

Abstract
Ischemic stroke is among the leading causes of morbidity and mortality worldwide. Improving the tolerance of neurons to ischemia and reperfusion injury could be a feasible strategy against ischemia. Adiponectin (APN) is a major adipokine that regulates glucose and lipid metabolism and plays an important role in the protection of the cerebral nervous system. We aimed to investigate the effects of APN on oxygen and glucose deprivation (OGD)-induced neuronal injury in hippocampal neuronal HT22 cells. APN displayed neuroprotective effects against OGD, evidenced by increased cell viability and decreased lactate dehydrogenase release and apoptotic rate. Additionally, APN also maintained mitochondrial ultrastructure and transmembrane potential, attenuated reactive oxygen species and malondialdehyde, and increased superoxide dismutase and glutathione peroxidase activity. Moreover, APN promoted Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) phosphorylation, enhanced STAT3 nuclear translocation, increased the Bcl-2/Bax ratio, and decreased cleaved caspase-3. The aforementioned APN-induced effects were almost reversed by a JAK2 inhibitor, AG490. APN may attenuate OGD-induced hippocampal HT22 neuronal impairment by protecting cells against mitochondrial oxidative stress and apoptosis, mediated by JAK2/STAT3 signaling.
Keywords: adiponectin, JAK2/STAT3, mitochondria, oxidative stress, oxygen and glucose deprivation
Introduction
Ischemic stroke is among the leading causes of morbidity and mortality worldwide1. Although intravenous administration of tissue plasminogen activator (t-PA) and endovascular approaches have been developed to achieve reperfusion, many patients still suffer from ischemia-reperfusion (IR) injury (IRI), or even worse miss the narrow time window or are unable to access these therapies1,2. Therefore, improving the tolerance of neurons to IRI could be a feasible strategy to combat ischemia. Oxygen and glucose deprivation (OGD) and subsequent in vitro restoration mimic the process of IR induced by blood flow blockage and recanalization, which are responsible for causing severe mitochondrial impairment, over-generation of reactive oxygen species (ROS), and definite cell death3. Consequently, as an effective cellular model, OGD in cells has been widely used in protective and mechanism studies3–6.
Adiponectin (APN) is an adipocyte-secreted circulating cytokine that contains a stalk with 22 collagen repeats and a highly conserved globular domain (gAd). There are three major oligomeric forms of APN in human and mouse plasma: trimers, hexamers, and a high-molecular-weight form7. The gAd is the functional area of APN and it has been reported that gAd exhibits much more extensive (over 20-fold) biological activity than the full-length form8,9. APN has a regulative effect on metabolism10. It also protects against metabolic syndrome11, liver fibrosis12, carcinogenesis13, and myocardial IRI4. Although, a meta-analysis has shown that APN is not associated with the risk of stroke; however, when controlling for risk factors that favorably correlate with APN14, there is evidence that lower APN may be related to a poorer prognosis after stroke. A clinical study has revealed that for humans a level of APN less than 4 µg/ml is independently associated with mortality, and low plasma adiponectin is related to an increased risk of 5-year mortality after the first ischemic stroke15. Additionally, APN has been reported as beneficial in attenuating cerebral IRI in type I diabetes mellitus16,17. Considering stroke with no complications, studies have shown that APN attenuates cerebral ischemic injury through an endothelial nitric oxide synthase-dependent mechanism18, and plays an anti-inflammatory role in IRI19. However, it is unclear whether APN has a direct effect on ischemic neurons.
Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) is an intracellular signal transduction pathway. JAK2 is a protein tyrosine kinase activated by cytokine receptors, such as the interleukin-6 receptor activated through glycoprotein 130 (gp130), which then phosphorylate and activate cytoplasmic STAT320. STAT3 has been reported to regulate gene transcription, involving genes for cell survival, proliferation, cell-cycle progression, and angiogenesis in cerebral development and disorders21. Studies have shown that JAK2/STAT3 activation protects myocardial H9c2 cells against hypoxia/re-oxygenation injury22, attenuates amyloid-β1-42-induced neurotoxicity in glioma SH-SY5Y cells23, and promotes neuroprotection and neuronal plasticity in murine stroke models6,24. Furthermore, APN has been shown to activate STAT3 in primary hepatocytes involved in the inhibition of hepatic gluconeogenesis10. It is also implicated in the activation of JAK2/STAT3, to prevent diabetic myocardial IRI25. Therefore, JAK2/STAT3 could also be a reasonable pathway that mediates the effects of APN in neurons.
Mitochondria are a major source of ROS, and are particularly vulnerable to hypoxia and ischemia. In ischemic stroke, mitochondrial dysfunction and oxidative stress form a wretched cycle, synergistically leading to neuronal death26–28. Additionally, studies have indicated that JAK2/STAT3 activation protects against apoptosis22, mitochondrial impairment22, and oxidative injury23 in different cell types. The JAK2/STAT3 pathway has been indicated to be associated with upregulation of Bcl-2, an anti-apoptotic protein, downregulation of apoptosis-related molecules, including Bax and cleaved-caspase-322,24, and maintaining mitochondrial adenosine triphosphate (ATP) generation29.
Therefore, we aimed to explore the direct effect of APN on hippocampal HT22 cells subjected to OGD and investigate protection against mitochondrial oxidative injury and apoptosis. We further aim to identify the involvement of JAK2/STAT3 signaling in causing this effect.
Materials and Methods
Materials
Recombinant human adiponectin was obtained from Sangon Biotech (Shanghai, China). AG490 was purchased from Selleck Chemicals (Houston, TX, USA). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Kumamoto, Japan). Lactate dehydrogenase (LDH) cytotoxicity assay kit and cell lysis buffer were purchased from Beyotime Biotechnology (Shanghai, China). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) kit, and protease and phosphatase inhibitors were obtained from Roche (Mannheim, Germany). Malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px) determination kits were purchased from the Institute of Jiancheng Bioengineering (Nanjing, Jiangsu, China). 2′,7′-dichlorofluorescein diacetate (H2DCF-DA), JC-1, and 4′,6-diamidino-2-phenylindole (DAPI) fluorescent probes were purchased from Sigma-Aldrich (St. Louis, MO, USA). The nuclear and cytoplasmic extraction kit was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was obtained from Bio-Rad (Hercules, CA, USA). Polyvinylidene difluoride (PVDF) membranes and enhanced chemiluminescence reagent were purchased from Millipore (Billerica, MA, USA). Antibodies against JAK2, p-JAK2 (Tyr1007/Try1008), STAT3, p-STAT3 (Try705), Bcl-2, Bax, cleaved-caspase-3, and histone H3 were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-β-actin and secondary HRP-labeled antibodies were obtained from Bioworld Technology (St. Louis Park, MN, USA). Fluorescent secondary antibody was purchased from Proteintech (Rosemont, IL, USA). Unless otherwise specified, the cell culture reagents were obtained from Gibco (Grand Island, NY, USA), and consumables from Jet Bio-Filtration (Guangzhou, China).
Cell Culture and Treatments
Mouse hippocampal neuron HT22 cells were cultured in Dulbecco’s modified eagle medium (DMEM) with high glucose and 10% fetal bovine serum (FBS; 37°C, 5% CO2), which was changed every 2 days. Culture media were substituted for phenol red-free DMEM 24 h before the experiment. APN was dissolved in DMEM (without FBS or phenol red) and diluted to working concentrations (25, 50, or 100 μmol/L) in advance. After a 4 h treatment with APN, cells were washed with phosphate buffered saline (PBS; 0.1 mol/L, pH = 7.4) and then subjected to OGD by culturing with glucose-free media in a hypoxic chamber (95% N2, 5% CO2, 37°C) for another 4 h, followed by a rapid restoration of glucose and oxygen, as previously described30. Cells were harvested after 24 h. In the inhibitor group, cells were treated with 50 μmol/L AG490 (dissolved in dimethyl sulfoxide [DMSO] and diluted with DMEM) for 16 h before all experiments. The control group was subjected to the same procedure, excluding APN or AG490 pretreatment.
CCK-8 Assay and LDH Release Assay
The CCK-8 and LDH release assays were performed to measure cell viability according to the manufacturers’ protocols. Briefly, cells were grown in 96-well plates at a density of 10,000 cells per well. After 24 h following OGD, media were replaced by phenol red-free DMEM with 10% CCK-8. Afterwards, cells were incubated at 37°C for 3 h, and transferred to a Spectra Max M5 microplate reader (Molecular Device, Sunnyvale, CA, USA) for optical density (OD) detection at 450 nm.
LDH is an intracellular enzyme, released after cell death. To detect LDH release, cells were cultured in a 96-well plate. After experimental interventions, the media were centrifuged and collected in a new 96-well plate, followed by incubation with 60 μl LDH detection buffer, per well, on a dark shaking table at room temperature for 30 min. The absorbance was then measured under 490 nm wavelength using the microplate reader, and the death rate was calculated in accordance with the manual provided.
TUNEL Staining
Apoptotic rate was estimated using TUNEL staining as previously described31. Cells cultured in confocal dishes were fixed in 4% paraformaldehyde for 30 min after all experimental interventions, and then incubated with TUNEL reaction mixture for 1 h and DAPI for 15 min at 37°C. Results were observed and photographed under a Nikon A1plus confocal microscope (Minato, Japan). Normal nuclei only stained with DAPI were visualized in a blue color, and apoptosis nuclei stained with DAPI and TUNEL were visualized in a green color. Apoptosis ratios were counted and calculated as an average of five random visual fields.
Determination of MDA, SOD, and GSH-Px
Cells were lysed on ice for 30 min and centrifuged at 4°C, 12,000 rpm for 15 min to obtain supernatant. After that, the MDA level and SOD and GSH-Px activities were assessed using corresponding kits, according to the manufacturer’s instructions.
ROS Detecting Assay
The ROS fluorescent probe H2DCF-DA was applied, as previously described31. After all interventions, the cells were incubated with 10 μmol/L H2DCF-DA in serum-free media for 30 min at 37°C, in a dark incubator, followed by visualization under a Nikon A1plus confocal microscope (excitation wavelength at 488 nm, emission wavelength at 525 nm). For precise quantification, cells were cultured in black 96-well plates at a density of 10,000 cells per well and were subjected to the same staining procedure. Afterwards, the fluorescent density of each group was measured with a fluorescence spectrophotometer 680 (Bio-Rad Laboratories Inc., Hercules, CA, USA). The excitation wavelength was 490 nm; emission wavelength was 530 nm.
Mitochondrial Membrane Potential Assay
The destruction of mitochondrial transmembrane potential (MMP) is a constituent of the earliest events in cell apoptosis32. The JC-1 fluorescent probe was employed to measure MMP, as previously described33. Briefly, after experimental interventions, cells were incubated with 10 μg/ml JC-1 buffer for 20 min at 37°C, and then scanned by a fluorescence spectrophotometer 680 (Bio-Rad Laboratories Inc.). JC-1 polymerized in the mitochondrial matrix (showed red fluorescence); however, when MMP reduced, JC-1 could not accumulate in the matrix and instead existed as a monomer (shown through green fluorescence). The reduction of MMP was calculated as a ratio of green fluorescence (excitation wavelength, 490 nm; emission wavelength, 530 nm) to red fluorescence (excitation wavelength, 525 nm; emission wavelength, 590 nm).
Mitochondrial Ultrastructure Scanning by Electron Microscopy
Cells were cultured on slides and treated in accordance with the groups, followed by fixation in 3% glutaraldehyde at 4°C overnight, and post-fixation in 1% osmic acid for 1 h. Afterwards, samples were dehydrated using ethanol solutions of gradually increasing concentrations, each for 15 min. Following drying in an HCP-2 critical-point dryer (Hitachi, Tokyo, Japan), samples were sputter-coated with 10 nm AuPd (60%/40% alloy) using a Denton DV-502A high-vacuum system (Moorestown, NJ, USA), and visualized using a Hitachi S-450 scanning electron microscope (Tokyo, Japan).
Immunofluorescence Staining
Slices were treated with 0.3% Triton X-100 for 30 min and 10% donkey serum for 2 h, followed by incubation with primary antibody (rabbit anti-p-STAT3, 1:100) at 4°C overnight, and secondary antibody (donkey anti-rabbit IgG, Alexa Fluor 488, 1:200) at 37°C for 4 h, subsequently followed by DAPI for 15 min. Negative controls went through the same procedure, except the primary antibody incubation.
Western Blot Analysis
Cells were harvested at 24 h after OGD using a cell scraper and lysed in 200 μl lysis buffer containing protease and phosphatase inhibitors. Nuclear and cytoplasmic proteins were extracted using a nuclear and cytoplasmic extraction kit. BCA quantification and western blot analysis were performed, as previously reported31. Briefly, identical amounts of protein samples were separated through SDS-PAGE, transferred onto PVDF membranes, and incubated with primary antibodies at 4°C overnight. Afterwards, corresponding secondary antibodies were applied at 25°C for 2 h. The antibodies employed were as follows: anti-p-JAK2 (1:1000), anti-JAK2 (1:1000), anti-p-STAT3 (1:1000), anti-STAT3 (1:1000), anti-β-actin (1:3000), anti-histone H3 (1:3000), anti-Bcl-2 (1:1000), anti-Bax (1:1000), anti-cleaved-caspase-3 (1:1000), and secondary HRP-labeled antibodies (1:20,000). The phosphorylated and total proteins were probed at different membranes, which were all normalized with loading control. The membranes were reacted with an enhanced chemiluminescence reagent and scanned with an imaging system (Bio-Rad Laboratories Inc.). Finally, data were analyzed with ImageJ software (version 1.46).
Statistical Analysis
GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA) was used to analyze the data. Results have been expressed as mean ± standard deviation (SD), unless otherwise noted. Differences in means between two groups were assessed by the two-tailed student t test. Comparisons among groups were assessed by one-way analysis of variance (ANOVA). Multiple comparisons were performed using the Mann–Whitney U test significant difference test for significant ANOVAs. Differences were considered statistically significant when P < 0.05.
Results
Effects of APN on HT22 cells, Without Other Treatments
Normally cultured HT22 cells were treated with 100 μmol/L APN (nearly human plasma level) for 4 h, and cell viability and plasma membrane permeability were detected using CCK-8 and LDH release assay 24 h later. APN did not affect cellular morphology, viability, or plasma membrane integrity (n = 6, P < 0.05, Figure 1(a)). Therefore, 100 μmol/L APN was used as the maximum concentration for the following experiments. Meanwhile, 100 μmol/L APN increased the expression and phosphorylation of JAK2 and STAT3 to 1.42 ± 0.28 (JAK2), 2.53 ± 0.57 (pY1007/Y1008-JAK2), 1.33 ± 0.11 (STAT3), and 1.62 ± 0.06 (pY705-STAT3) in HT22 cells (n = 6, P < 0.05, Figure 1(b)).
Figure 1.

Effects of APN on HT22 cells. HT22 cells were incubated with 100 µmol/L APN for 4 h. (a) Cell morphology, viability, and LDH release were measured 24 h later. Cells were photographed under an inverted/phase-contrast microscope. Viability and LDH release are expressed as optical density (OD) and absorbance, respectively. (b) Effects of 100 µmol/L APN on the expression and phosphorylation of JAK2 and STAT3 were evaluated using western blot analysis. β-actin was used as a loading control, and the levels of total JAK2, p-JAK2 (Y1007/Y1008), total STAT3, and p-STAT3 (Y705) were normalized to control. n = 6. *P < 0.05, compared with control.
Effects of APN on OGD-Induced Cell Death
Cells were subjected to treatments of vehicle or APN at 25, 50, or 100 μmol/L for 4 h, followed by OGD for another 4 h. After 24 h, OGD induced obvious cell shrinkage and fracture, decreased cell viability to 0.54 ± 0.08, and increased LDH release to 0.61 ± 0.15, compared to cells in the control group (0.97 ± 0.02, 0.10 ± 0.06). APN pretreatment significantly improved cell viability to 0.73 ± 0.07 (50 μmol/L) and 0.79 ± 0.10 (100 μmol/L), and attenuated LDH release to 0.28 ± 0.11 (100 μmol/L; n = 6, P < 0.05, Figure 2(a)). Apoptosis is a major constituent of OGD-induced neuronal death. APN of 50 and 100 μmol/L significantly decreased apoptosis rate to 0.60 ± 0.04 and 0.54 ± 0.06, respectively, compared to OGD alone (0.84 ± 0.04) (n = 6, P < 0.05; Figure 2(b)).
Figure 2.

The protective effect of APN against OGD. HT22 cells were incubated with or without APN (25, 50, or 100 µmol/L) for 4 h and then subjected to OGD. (a) Cell viability and LDH release (expressed as OD value and absorbance, respectively) were evaluated 24 h later. (b) Apoptotic cells stained with TUNEL shown in green, and all nuclei stained with DAPI shown in blue. Apoptosis rates were expressed as a ratio of TUNEL-positive cells (green). n = 6. *P < 0.05, compared with control. # P < 0.05, compared with OGD.
Effects of APN on OGD-Induced Mitochondrial Oxidative Injury
To observe the effects of APN on mitochondria, subcellular structure was imaged under a scanning electron microscope. OGD significantly changed mitochondrial ultrastructure compared to the control group, including changes such as mitochondrial swelling, vanished cristae, and electron density reduction; APN relieved these structural damages to some extent (Figure 3(a)). MMP reduction is a hallmark of the early stage of apoptosis, indicating an increase in membrane permeability. OGD markedly reduced MMP to 0.23 ± 0.12, whereas APN pretreatment at 50 and 100 μmol/L improved MMP to 0.56 ± 0.13 and 0.55 ± 0.15, respectively (n = 6, P < 0.05, Figure 3(b)). In addition, OGD noticeably increased the ROS level in HT22 cells, and APN decreased H2DCF-DA-positive cells (Figure 3(c)). Furthermore, MDA, SOD, and GSH-Px levels were detected to precisely measure the oxidative stress level. MDA is a frequently used membrane lipid peroxidation hallmark, whereas SOD and GSH-Px are intracellular anti-oxidative enzymes. OGD increased MDA level to 0.65 ± 0.12, compared to the control group at 0.10 ± 0.05, and decreased SOD activity to 10.00 ± 2.45, compared to the control group at 24.60 ± 2.86, apart from decreasing GSH-Px activity to 2.02 ± 0.27, compared to the control group at 3.82 ± 0.25. However, 100 μmol/L APN pretreatment decreased MDA to 0.38 ± 0.11, and increased SOD and GSH-Px to 19.23 ± 2.60 and 2.68 ± 0.27, respectively (n = 6, P < 0.05, Figure 3(d)).
Figure 3.

The protective effect of APN against mitochondrial oxidative injury. HT22 cells were exposed to OGD following treatments of APN (25, 50, or 100 µmol/L) for 4 h. (a) Mitochondrial morphology was visualized by scanning under an electron microscope. Representative mitochondria marked by black arrows. Significant mitochondrial swelling, cristae vanishing, and a reduction in electron-dense substances were observed in OGD compared to control, and these noticeably improved in the APN pretreatment group. (b) MMP was determined by JC-1 staining. The reduction of MMP indicates mitochondrial apoptosis. (c) Oxidative stress level was determined by H2DCF-DA staining. Intracellular ROS were stained with green fluorescence, and the level of ROS was expressed as fluorescent density. (d) Intracellular MDA, SOD activities, and GSH-Px levels were determined using corresponding kits. n = 6. *P < 0.05, compared with control. # P < 0.05, compared with OGD.
Effects of APN on JAK2/STAT3 Signaling and Expression of Bcl-2, Bax, Cleaved-Caspase-3, in the OGD Condition
The changes in JAK2/STAT3, Bcl-2, Bax, and cleaved-caspase-3, with APN pretreatment, were estimated by western blot analysis. Treatment with APN before OGD significantly increased phosphorylation of JAK2 at Tyr1007/1008 and STAT3 at Tyr705, and promoted nuclear translocation of STAT3, compared to OGD alone (n = 6, P < 0.05, Figure 4(a,b)). OGD decreased Bcl-2 expression and increased Bax expression, resulting in a reduced ratio of Bcl-2/Bax, and increased cleaved-caspase-3 in the OGD group. APN pretreatment significantly reversed the OGD-induced decrease in the ratio of Bcl-2/Bax, and attenuated the level of cleaved-caspase-3 (n = 6, P < 0.05; Figure 4(c)).
Figure 4.

APN activates JAK2/STAT3 signaling, increases the ratio of Bcl-2 to Bax, and decreases cleaved-caspase-3 in OGD. HT22 cells were exposed to OGD following 4 h of treatment with APN (25, 50, or 100 µmol/L). (a) Effects of APN on phosphorylation of JAK2 and STAT3 were evaluated using western blot analysis. β-actin was used as a loading control, and the levels of total JAK2, p-JAK2 (Y1007/Y1008), total STAT3, and p-STAT3 (Y705) were normalized to control. (b) Nuclear and cytoplasmic STAT3 were extracted using the nuclear protein extraction kit and evaluated using western blot analysis. Histone H3 and β-actin were used as a loading control, and the levels of nuclear and cytoplasmic STAT3 were normalized to control. (c) Bcl-2, Bax, and cleaved-caspase-3 were evaluated using western blot analysis, which were normalized to control, and β-actin was used as a loading control. n = 6. *P < 0.05, compared with control. # P < 0.05, compared with OGD. & P < 0.05, compared with OGD + 25 µmol/L APN.
AG490 abolishes the effects of APN on cell viability, apoptosis, mitochondrial injury, and oxidative stress in the OGD condition. JAK2-specific inhibitor, AG490, was used to verify whether JAK2/STAT3 signaling mediates the protection of APN against OGD. AG490 pretreatment significantly abolished APN-induced improvement in cell viability, and decrease in LDH release and apoptosis rate, compared to the APN + OGD group (n = 6, P < 0.05; Figure 5(a)). In addition, MMP reduction rate was decreased to 1.27 ± 0.04 in the AG490 + APN + OGD group, compared to 1.58 ± 0.08 in the APN + OGD group (n = 6, P < 0.05, Figure 5(b)). Furthermore, AG490 increased the ROS level and significantly decreased the APN-induced increase in SOD activity (n = 6, P < 0.05; Figure 5(c,d)).
Figure 5.

AG490 abolishes effects of APN on OGD-induced cell death and mitochondrial oxidative injury. HT22 cells were incubated with 50 µmol/L AG490 for 16 h in advance, and then treated with APN, followed by exposure to OGD. (a) Cell viability, LDH release, and apoptosis rate were measured 24 h later. Results are given in OD value, absorbance, and ratio of TUNEL-positive cells, respectively. (b) MMP was determined by JC-1 staining, and fluorescence was detected using a spectrophotometer. Reduction rate was determined as the ratio of red and green fluorescence. (c) ROS level was determined using H2DCF-DA staining. Intracellular ROS was stained with green fluorescence, and the level of ROS was expressed as fluorescent density. (d) Intracellular MDA, SOD activities, and GSH-Px levels were normalized to OGD. n = 6. # P < 0.05, compared with OGD. & P < 0.05, compared with AG490 + OGD. $ P < 0.05, compared with APN + OGD.
AG490 decreases APN-induced activation of JAK2/STAT3 signaling and reverses the effects of APN on Bcl-2, Bax, and cleaved-caspase-3. Interestingly, AG490 significantly inhibited phosphorylation and nuclear translocation of STAT3 in the AG490 + APN + OGD group compared to that in the APN + OGD group. However, it showed no effect on the protein level of pY1007/1008-JAK2, and JAK2 (n = 6, P < 0.05, Figure 6(a,b)). Meanwhile, the nuclear proportion of pY705-STAT3 after OGD decreased in the AG490 + APN group compared to that in the APN group when tested using immunofluorescence detection (Figure 7). The increased ratio of Bcl-2/Bax, and the attenuated level of cleaved-caspase-3 were also reversed by AG490, compared to the APN + OGD group (n = 6, P < 0.05, Figure 6(c)).
Figure 6.

AG490 inhibits JAK2/STAT3 signaling and reverses the effects of APN on the ratio of Bcl-2 to Bax and the level of cleaved-caspase-3. HT22 cells were incubated with 50 µmol/L AG490 for 16 h in advance, and then treated with APN, followed by exposure to OGD. (a) Phosphorylation levels of JAK2 and STAT3 were evaluated using western blot analysis. β-actin was used as a loading control, and the data in the OGD group were defined as 1.0. (b) Nuclear and cytoplasmic STAT3 were extracted using the nuclear protein extraction kit and evaluated using western blot analysis. Histone H3 and β-actin were used as a loading control, respectively, for nuclear and cytoplasmic proteins, and the nuclear translocation of STAT3 in the OGD group was defined as 1.0. (c) Bcl-2, Bax, and cleaved-caspase-3 were evaluated using western blot analysis, which were normalized to the OGD group, and β-actin was used as a loading control. n = 6. # P < 0.05, compared with OGD. & P < 0.05, compared with AG490 + OGD. $ P < 0.05, compared with APN + OGD.
Figure 7.

Effects of APN on nuclear translocation of p-STAT3 after OGD, with or without AG490 pretreatment. HT22 cells were incubated with 50 µmol/L AG490 for 16 h in advance, and then treated with APN, followed by exposure to OGD. (a) After experimental interventions, cells went through immunofluorescence staining with pY705-STAT3 (green) antibody and DAPI (blue).
Discussion
APN is an endogenous adipokine that exerts multiple biological effects in different tissues and cells11–13. Li et al. suggested that ischemic post-conditioning exerts myocardial protection through the APN/AdipoR1 (APN receptor)/Caveolin-3 pathway; this protection is lost in diabetes mellitus, owing to impaired APN/AdipoR1/Caveolin-3 signaling4. Specifically, Song et al. have reported that APN, dependent on adipoR1, attenuates cerebral IRI in type 1 diabetes mellitus17. Although Arregui et al. have declared in a meta-analysis that APN is not associated with the risk of stroke, except when controlled for risk factors that favorably correlate with APN, such as diabetes mellitus, studies have reported that APN may be related to the prognosis of stroke by attenuating cerebral ischemic injury via activation of endothelial nitric oxide synthase18 and inhibiting inflammation19. APN was found at relatively high total levels in human serum, ranging in concentration from 5 to 30 μg/ml, although cerebrospinal fluid (CSF) APN in humans was approximately 0.1% of the serum concentration due to the restricted permeability of the blood–brain barrier (BBB) to macromolecules34. Our team developed a peptide that could effectively pass through the BBB and simulate and maintain the function of APN35. APN exerts multiple vasoprotective effects via its action on the vascular system, including endothelial cells, monocytes, macrophages, leukocytes, platelets and smooth muscle cells, plaque formation, and development of thrombosis36. However, the effect of APN on neurons is unclear. In this study, APN attenuated OGD-induced apoptosis and LDH release in murine hippocampal neuronal HT22 cells, and did not show toxicity at a concentration of 100 μmol/L, indicating a protective effect against OGD (mimicking the IRI state, in vitro).
JAK2 is a protein tyrosine kinase activated by a cytokine receptor. Activated JAK2 phosphorylates and activates cytoplasmic STAT320, thereby regulating expression of genes involved in cell survival, proliferation, cell-cycle progression, and angiogenesis in cerebral development and disorders21. APN has been reported to activate STAT3 for the inhibition of hepatic gluconeogenesis in primary hepatocytes10, and for the utilization of the JAK2/STAT3 pathway to protect against diabetic myocardial IRI25. Furthermore, Wang et al. have reported that adiponectin-derived active peptide, ADP355, exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury, associated with the promotion of AMP-activated protein kinase (AMPK) and STAT3 signaling12. In the central nervous system, studies have reported that activated JAK2/STAT3 signaling attenuates amyloid-β1-42-induced neurotoxicity in glioma SH-SY5Y cells23, and promotes neuronal survival and plasticity in murine models of stroke6,24. Therefore, protection of APN against OGD in the neurons may be mediated by the JAK2/STAT3 pathway. In this study, APN reduced OGD-induced cell impairment and increased phosphorylation of JAK2 and STAT3, as well as the nuclear translocation of STAT3. Importantly, the APN-induced improvement in cell viability, reduction in LDH release, and a reduction in neuronal apoptosis were all reversed by the JAK2 inhibitor, AG490. These indicate a JAK2/STAT3-dependent protective effect of APN against OGD in neurons.
Considering further intracellular mechanisms, studies indicate that JAK2/STAT3 activation is associated with upregulation of anti-apoptotic Bcl-2, downregulation of Bax and cleaved-caspase-322,24, and maintenance of mitochondrial ATP generation29, all of which possess the properties of protection against mitochondrial impairment22, oxidative injury23, and cell apoptosis22. Therefore, the effector mechanism of APN was speculated to involve the attenuation of mitochondrial injury, oxidative stress, and apoptosis. In this study, APN attenuated OGD-induced mitochondrial swelling, cristae vanishing, and electron density reduction observed through scanning electron microscopy, and maintained the MMP. However, inhibition of JAK2 by AG490 significantly decreased these protective effects on mitochondria. Furthermore, APN attenuated oxidative stress, as indicated by decreased ROS and MDA levels, as well as increased SOD and GSH-Px activities; in contrast, the protection of APN against oxidative stress on HT22 cells was also reversed by AG490. In addition, the APN-induced increase in the ratio of Bcl-2 to Bax, and a reduction in the level of cleaved-caspase-3, were reversed by AG490 as well.
Conclusions
APN protects HT22 neuronal cells from OGD-induced mitochondrial oxidative stress and apoptosis via the phosphorylated activation of JAK2/STAT3 signaling, which increases the ratio of anti-apoptotic Bcl-2 to apoptosis-related Bax, and decreases the level of cleaved-caspase-3 (Figure 8). These results suggest that endogenous APN reduction may play a role in the IRI process; therefore, exogenously supplying APN could improve the tolerance of vulnerable neurons to ischemia-reperfusion. This may be a promising approach to treating ischemic stroke accompanying t-PA and endovascular treatment. Further studies on the mechanism and application of APN in the central nervous system, in order to fully understand the scope using APN in treatment, are warranted.
Figure 8.
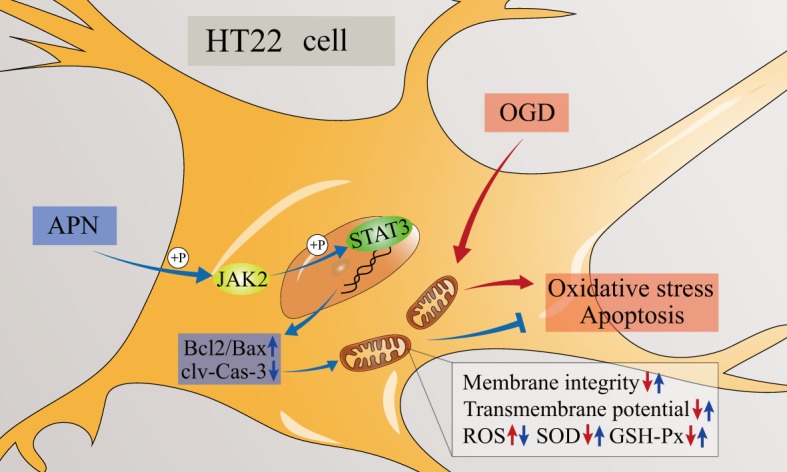
JAK2/STAT3 mediated protection of APN against OGD. OGD damages mitochondrial structure and function, which induces oxidative stress and apoptosis. APN promotes the phosphorylated activation of JAK2/STAT3 signaling, and may further upregulate Bcl-2 and decrease cleaved-caspase-3 expression, which improve mitochondrial membrane integrity, maintain MMP, attenuate ROS generation, and increase anti-oxidative enzymes. Taken together, APN attenuates mitochondrial vulnerability through the JAK2/STAT3 pathway, performing an anti-oxidative and anti-apoptotic effect in HT22 neuronal cells subjected to OGD.
Footnotes
Authors’ Contributions: Bodong Wang, Hao Guo, and Xia Li contributed equally to this work.
Ethical Approval: This study was approved by our institutional review board.
Statement of Human and Animal Rights: This article does not contain any studies with human or animal subjects.
Statement of Informed Consent: There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this study was funded by the National Natural Science Foundation of China (Nos. 81571215, 81630027); the New Century Talent Supporting Project, supported by the Chinese Education Ministry (No. NCET-12-1004); and Leading Talents of Middle-age and Young in S&T Innovation, supported by the Chinese Science and Technology Ministry (No. 2013RA2181).
References
- 1. Khandelwal P, Yavagal DR, Sacco RL. Acute ischemic stroke intervention. J Am Coll Cardiol. 2016;67(22):2631–2644. [DOI] [PubMed] [Google Scholar]
- 2. Catanese L, Tarsia J, Fisher M. Acute ischemic stroke therapy overview. Circ Res. 2017;120(3):541–558. [DOI] [PubMed] [Google Scholar]
- 3. Andreev DE, O’Connor PB, Zhdanov AV, Dmitriev RI, Shatsky IN, Papkovsky DB, Baranov PV. Oxygen and glucose deprivation induces widespread alterations in mRNA translation within 20 minutes. Genome Biol. 2015;16(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li H, Yao W, Liu Z, Xu A, Huang Y, Ma XL, Irwin MG, Xia Z. Hyperglycemia abrogates ischemic postconditioning cardioprotection by impairing AdipoR1/Caveolin-3/STAT3 signaling in diabetic rats. Diabetes. 2016;65(4):942–955. [DOI] [PubMed] [Google Scholar]
- 5. Ryou MG, Liu R, Ren M, Sun J, Mallet RT, Yang SH. Pyruvate protects the brain against ischemia-reperfusion injury by activating the erythropoietin signaling pathway. Stroke. 2012;43(4):1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shyu WC, Lin SZ, Chiang MF, Chen DC, Su CY, Wang HJ, Liu RS, Tsai CH, Li H. Secretoneurin promotes neuroprotection and neuronal plasticity via the Jak2/Stat3 pathway in murine models of stroke. J Clin Invest. 2008;118(1):133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure–function studies of the adipocyte-secreted hormone Acrp30/adiponectin: implications for metabolic regulation and bioactivity. J Biol Chem. 2003;278(11):9073–9085. [DOI] [PubMed] [Google Scholar]
- 8. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–1295. [DOI] [PubMed] [Google Scholar]
- 9. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–946. [DOI] [PubMed] [Google Scholar]
- 10. Ding Y, Zhang D, Wang B, Zhang Y, Wang L, Chen X, Li M, Tang Z, Wang C. APPL1-mediated activation of STAT3 contributes to inhibitory effect of adiponectin on hepatic gluconeogenesis. Mol Cell Endocrinol. 2016;433:12–19. [DOI] [PubMed] [Google Scholar]
- 11. Sun H, Zhang Y, Gao P, Li Q, Sun Y, Zhang J, Xu C. Adiponectin reduces C-reactive protein expression and downregulates STAT3 phosphorylation induced by IL-6 in HepG2 cells. Mol Cell Biochem. 2011;347(1–2):183–189. [DOI] [PubMed] [Google Scholar]
- 12. Wang H, Zhang H, Zhang Z, Huang B, Cheng X, Wang D, La Gahu Z, Xue Z, Da Y, Li D, Yao Z, Gao F, Xu A, Zhang R. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci Rep. 2016;6:19445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moon HS, Mantzoros CS. Adiponectin and metformin additively attenuate IL1beta-induced malignant potential of colon cancer. Endocr Relat Cancer. 2013;20(6):849–859. [DOI] [PubMed] [Google Scholar]
- 14. Arregui M, Buijsse B, Fritsche A, di Giuseppe R, Schulze MB, Westphal S, Isermann B, Boeing H, Weikert C. Adiponectin and risk of stroke: prospective study and meta-analysis. Stroke. 2014;45(1):10–17. [DOI] [PubMed] [Google Scholar]
- 15. Efstathiou SP, Tsioulos DI, Tsiakou AG, Gratsias YE, Pefanis AV, Mountokalakis TD. Plasma adiponectin levels and five-year survival after first-ever ischemic stroke. Stroke J Cerebral Circ. 2005;36(9):1915–1919. [DOI] [PubMed] [Google Scholar]
- 16. Duan J, Yin Y, Cui J, Yan J, Zhu Y, Guan Y, Wei G, Weng Y, Wu X, Guo C, Wang Y, Xi M, Wen A. Chikusetsu saponin IVa ameliorates cerebral ischemia reperfusion injury in diabetic mice via adiponectin-mediated AMPK/GSK-3beta pathway in vivo and in vitro. Mol Neurobiol. 2016;53(1):728–743. [DOI] [PubMed] [Google Scholar]
- 17. Song W, Guo F, Zhong H, Liu L, Yang R, Wang Q, Xiong L. Therapeutic window of globular adiponectin against cerebral ischemia in diabetic mice: the role of dynamic alteration of adiponectin/adiponectin receptor expression. Sci Rep. 2015;5:17310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo C, Shin HK, Moskowitz MA, Ouchi N. Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase dependent mechanisms. Circulation. 2008;117(2):216–223. [DOI] [PubMed] [Google Scholar]
- 19. Chen B, Liao WQ, Xu N, Xu H, Wen JY, Yu CA, Liu XY, Li CL, Zhao SM, Campbell W. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res. 2009;1273:129–137. [DOI] [PubMed] [Google Scholar]
- 20. Wormann SM, Song L, Ai J, Diakopoulos KN, Gorgulu K, Ruess D, Campbell A, Doglioni C, Jodrell D, Neesse A, Demir IE, Karpathaki AP, Barenboim M, Hagemann T, Rose-John S, Sansom O, Schmid RM, Protti MP, Lesina M, Algül H. Loss of P53 function activates JAK2–STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology 2016;151(1):180–193. [DOI] [PubMed] [Google Scholar]
- 21. Raible DJ, Frey LC, Brooks-Kayal AR. Effects of JAK2–STAT3 signaling after cerebral insults. JAKSTAT. 2014;3(2): e29510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y, Shi X, Li J, Zhang M, Yu B. Knockdown of KLF11 attenuates hypoxia/reoxygenation injury via JAK2/STAT3 signaling in H9c2. Apoptosis 2016;22(4):510–518. [DOI] [PubMed] [Google Scholar]
- 23. Wang K, Xie M, Zhu L, Zhu X, Zhang K, Zhou F. Ciliary neurotrophic factor protects SH-SY5Y neuroblastoma cells against Abeta1-42-induced neurotoxicity via activating the JAK2/STAT3 axis. Folia Neuropathol. 2015;53(3):226–235. [DOI] [PubMed] [Google Scholar]
- 24. Liu X, Zhang X, Zhang J, Kang N, Zhang N, Wang H, Xue J, Yu J, Yang Y, Cui H, Cui L, Wang L, Wang X. Diosmin protects against cerebral ischemia/reperfusion injury through activating JAK2/STAT3 signal pathway in mice. Neuroscience. 2014;268:318–327. [DOI] [PubMed] [Google Scholar]
- 25. Wang T, Mao X, Li H, Qiao S, Xu A, Wang J, Lei S, Liu Z, Ng KF, Wong GT, Vanhoutte PM, Irwin MG, Xia Z. N-acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med. 2013;63:291–303. [DOI] [PubMed] [Google Scholar]
- 26. Gao Y, Chen T, Lei X, Li Y, Dai X, Cao Y, Ding Q, Lei X, Li T, Lin X. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep. 2016;14(6):5481–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ham PB, III, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol. 2017;157:92–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim JJ, Kang YJ, Shin SA, Bak DH, Lee JW, Lee KB, Yoo YC, Kim DK, Lee BH, Kim DW, Lee J, Jo EK, Yuk JM. Phlorofucofuroeckol improves glutamate-induced neurotoxicity through modulation of oxidative stress-mediated mitochondrial dysfunction in PC12 cells. PLoS One. 2016;11(9): e0163433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitra MS, Donthamsetty S, White B, Mehendale HM. High fat diet-fed obese rats are highly sensitive to doxorubicin-induced cardiotoxicity. Toxicol Appl Pharmacol. 2008;231(3):413–422. [DOI] [PubMed] [Google Scholar]
- 30. Wang CP, Shi YW, Tang M, Zhang XC, Gu Y, Liang XM, Wang ZW, Ding F. Isoquercetin ameliorates cerebral impairment in focal ischemia through anti-oxidative, anti-inflammatory, and anti-apoptotic effects in primary culture of rat hippocampal neurons and hippocampal CA1 region of rats. Mol Neurobiol 2016;54(3):2126–2142. [DOI] [PubMed] [Google Scholar]
- 31. Wang B, Liu H, Yue L, Li X, Zhao L, Yang X, Wang X, Yang Y, Qu Y. Neuroprotective effects of pterostilbene against oxidative stress injury: involvement of nuclear factor erythroid 2-related factor 2 pathway. Brain Res. 2016;1643:70–79. [DOI] [PubMed] [Google Scholar]
- 32. Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311(5762):847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y, Fan C, Wang B, Ma Z, Wang D, Gong B, Di S, Jiang S, Li Y, Li T, Yang Z, Luo E. Pterostilbene attenuates high glucose-induced oxidative injury in hippocampal neuronal cells by activating nuclear factor erythroid 2-related factor 2. Biochim Biophys Acta 2017;1863(4):827–837. [DOI] [PubMed] [Google Scholar]
- 34. Neumeier M, Weigert J, Buettner R, Wanninger J, Schaffler A, Muller AM, Killian S, Sauerbruch S, Schlachetzki F, Steinbrecher A, Aslanidis C, Schölmerich J, Buechler C. Detection of adiponectin in cerebrospinal fluid in humans. Am J Physiol Endocrinol Metab. 2007;293(4): E965–E969. [DOI] [PubMed] [Google Scholar]
- 35. Yue L, Zhao L, Liu H, Li X, Wang B, Guo H, Gao L, Feng D, Qu Y. Adiponectin protects against glutamate-induced excitotoxicity via activating SIRT1-dependent PGC-1alpha expression in HT22 hippocampal neurons. Oxid Med Cell Longev. 2016;2016:2957354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ebrahimi-Mamaeghani M, Mohammadi S, Arefhosseini SR, Fallah P, Bazi Z. Adiponectin as a potential biomarker of vascular disease. Vasc Health Risk Manag. 2015;11:55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]