonal antibody 2C11 (0·5 μg/ml) (eBioscience, San Diego, CA, USA). The supernatants were collected at 48 h and kept frozen at –80°C until use. Pro‐ or anti‐arthritic T helper cytokines (IL‐17A, TNF‐α, IFN‐γ, IL‐4 and IL‐10) in the culture supernatants were measured with the ELISA kit Ready‐Set Go! (eBioscience) according to the manufacturer’s instructions.
RNA extraction and real‐time PCR analysis of PLNs
At the end of the animal experiments, PLNs were taken from each mouse, snap‐frozen in liquid nitrogen and kept at –80°C until use. Eight hundred microliters of homogenizing reagent RNAiso (Takara Bio Inc., Otsu, Japan) was added to each sample. After homogenization, a 0·2‐fold volume of chloroform was added and the samples were centrifuged at 10 000 g for 15 min. The aqueous upper phase was collected and RNA was precipitated by adding 500 μl of isopropanol. After centrifugation, the RNA pellet was rinsed with 70% ethanol, centrifuged, air‐dried and redissolved in RNAase‐free water. The purified RNA samples were reverse‐transcribed using the PrimeScript RT reagent kit with gDNA Eraser (Takara Bio). The cDNA samples were used for real‐time PCR analysis with a StepOne Plus thermal cycler (Applied Biosystems/Life Technologies Corporation, Carlsbad, CA, USA) and SYBR Premix Ex Taq II kit (Takara Bio). The sequences of each primer were based on commercially available pre‐made primer sets (Takara Bio Perfect Real Time Primer Support System) and summarized in Table 1. The housekeeping gene β‐actin was used to normalize the expression levels of each gene. Relative gene expression levels were calculated based on standard curves created by serial dilutions of highly expressing samples for each gene.
Table 1.
Sequences of primers used for real‐time polymerase chain reaction (PCR) analysis (5′–3′)
| Gene | Forward | Reverse |
|---|---|---|
| β‐actin | CATCCGTAAAGACCTCTATGCCAAC | ATGGAGCCACCGATCCACA |
| IL‐1β | TCCAGGATGAGGACATGAGCAC | GAACGTCACACACCAGCAGGTTA |
| IL‐6 | CCACTTCACAAGTCGGAGGCTTA | CCAGTTTGGTAGCATCCATCATTTC |
| RANKL | AAACTGGTCGGGCAATTCTG | AGGGTTGGACACCTGAATGCTA |
| IL‐21 | CAGGCTAAGAGCTTGTATCGTTTGG | AGGACTGGCTGAGTCTTGAGCAC |
| TNF‐α | TATGGCCCAGACCCTCACA | GGAGTAGACAAGGTACAACCCATC |
IL = interleukin; RANKL = receptor activator of nuclear factor kappaB ligand; TNF = tumor necrosis factor.
Statistical analyses
Differences in disease severity and immunological parameters between experimental groups were tested using the Mann–Whitney U‐test. Differences in anti‐IIC antibody levels were analyzed with the unpaired t‐test. Correlations between arthritis scores/the numbers of ankylosing paws and cytokine expression levels were analyzed based on Spearman’s rank correlation coefficients (shown in bold type in Table 2). Correlations among cytokine expression levels were analyzed based on Pearson’s correlation coefficients. Statistical analysis was performed with StatView version 5.0 software.
Table 2.
Correlations between cytokines and arthritis severity in STAT6 KO mice
| IL‐6 | 0.704*** | ||||
| RANKL | 0.532** | 0.729*** | |||
| IL‐21 | 0.394 | 0.421* | 0.454* | ||
| TNF‐α | –0.220 | –0.282 | –0.189 | –0.609*** | |
| Score | 0.603** | 0.470* | 0.277 | 0.465* | –0.279 |
| Ankylosis | 0.646*** | 0.511** | 0.304 | 0.488* | –0.353 |
| IL‐1β | IL‐6 | RANKL | IL‐21 | TNF‐α | |
Bold letters represent Spearman’s rank correlation coefficients. Other values are Pearson’s correlation coefficients.
Results
CIA is weakened in STAT6 KO mice and promoted in IL‐10 KO mice
Disease severity was evaluated by edematous change as measured by the arthritis score, shown in Fig. 1a, and paw thickness, shown in Fig. 1b. STAT6 KO mice and IL‐10 KO mice developed CIA following the immunization protocol usually used for DBA/1 (WT) mice. Compared with WT mice, edematous change was significantly reduced in STAT6 KO mice. In contrast, edema was increased in IL‐10 KO mice. These results demonstrated that CIA was weakened in STAT6 KO mice and promoted in IL‐10 KO mice.
Figure 1.
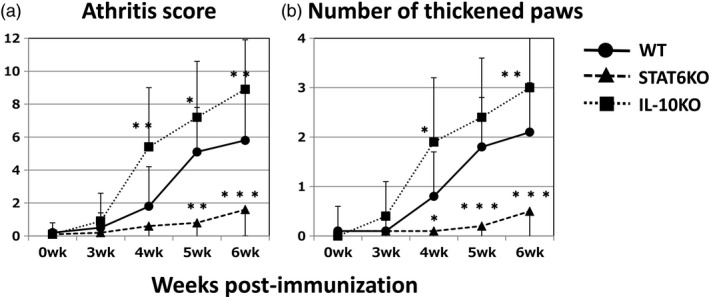
Genetic absence of signal transducer and activator of transcription 6 (STAT6) or interleukin (IL‐10) affects the development of collagen‐induced arthritis (CIA) in DBA/1 mice. Circles, triangles and rectangles represent type‐II collagen (IIC)‐immunized wild‐type (WT), STAT6 knock‐out (KO) and IL‐10 KO mice, respectively. Data are expressed as the mean ± standard deviation (s.d.) of total scores for four limbs (a) or total number of arthritic paws (thickness ≥ 2·2 mm) for each mouse (b). *P < 0.05 and **P < 0.01, respectively (Mann–Whitney U‐test). The combined results of four independent experiments are shown.
Sm infection suppresses CIA in WT mice but exacerbates CIA in STAT6 KO mice
Consistent with our previous report 7, prior Sm infection significantly reduced the severity of CIA in WT DBA/1 mice (Fig. 2a,d). Conversely, in the genetic absence of STAT6, Sm markedly and significantly exacerbated CIA (Fig. 2b,e). These effects were also supported by the data of ankylosis (Fig. 3a,b). Based on histopathological observation, Sm infection reduced cell infiltration and bone destruction (indicated by arrows) in WT mice (Fig. 4a,d). However, Sm largely increased these inflammatory changes in STAT6 KO mice (Fig. 4b,e). The numbers of recovered adult worms were comparable between WT mice and STAT6 KO mice (data not shown).
Figure 2.
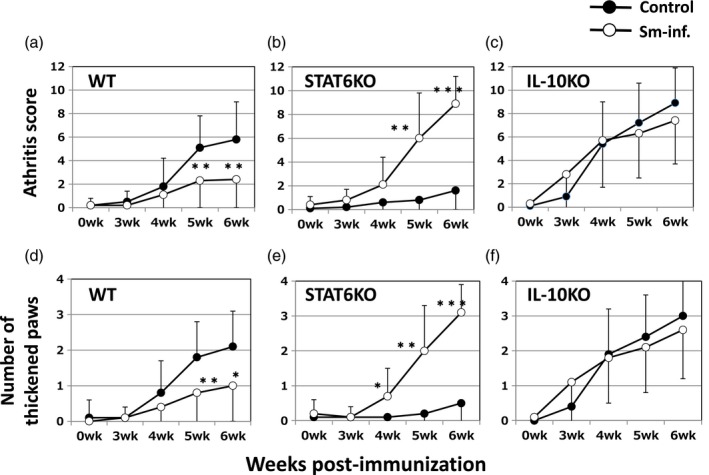
Effects of Schistosoma mansoni (Sm) on edematous changes in CIA differ among WT (a,d), STAT6 KO (b,e) and IL‐10 KO (c,f) mice. The open and closed circles represent IIC immunized mice with and without Sm infection, respectively. Mice were infected with Sm 3 weeks prior to IIC‐immunization. Data are expressed as the mean ± standard deviation (s.d.) of total arthritis scores for four limbs (a–c) or total numbers of arthritic paws for each mouse (d–f). The values for control mice (closed circles) are the same as those in Fig. 1. *P < 0·05 and **P < 0·01, respectively (Mann–Whitney U‐test). The combined results of four independent experiments are shown.
Figure 3.
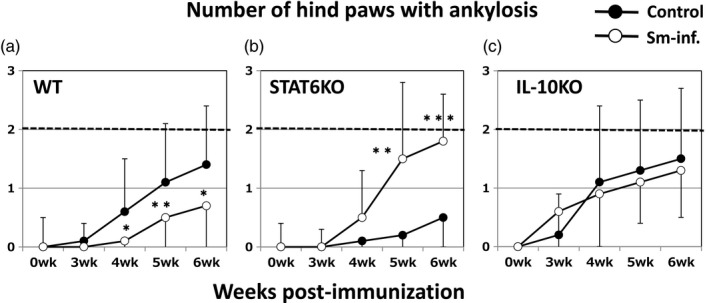
The effects of Sm on joint ankylosis in WT (a), signal STAT6 KO (b) and IL‐10 KO (c) mice are consistent with the effects of Sm on edematous changes. The open and closed symbols represent IIC‐immunized mice with and without Sm infection, respectively. Data are the mean ± standard deviation (s.d.) of the total number of hind paws with ankylosis for each mouse. *P < 0·05 and **P < 0·01, respectively (Mann–Whitney U‐test). The combined results of four independent experiments are shown.
Figure 4.
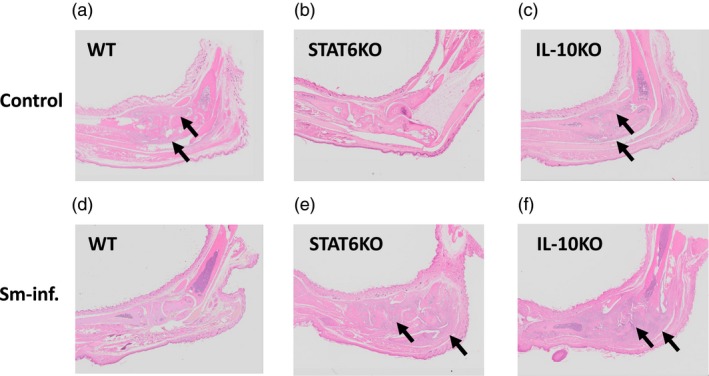
The effects of Sm on joint pathology in WT (a), STAT6 KO (b) and IL‐10 KO (c) mice are consistent with the effects of Sm on edematous changes. Images are hematoxylin and eosin (H&E) staining of ankle joints of CIA mice at 6 weeks post‐immunization. The arrows indicate the presence of joint destruction and inflammation with a marked influx of inflammatory cells. Note that Sm infection ameliorated or exacerbated joint inflammation in WT or STAT6 KO mice, respectively. In IL‐10 KO mice, severe joint inflammation was observed in both the control and infected mice.
Sm infection does not affect the severity of CIA in IL‐10 KO mice
In IL‐10 KO mice, disease severity evaluated by edematous change was comparable between Sm‐infected mice and non‐infected control mice until 6 weeks post‐immunization (Fig. 2c,f). The numbers of ankylosis‐positive hind paws in each group (Fig. 3c) coincided with the results for edema. These results were supported by histopathological observations (Fig. 4c,f). Marked cell infiltration and bone destruction was observed both in control mice and Sm‐infected mice (indicated by arrows). The numbers of adult worms recovered from the portal veins were comparable between WT mice and IL‐10 KO mice (data not shown).
Sm infection reduces anti‐IIC IgG production in WT mice but not in STAT6 KO or IL‐10 KO mice
Sm infection reduced the serum IIC IgG levels in the immunized WT mice (bars of WT in Fig. 5a–c). Worm infection attenuated production of all whole IgG (significant), IgG1 (significant) and IgG2a (not significant). In contrast, we did not observe significant attenuation of anti‐IIC IgG/IgG1/IgG2a production in STAT6 KO or IL‐10 KO mice (bars of STAT6 KO and IL‐10 KO in Fig. 5a–c).
Figure 5.
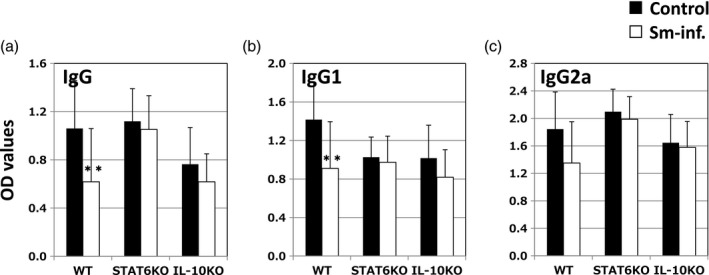
Sm lowers serum anti‐type‐II collagen (IIC) immunoglobulin (Ig)G levels in wild‐type (WT) mice but not in signal transducer and activator of transcription 6 (STAT6) knock‐out (KO) or interleukin (IL)‐10 KO mice. (a–c) IgG, IgG1 and IgG2a, respectively. Closed and open bars represent non‐infected mice and Sm‐infected mice, respectively. Means ± standard deviation (s.d.) [of optical density (OD) 415] for each mouse are shown. *P < 0·05 and **P < 0·01, respectively (Student’s t‐test). The combined results of four independent experiments are shown.
Sm infection down‐regulates Th1/Th17 cytokines and up‐regulates Th2 cytokines in WT mice
Since the 1990s, Sm has been known to induce robust Th2‐polarization by laying a large number of eggs in the host blood vessels 21. Th2 polarization regulates both parasite antigens and unrelated foreign antigens 22. Moreover, infection attenuated the production of IL‐17A and TNF‐α 7, 8, which are essential cytokines for the pathogenesis of autoimmune arthritis 23, 24, 25, 26, 27. We confirmed the cytokine‐altering effects of Sm, i.e. down‐regulation of Th1/Th17 cytokines (IL‐17A, TNF‐α and IFN‐γ) and up‐regulation of Th2 cytokines (IL‐4 and IL‐10) in WT mice (Fig. 6a).
Figure 6.
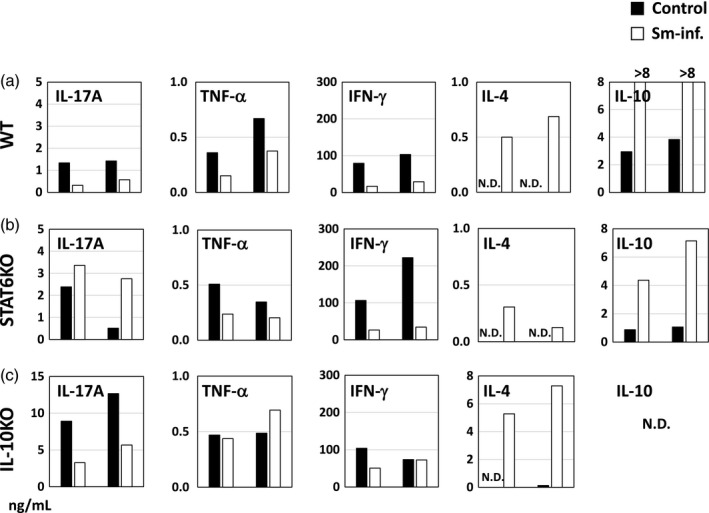
Sm‐induced regulation of pro/anti‐inflammatory cytokines is altered by the genetic absence of STAT6 or IL‐10. (a–c) Data from WT, STAT6 KO and IL‐10 KO mice, respectively. Spleen cells from mice in the each treatment group were pooled and cultured upon stimulation with anti‐CD3 monoclonal antibody. Closed and open bars represent non‐infected control mice and Sm‐infected mice, respectively. Data from two representative experiments are shown as two sets of bars in each panel.
STAT6 is essential for Sm‐induced down‐regulation of IL‐17A
The genetic absence of STAT6 abolished Sm‐induced down‐regulation of IL‐17A (Fig. 6b). Conversely, IL‐17A production was somewhat increased by the infection; however, down‐regulation of TNF‐α and IFN‐γ by the infection remained in STAT6 KO mice. The infection‐induced up‐regulation of IL‐4 and IL‐10 also remained, but their increase was attenuated by the absence of STAT6.
IL‐10 regulates the basal level of IL‐17A, and is essential for the Sm‐induced down‐regulation of TNF‐α
One notable difference between non‐infected WT mice and IL‐10 KO mice was the basal IL‐17A level (Fig. 6a,c, IL‐17A panels). A markedly high basal IL‐17A production (ca. 10 ng/ml) was observed in non‐infected IL‐10 KO mice. Therefore, even after down‐regulation by Sm, the IL‐17A level was still higher than that in non‐infected WT mice (1–2 ng/ml). In addition, Sm‐induced down‐regulation of TNF‐α was abolished and IFN‐γ was not consistently down‐regulated (Fig. 6c, TNF‐α and IFN‐γ panels).
Sm increases IL‐1β expression in PLNs of STAT6 KO mice
To clarify the cytokine profiles in the draining lymph nodes of inflamed paws, we measured cytokine gene expression in PLNs from the hind paws. In WT mice, there were no significant changes in the expression of arthritogenic cytokines by the infection (Fig. 7a), whereas in the infected STAT6 KO mice, we observed a significantly higher expression of IL‐1β than that in non‐infected control mice (Fig. 7b, IL‐1β panel). In IL‐10 KO mice most cytokines, except receptor activator of nuclear factor kappaB ligand (RANKL), were not mobilized by the infection (Fig. 7c).
Figure 7.
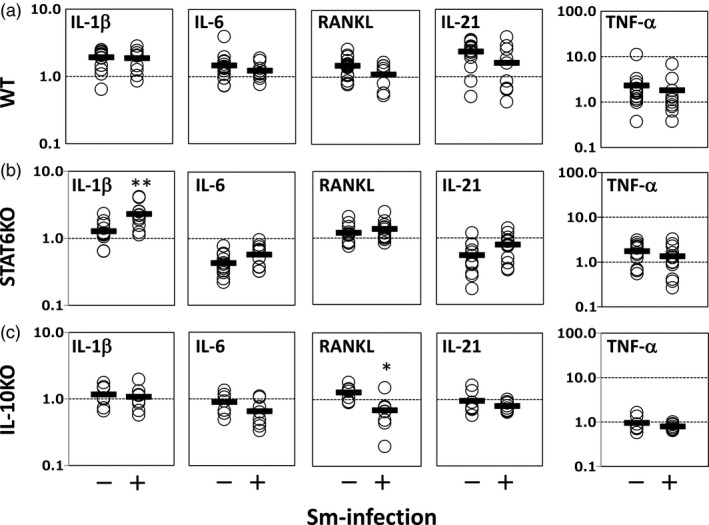
Sm increases IL‐1β expression in popliteal lymph nodes (PLNs) of STAT6 KO mice. (a,b,c) Data from WT, STAT6 KO and IL‐10 KO mice, respectively. Values along the vertical axis represent the fold increases in gene expression over normal mouse controls. Expression levels were normalized with β‐actin. Each dot represents an individual mouse. The means of each group are shown as short horizontal bars. *P < 0·05 and **P < 0·01, respectively (Mann–Whitney U‐test). The combined results of four independent experiments are shown.
Arthritis severity is correlated with PLN IL‐1β expression in STAT6 KO mice
To evaluate the correlation between arthritis severity in the hind paws and LN gene expression levels in STAT6 KO mice, we calculated correlation coefficients. Data for non‐infected mice and infected mice were pooled, and correlations among each parameter were analyzed. As shown in Table 2, IL‐1β expression was correlated highly with the arthritis score and ankylosis. The other arthritogenic cytokines IL‐6 and IL‐21 were also correlated positively with the score and ankylosis. TNF‐α did not correlate with the other cytokine levels or arthritis severity.
Discussion
Parasitic helminths are known to evoke Th2‐biased immunity characterized by hyper‐IgE and eosinophilia. Helminthic infections also induce Treg cells, IL‐10‐producing B (Breg) cells and M2 macrophages. These immunological changes by helminths have been reported to be involved in the suppression of inflammatory and/or allergic diseases 28. Among helminth‐induced regulatory cytokines, IL‐4, IL‐13 and IL‐10 are considered to be key players in their anti‐arthritic effects because these cytokines may be negative regulators of arthritis development 29, 30, 31, 32, 33, 34, 35, 36, 37. As STAT6 is an adaptor molecule essential for signaling IL‐4 and IL‐13, we used STAT6 KO mice and IL‐10 KO mice to evaluate the possible roles of these cytokines in the anti‐arthritic effects of Sm.
Before evaluation of Sm infection on CIA, we first examined the progression of CIA in the two gene‐deficient mouse strains without infection. As shown in Fig. 1a,b, CIA was exacerbated in IL‐10 KO mice. This phenomenon was reported previously 35, 38, and is theorized to be due to the regulatory roles of IL‐10 for Th17 and CIA 36, 37, 38. Splenic IL‐17A production was markedly enhanced by the absence of IL‐10 (Fig. 6c), suggesting the essential role of this cytokine for controlling basal IL‐17A production. In contrast to IL‐10 KO mice, CIA development was attenuated significantly in STAT6 KO mice (Fig. 1a,b). Unlike IL‐10, the roles of IL‐4 in CIA are controversial, because this cytokine plays both promoting and suppressive roles 30, 31, 32, 33, 34, 39, 40, 41, 42. According to previous reports, IL‐4 promotes CIA onset at the early phase of arthritis and suppresses the established arthritis. Therefore, the absence of IL‐4 signaling in the early phase of CIA in STAT6 KO mice may explain the slower arthritis development in our experiments.
Next, we compared the effects of Sm infection between WT mice and two KO mice strains. We found that the effects of Sm infection on CIA were largely affected by the genetic absence of STAT6 or IL‐10. As shown in Fig. 2a versus b, d versus e and 3a versus b, STAT6 deficiency reversed the anti‐arthritic effects of Sm on CIA, and arthritis was markedly exacerbated by the infection. Arthritis prevention in WT mice and exacerbation in STAT6 KO mice was also confirmed by histopathological observation (Fig. 4a versus d, b versus e). These results suggest that Sm has both STAT6‐dependent anti‐arthritic mechanisms and STAT6‐independent pro‐arthritic mechanisms.
Anti‐IIC antibodies are considered to promote arthritis via destruction of cartilage 43. As shown in Fig. 5, Sm infection decreased the levels of anti‐IIC IgG (a)/IgG1 (b)/IgG2a (c). Moreover, we found that the lowering effects on anti‐IIC IgG production were abrogated or reduced in both STAT6 KO and IL‐10 KO mice. As this was equally observed for whole IgG, IgG1 and IgG2a, ‘helminth‐induced Th2‐polarization’ cannot explain the down‐regulation of IgG. Although the underlying mechanism is not clear, the loss of the IgG‐lowering effects of Sm in STAT6 KO and IL‐10 KO mice may be related, in part, to the loss of the anti‐arthritic effects in these KO mice.
Consistent with arthritis exacerbation, the splenic potential of IL‐17A production was increased in the infected STAT6 KO mice (Fig. 6b). One possible explanation is that down‐regulation of IFN‐γ by the helminth renders Th17 cells more activated/expanded in STAT6 KO mice. Different from IL‐17A, helminth‐induced suppression of splenic IFN‐γ production remained in STAT6 KO mice. IFN‐γ was reported to negatively regulate Th17 development 42 and the action of IL‐17 on target cells 44. Although both IFN‐γ and IL‐4 are known to be anti‐arthritic cytokines, IFN‐γ is primarily important for negative regulation of Th17 and arthritis development 42. Indeed, in CIA non‐susceptible C57BL/6 mice, genetic absence of IFN‐γ or its receptor rendered the mice more susceptible to CIA 44, 45. The importance of IFN‐γ in the negative regulation of Th17‐dependent arthritis is consistent with previous findings that ‘Th1‐polarizing’ protozoan parasites ameliorate arthritis. Trypanosoma brucei protects rats from CIA 46 and Toxoplasma gondii ameliorates spontaneous arthritis in IL‐1 receptor antagonist‐deficient mice 47. Another possible explanation for the exacerbation in STAT6 KO mice is IL‐21 induction by Sm infection. Sm infection induces IL‐21 expression in the liver 48. As IL‐21 is known to promote autoimmune inflammation, including CIA 49, 50, 51, this cytokine may be partially involved in the exacerbation of CIA in the absence of STAT6. Although the infection‐induced increase in IL‐21 expression in STAT6 KO mice was not significant (Fig. 7b, IL‐21 panel), it was correlated positively with the arthritis score and ankylosis (Table 2).
In IL‐10 KO mice, Sm infection did not significantly alter arthritis development (Figs 2c and 3c). IL‐17A down‐regulation by Sm remained in IL‐10 KO mice (Fig. 6c, IL‐17A panel), indicating that IL‐10 is not essential for the down‐regulation of Th17 response. The inconsistency as to why Sm was unable to affect arthritis in IL‐10 KO mice may be explained by the marked increase in basal IL‐17A production level and consequent arthritis symptoms in the genetic absence of IL‐10. Even with the down‐regulation of IL‐17A by Sm infection, the cytokine amount may have been sufficient to induce severe arthritis. In other words, arthritic inflammation induced in IL‐10 KO mice was too severe to be regulated by Sm‐mediated IL‐17A down‐regulation. In contrast to IL‐17A, the down‐regulation of TNF‐α observed in WT mice was abolished in IL‐10 KO mice (Fig. 6c, TNF‐α panel).
We next measured the expression of arthritogenic cytokines in PLNs. As shown in Fig. 7, there were only a few significant changes due to Sm infection: the increase of IL‐1β in the infected STAT6 KO mice (Fig. 7b, IL‐1β panel) and the decrease of RANKL in the infected IL‐10 KO mice (Fig. 7c, RANKL panel). The former was consistent with the change in disease severity, suggesting that IL‐1β augmentation may be related to arthritis exacerbation. As IL‐1β production is negatively regulated by IFN‐γ 45, 52, helminth‐induced down‐regulation of IFN‐γ (Fig. 6b, IFN‐γ panel) may be responsible for this IL‐1β augmentation. Moreover, we calculated correlation coefficients for arthritis severity and PLN cytokine expression in STAT6 KO mice (Table 2). Of note, IL‐1β expression was correlated most highly with the arthritis score and ankylosis in STAT6 KO mice. This result supports the relationship between IL‐1β augmentation and arthritis exacerbation in STAT6 KO mice.
Regarding the involvement of Th2 cytokines in the protection against arthritis, Shi et al. 17 found that the genetic absence of IL‐4R or IL‐10 abrogated the anti‐arthritic effects of Hd for adjuvant‐induced monoarthritis in mice. The Nb‐induced protection from SIA was IL‐4/IL‐13/STAT6‐dependent 18. Our current study results are consistent with these previous reports demonstrating the requirement of these cytokines for the anti‐arthritic effects of helminths. However, it is premature to conclude that the anti‐arthritic effects of helminths are generally Th2‐dependent. In our experiments, down‐regulation of IFN‐γ by Sm was not STAT6‐dependent (Fig. 6), suggesting that Sm might be effective against ‘Th1‐type arthritis’, such as proteoglycan‐induced arthritis 53, via STAT6‐independent mechanisms. In addition, we recently found that Trichinella spiralis (Ts) suppressed CIA in a STAT6‐ and IL‐10‐independent manner (manuscript in preparation). Therefore, the anti‐arthritic mechanisms of helminths should be investigated carefully for each parasite and arthritis model to avoid a rough generalization of study results.
According to our study, Sm may have pro‐arthritic mechanisms in addition to anti‐arthritic mechanisms. There are several previous reports of arthritis‐promoting effects of helminths, including Sm. For example, experimental arthropathy 54 and human polyarthritis 55 are caused by Sm infection. Sm increases the serum levels of rheumatoid factor and anti‐dsDNA in IL‐1 receptor antagonist‐deficient mice that develop spontaneous autoimmune arthritis 8. Graepel et al. 56 reported that the intestinal tapeworm Hd exacerbates K/B×N SIA. These studies demonstrate the complexity of helminths and difficulty of therapeutic usage of viable worms.
In schistosomiasis, the egg embolization causes liver fibrosis and pulmonary hypertension in a Th2‐dependent manner 57, 58. Therefore, it is not possible to use viable schistosome or its eggs for therapeutic purposes. An alternative is the isolation of anti‐arthritic products from helminths. In clinical application of effector molecules the pathogenicity of helminths does not need to be considered, and anti‐arthritic effects and pro‐arthritic effects can be separated by isolation of effector molecules. Currently, several helminth‐derived products, cystatin 59, rSj16 60 and SJMHE1 61 of Sj, the filarial product ES‐62 62, 63, 64, crude antigens from Sm and Ts 65, Fasciola sp. 66, 67 and Ascaris suum 68 have been reported to protect against experimental arthritis.
In conclusion, we found that STAT6‐mediated signaling, i.e. IL‐4 and/or IL‐13, is essential for the down‐regulation of IL‐17A and arthritis prevention by Sm infection. IL‐10 regulated the basal IL‐17A level and arthritis severity, and may therefore be indirectly involved in the anti‐arthritic effects of Sm. Furthermore, the Sm‐induced arthritis exacerbation in STAT6 KO mice suggests the presence of pro‐arthritic mechanisms, in addition to anti‐arthritic mechanisms, in this parasite.
Disclosure
The authors have no conflicts of interest.
Acknowledgements
This work was supported by Grants‐in‐Aids for Scientific Research from Japan Society for Promotion of Science (No.25460523). We are grateful to the other laboratory members for their valuable scientific advice. We acknowledge clerical assistance of Ms Satomi Ito and Ms Mamiko Mikuma.
References
- 1. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 2002;347:911–20. [DOI] [PubMed] [Google Scholar]
- 2. Shapira Y, Agmon‐Levin N, Shoenfeld Y. Defining and analyzing geoepidemiology and human autoimmunity. J Autoimmun 2010;34:J168–77. [DOI] [PubMed] [Google Scholar]
- 3. Leonardi‐Bee J, Pritchard D, Britton J. Asthma and current intestinal parasite infection: systematic review and meta‐analysis. Am J Respir Crit Care Med 2006;174:514–23. [DOI] [PubMed] [Google Scholar]
- 4. Lynch NR, Palenque M, Hagel I, DiPrisco MC. Clinical improvement of asthma after anthelminthic treatment in a tropical situation. Am J Respir Crit Care Med 1997;156:50–4. [DOI] [PubMed] [Google Scholar]
- 5. Osada Y, Fujiyama T, Kamimura N, et al. Dual genetic absence of STAT6 and IL‐10 does not abrogate anti‐hyperglycemic effects of Schistosoma mansoni in streptozotocin‐treated diabetic mice. Exp Parasitol 2017;177:1–12. [DOI] [PubMed] [Google Scholar]
- 6. Osada Y, Yamada S, Nabeshima A, et al. Heligmosomoides polygyrus infection reduces severity of type 1 diabetes induced by multiple low‐dose streptozotocin in mice via STAT6‐ and IL‐10‐independent mechanisms. Exp Parasitol 2013;135:388–96. [DOI] [PubMed] [Google Scholar]
- 7. Osada Y, Shimizu S, Kumagai T, Yamada S, Kanazawa T. Schistosoma mansoni infection reduces severity of collagen‐induced arthritis via down‐regulation of pro‐inflammatory mediators. Int J Parasitol 2009;39:457–64. [DOI] [PubMed] [Google Scholar]
- 8. Osada Y, Yamada S, Nakae S, Sudo K, Kanazawa T. Reciprocal effects of Schistosoma mansoni infection on spontaneous autoimmune arthritis in IL‐1 receptor antagonist‐deficient mice. Parasitol Int 2015;64:13–7. [DOI] [PubMed] [Google Scholar]
- 9. Pearson DJ, Taylor G. The influence of the nematode Syphacia oblevata on adjuvant arthritis in the rat. Immunology 1975;29:391–6. [PMC free article] [PubMed] [Google Scholar]
- 10. Salinas‐Carmona MC, de la Cruz‐Galicia G, Perez‐Rivera I, et al. Spontaneous arthritis in MRL/lpr mice is aggravated by Staphylococcus aureus and ameliorated by Nippostrongylus brasiliensis infections. Autoimmunity 2009;42:25–32. [DOI] [PubMed] [Google Scholar]
- 11. He Y, Li J, Zhuang W, et al. The inhibitory effect against collagen‐induced arthritis by Schistosoma japonicum infection is infection stage‐dependent. BMC Immunol 2010;11: 28, 2172–11–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song X, Shen J, Wen H, et al. Impact of Schistosoma japonicum infection on collagen‐induced arthritis in DBA/1 mice: a murine model of human rheumatoid arthritis. PLOS ONE 2011;6:e23453.21858123 [Google Scholar]
- 13. Sewell D, Qing Z, Reinke E, et al. Immunomodulation of experimental autoimmune encephalomyelitis by helminth ova immunization. Int Immunol 2003;15:59–69. [DOI] [PubMed] [Google Scholar]
- 14. Finlay CM, Stefanska AM, Walsh KP, et al. Helminth products protect against autoimmunity via innate type 2 cytokines IL‐5 and IL‐33, which promote eosinophilia. J Immunol 2016;196:703–14. [DOI] [PubMed] [Google Scholar]
- 15. Liu Q, Sundar K, Mishra PK, et al. Helminth infection can reduce insulitis and type 1 diabetes through CD25‐ and IL‐10‐independent mechanisms. Infect Immun 2009;77:5347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mishra PK, Patel N, Wu W, Bleich D, Gause WC. Prevention of type 1 diabetes through infection with an intestinal nematode parasite requires IL‐10 in the absence of a Th2‐type response. Mucosal Immunol 2013;6:297–308. [DOI] [PubMed] [Google Scholar]
- 17. Shi M, Wang A, Prescott D, et al. Infection with an intestinal helminth parasite reduces Freund's complete adjuvant‐induced monoarthritis in mice. Arthritis Rheum 2011;63:434–44. [DOI] [PubMed] [Google Scholar]
- 18. Chen Z, Andreev D, Oeser K, et al. Th2 and eosinophil responses suppress inflammatory arthritis. Nat Commun 2016;7:11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeda K, Tanaka T, Shi W, et al. Essential role of Stat6 in IL‐4 signalling. Nature 1996;380:627–30. [DOI] [PubMed] [Google Scholar]
- 20. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin‐10‐deficient mice develop chronic enterocolitis. Cell 1993;75:263–74. [DOI] [PubMed] [Google Scholar]
- 21. Grzych JM, Pearce E, Cheever A, et al. Egg deposition is the major stimulus for the production of Th2 cytokines in murine Schistosomiasis mansoni . J Immunol 1991;146:1322–7. [PubMed] [Google Scholar]
- 22. Kullberg MC, Pearce EJ, Hieny SE, Sher A, Berzofsky JA. Infection with Schistosoma mansoni alters Th1/Th2 cytokine responses to a non‐parasite antigen. J Immunol 1992;148:3264–70. [PubMed] [Google Scholar]
- 23. Williams RO, Marinova‐Mutafchieva L, Feldmann M, Maini RN. Evaluation of TNF‐alpha and IL‐1 blockade in collagen‐induced arthritis and comparison with combined anti‐TNF‐alpha/anti‐CD4 therapy. J Immunol 2000;165:7240–5. [DOI] [PubMed] [Google Scholar]
- 24. Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen‐induced arthritis in IL‐17‐deficient mice. J Immunol 2003;171:6173–7. [DOI] [PubMed] [Google Scholar]
- 25. Lubberts E, van den Bersselaar L, Oppers‐Walgreen B, et al. IL‐17 promotes bone erosion in murine collagen‐induced arthritis through loss of the receptor activator of NF‐kappa B ligand/osteoprotegerin balance. J Immunol 2003;170:2655–62. [DOI] [PubMed] [Google Scholar]
- 26. Lubberts E, Koenders MI, Oppers‐Walgreen B, et al. Treatment with a neutralizing anti‐murine interleukin‐17 antibody after the onset of collagen‐induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 2004;50:650–9. [DOI] [PubMed] [Google Scholar]
- 27. Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006;203:2673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osada Y, Kanazawa T. Parasitic helminths: new weapons against immunological disorders. J Biomed Biotechnol 2010;2010:743758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bessis N, Boissier MC, Ferrara P, Blankenstein T, Fradelizi D, Fournier C. Attenuation of collagen‐induced arthritis in mice by treatment with vector cells engineered to secrete interleukin‐13. Eur J Immunol 1996;26:2399–403. [DOI] [PubMed] [Google Scholar]
- 30. Bessis N, Honiger J, Damotte D, et al. Encapsulation in hollow fibres of xenogeneic cells engineered to secrete IL‐4 or IL‐13 ameliorates murine collagen‐induced arthritis (CIA). Clin Exp Immunol 1999;117:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horsfall AC, Butler DM, Marinova L, et al. Suppression of collagen‐induced arthritis by continuous administration of IL‐4. J Immunol 1997;159:5687–96. [PubMed] [Google Scholar]
- 32. Joosten LA, Lubberts E, Helsen MM, et al. Protection against cartilage and bone destruction by systemic interleukin‐4 treatment in established murine type II collagen‐induced arthritis. Arthritis Res 1999;1:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kageyama Y, Koide Y, Uchijima M, et al. Plasmid encoding interleukin‐4 in the amelioration of murine collagen‐induced arthritis. Arthritis Rheum 2004;50:968–75. [DOI] [PubMed] [Google Scholar]
- 34. Kim SH, Kim S, Evans CH, Ghivizzani SC, Oligino T, Robbins PD. Effective treatment of established murine collagen‐induced arthritis by systemic administration of dendritic cells genetically modified to express IL‐4. J Immunol 2001;166:3499–505. [DOI] [PubMed] [Google Scholar]
- 35. Finnegan A, Kaplan CD, Cao Y, Eibel H, Glant TT, Zhang J. Collagen‐induced arthritis is exacerbated in IL‐10‐deficient mice. Arthritis Res Ther 2003;5:R18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang M, Deng J, Liu Y, et al. IL‐10‐producing regulatory B10 cells ameliorate collagen‐induced arthritis via suppressing Th17 cell generation. Am J Pathol 2012;180:2375–85. [DOI] [PubMed] [Google Scholar]
- 37. Carter NA, Rosser EC, Mauri C. Interleukin‐10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen‐induced arthritis. Arthritis Res Ther 2012;14:R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye L, Wen Z, Li Y, et al. Interleukin‐10 attenuation of collagen‐induced arthritis is associated with suppression of interleukin‐17 and retinoid‐related orphan receptor gamma T production in macrophages and repression of classically activated macrophages. Arthritis Res Ther 2014;16:R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nandakumar KS, Holmdahl R. Arthritis induced with cartilage‐specific antibodies is IL‐4‐dependent. Eur J Immunol 2006;36:1608–18. [DOI] [PubMed] [Google Scholar]
- 40. Svensson L, Nandakumar KS, Johansson A, Jansson L, Holmdahl R. IL‐4‐deficient mice develop less acute but more chronic relapsing collagen‐induced arthritis. Eur J Immunol 2002;32:2944–53. [DOI] [PubMed] [Google Scholar]
- 41. Ortmann RA, Shevach EM. Susceptibility to collagen‐induced arthritis: cytokine‐mediated regulation. Clin Immunol 2001;98:109–18. [DOI] [PubMed] [Google Scholar]
- 42. Sarkar S, Cooney LA, White P, et al. Regulation of pathogenic IL‐17 responses in collagen‐induced arthritis: roles of endogenous interferon‐gamma and IL‐4. Arthritis Res Ther 2009;11:R158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Croxford AM, Whittingham S, McNaughton D, Nandakumar KS, Holmdahl R, Rowley MJ. Type II collagen‐specific antibodies induce cartilage damage in mice independent of inflammation. Arthritis Rheum 2013;65:650–9. [DOI] [PubMed] [Google Scholar]
- 44. Kelchtermans H, Schurgers E, Geboes L, et al. Effector mechanisms of interleukin‐17 in collagen‐induced arthritis in the absence of interferon‐gamma and counteraction by interferon‐gamma. Arthritis Res Ther 2009;11:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guedez YB, Whittington KB, Clayton JL, et al. Genetic ablation of interferon‐gamma up‐regulates interleukin‐1beta expression and enables the elicitation of collagen‐induced arthritis in a nonsusceptible mouse strain. Arthritis Rheum 2001;44:2413–24. [DOI] [PubMed] [Google Scholar]
- 46. Mattsson L, Larsson P, Erlandsson‐Harris H, Klareskog L, Harris RA. Parasite‐mediated down‐regulation of collagen‐induced arthritis (CIA) in DA rats. Clin Exp Immunol 2000;122:477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Washino T, Moroda M, Iwakura Y, Aosai F. Toxoplasma gondii infection inhibits Th17‐mediated spontaneous development of arthritis in interleukin‐1 receptor antagonist‐deficient mice. Infect Immun 2012;80:1437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pesce J, Kaviratne M, Ramalingam TR, et al. The IL‐21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest 2006;116:2044–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spolski R, Leonard WJ. Interleukin‐21: a double‐edged sword with therapeutic potential. Nat Rev Drug Discov 2014;13:379–95. [DOI] [PubMed] [Google Scholar]
- 50. Young DA, Hegen M, Ma HL, et al. Blockade of the interleukin‐21/interleukin‐21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum 2007;56:1152–63. [DOI] [PubMed] [Google Scholar]
- 51. Sakuraba K, Oyamada A, Fujimura K, et al. Interleukin‐21 signaling in B cells, but not in T cells, is indispensable for the development of collagen‐induced arthritis in mice. Arthritis Res Ther 2016;18:016 1086-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Page CE, Smale S, Carty SM, et al. Interferon‐gamma inhibits interleukin‐1beta‐induced matrix metalloproteinase production by synovial fibroblasts and protects articular cartilage in early arthritis. Arthritis Res Ther 2010;12:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Doodes PD, Cao Y, Hamel KM, et al. Development of proteoglycan‐induced arthritis is independent of IL‐17. J Immunol 2008;181:329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ismail HI, Sallam FA, Sheer HA. The pathogenesis of arthropathy in experimental schistosomiasis mansoni. J Egypt Soc Parasitol 2000;30:943–9. [PubMed] [Google Scholar]
- 55. Atkin SL, Kamel M, el‐ Hady AM, el‐ Badawy SA, el‐ Ghobary A, Dick WC. Schistosomiasis and inflammatory polyarthritis: a clinical, radiological and laboratory study of 96 patients infected by S. mansoni with particular reference to the diarthrodial joint. Q J Med 1986;59:479–87. [PubMed] [Google Scholar]
- 56. Graepel R, Leung G, Wang A, et al. Murine autoimmune arthritis is exaggerated by infection with the rat tapeworm. Hymenolepis diminuta Int J Parasitol 2013;43:593–601. [DOI] [PubMed] [Google Scholar]
- 57. Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL‐13 inhibitor blocks the development of hepatic fibrosis during a T‐helper type 2‐dominated inflammatory response. J Clin Invest 1999;104:777–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kumar R, Mickael C, Chabon J, et al. The causal role of IL‐4 and IL‐13 in Schistosoma mansoni pulmonary hypertension. Am J Respir Crit Care Med 2015;192:998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu F, Cheng W, Pappoe F, et al. Schistosoma japonicum cystatin attenuates murine collagen‐induced arthritis. Parasitol Res 2016;115:3795–806. [DOI] [PubMed] [Google Scholar]
- 60. Sun X, Liu YH, Lv ZY, et al. rSj16, a recombinant protein of Schistosoma japonicum‐derived molecule, reduces severity of the complete Freund’s adjuvant‐induced adjuvant arthritis in rats’ model. Parasite Immunol 2010;32:739–48. [DOI] [PubMed] [Google Scholar]
- 61. Wang X, Li L, Wang J, et al. Inhibition of cytokine response to TLR stimulation and alleviation of collagen‐induced arthritis in mice by Schistosoma japonicum peptide SJMHE1. J Cell Mol Med 2017;21:475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pineda MA, Al‐Riyami L, Harnett W, Harnett MM. Lessons from helminth infections: ES‐62 highlights new interventional approaches in rheumatoid arthritis. Clin Exp Immunol 2014;177:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McInnes IB, Leung BP, Harnett M, Gracie JA, Liew FY, Harnett W. A novel therapeutic approach targeting articular inflammation using the filarial nematode‐derived phosphorylcholine‐containing glycoprotein ES‐62. J Immunol 2003;171:2127–33. [DOI] [PubMed] [Google Scholar]
- 64. Pineda MA, McGrath MA, Smith PC, et al. The parasitic helminth product ES‐62 suppresses pathogenesis in collagen‐induced arthritis by targeting the interleukin‐17‐producing cellular network at multiple sites. Arthritis Rheum 2012;64:3168–78. [DOI] [PubMed] [Google Scholar]
- 65. Eissa MM, Mostafa DK, Ghazy AA, El Azzouni MZ, Boulos LM, Younis LK. Anti‐arthritic activity of Schistosoma mansoni and Trichinella spiralis derived‐antigens in adjuvant arthritis in rats: role of Foxp3+ Treg cells. PLOS ONE 2016;11:e0165916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Khan YA, Umar S, Abidi SM. Somatic antigens of tropical liver flukes ameliorate collagen‐induced arthritis in Wistar rats. PLOS ONE 2015;10:e0126429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carranza F, Falcon CR, Nunez N, et al. Helminth antigens enable CpG‐activated dendritic cells to inhibit the symptoms of collagen‐induced arthritis through Foxp3+ regulatory T cells. PLOS ONE 2012;7:e40356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rocha FA, Leite AK, Pompeu MM, et al. Protective effect of an extract from Ascaris suum in experimental arthritis models. Infect Immun 2008;76:2736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]