

Abstract
The nuclear hormone receptors liver X receptor α (LXRα) and peroxisome proliferator-activated receptor γ (PPARγ) play key roles in the development of fatty liver. To determine the link between hepatic PPARγ and LXRα signaling and the development of fatty liver, a LXRα-specific ligand, T0901317, was administered to normal OB/OB and genetically obese (ob/ob) mice lacking hepatic PPARγ (Pparγ∆H). In ob/ob-Pparγ∆H and OB/OB-Pparγ∆H mice, as well as ob/ob-PparcWT and OB/OB-PparγWT mice, the liver weights and hepatic triglyceride levels were markedly increased in response to T0901317 treatment. These results suggest that hepatic PPARγ and LXRα signals independently contribute to the development of fatty liver.
Keywords: Liver X receptor α, Peroxisome proliferator-activated receptor, γ, Fatty liver
1. Introduction
Liver X receptor (LXR) α and b are members of a family of ligand-dependent nuclear receptors. LXRs heterodimerize with retinoid X receptors (RXRs) and regulate the transcription of target genes by binding to LXR-responsive elements in the 5ʹ regions upstream of target genes. LXRs play important roles in the regulation of genes associated with lipid metabolism [1]. Genes that encode proteins involved in sterol metabolism including ATP-binding cassette transporters ABCA1, ABCG1, ABCG5, and ABCG8, as well as apolipoprotein E and CYP7a1 are LXR targets. Further, LXRα also plays important roles in the regulation of hepatic fat accumulation.
Two transcription factors, sterol regulatory element-binding protein-1c (SREBP-1c) and carbohydrate response element-binding protein (ChREBP), play key roles in LXRα-mediated hepatic lipogenesis. SREBP-1c is a basic helix-loop-helix family transcription factor and a direct target of the LXRα/RXRα heterodimer [1]. SREBP-1c controls the expression of nearly all genes integral to lipogenesis, including fatty acid synthase (Fas), acetyl-CoA carboxylase (Acc), and stearoyl-CoA desaturase 1 (Scd1) [1]. ChREBP is also an LXR target gene [2]. ChREBP is a glucose-sensitive transcription factor that promotes the hepatic conversion of excess carbohydrates to lipids [3]. Studies using mice lacking ChREBP expression revealed that ChREBP regulates Fas, Acc, Scd1, and SREBP-1c [4]. LXRa signaling mediated by SREBP-1c and ChREBP contributes to the increase in hepatic triglyceride content by upregulating lipogenic genes. Moreover, administration of the LXR ligand T0901317 to mice induces severe fatty liver [5]. Therefore, LXRα signaling is associated with the development of pathogenic conditions such as hepatic steatosis.
The leptin-deficient obese genetic background (ob/ob) mouse, a typical type 2 diabetes model with severe fatty liver, was previously generated with hepatocyte-specific disruption of the gene encoding peroxisome proliferator-activated receptor γ (PPARγ, designated ob/ob-Pparγ∆H. The ob/ob-Pparγ∆H) mice showed recovery from the severe fatty liver disease that was found in their PPARγ-expressing ob/ob-PparγWT counterparts, suggesting that hepatic PPARγ promotes the development of fatty liver [6]. To determine the mechanism of PPARγ-dependent development of fatty liver, the levels of mRNAs encoded by lipogenic genes in the livers of ob/ob-Pparγ∆H and ob/ob-PparγWT and non-obese counter-parts OB/OB-Pparγ∆H and OB/OB-PparγWT were compared [6]. The results showed that Fas, Acc, Scd1, malic enzyme, ATP-citrate lyase, and glycerol-3-phosphate acyltransferase (Gpat) were lower in ob/ob-Pparγ∆H livers than in those of ob/ob-PparγWT mice, suggesting that PPARγ promotes the development of fatty liver in ob/ob mice by upregulating the expression of lipogenic genes. Notably, these genes are also typical LXRα-SREBP-1c or LXRα-ChREBP targets. Thus, these findings point toward a possible link between LXRα and PPARα signaling in ob/ob fatty liver. Further, PPARγ in macrophages was shown to regulate directly the expression of LXRα [7,8]. However, whether PPARγ regulates LXRα expression in the liver is not known.
In the present study, the role of hepatic PPARγ in LXRα-associated fat accumulation was determined using ob/ob-Pparγ∆H mice. Our results demonstrated that hepatic PPARγ and LXRα independently regulate the expression of lipogenic genes and development of fatty liver in ob/ob mice. These finding suggests the presence of two major signaling pathways involving two nuclear receptors, which results in the development of fatty liver.
2. Materials and Methods
2.1. Mice
Hepatocyte-specific PPARγ knockout mice were generated in ob/ob (ob/ob-Pparγ∆H) and normal genetic backgrounds (OB/OB-Pparγ∆H) using a Pparγ-floxed allele and Cre recombinase transgene under control of the albumin promoter as described previously [6]. Mouse experiments were performed under the guidelines of the center for experimental animals of the Fukuoka University.
2.2. Administration of LXR ligand
The LXR ligand, T0901317 (Sigma–Aldrich, Japan) was administered to the T0901317 group as described previously [5]. The control group was not administered T0901317. Diets containing 0.025% T0901317 were prepared by mixing the ligand with powdered chow (CE-2, CLEA, Japan).
2.3. Measurement of lipids
Total triglyceride and cholesterol contents were measured as described previously [9].
2.4. RNA extraction and quantitative PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, Carls-bad, CA, USA). Quantitative polymerase chain reaction (qPCR) was performed using cDNA that was synthesized from 1 μg of total RNA by using the AffinityScript QPCR cDNA Synthesis kit (Agilent Technologies). Sequences of the primers used were as follows: LXRa forward, 5ʹ -ATCGCCTTGCTGAAGACCTCTG-3ʹ and reverse, 5ʹ -GATGGGGTTGATGAACTCCACC-3ʹ; PPARγ forward, 5ʹ -CATGGCC ATTGAGTGCCGAGT-3ʹ and reverse, 5ʹ -ACATCCCCACAGCAAGGCAC-3ʹ; Scd1 forward, 5ʹ -CGTCTGGAGGAACATCATTCT-3ʹ and reverse, 5ʹ -CAGAGCGCTGGTCATGTAGT-3ʹ; Fas forward, 5ʹ -GGAGGTGGTGATA GCCGGTAT-3ʹ and reverse, 5ʹ -TGGGTAATCCATAGAGCCCAG-3ʹ; SREBP-1c forward, 5ʹ -GGAGCCATGGATTGCACATT-3ʹ and reverse, 5ʹ -AGG AAGGCTTCCAGAGAGGA-3ʹ; Gpat forward, 5ʹ -TTGGGACTTGCACGT TCTG-3ʹ and reverse, 5ʹ -TAGGTTTGAACCCACAGTCAAC-3ʹ; ChREBP forward, 5ʹ -ATTTCCTGGCTCCCAAGC-3ʹ and reverse, 5ʹ -GAAGCAG TCCAGGTCTAGAAGC-3ʹ; 36B4 forward, 5ʹ -AAACTGCTGCCTCACAT CCG-3ʹ and reverse, 5ʹ -TGGTGCCTCTGGAGATTTTCG-30; Lpk forward, 50 -CATTGTGCTGACAAAGACTGG-3ʹ and reverse, 5ʹ -AGCCTGTCACCA CAATCACC-3ʹ; adipocyte fatty acid-binding protein (Ap2) forward, 5ʹ - GATGCCTTTGTGGGAACCTG-3ʹ and reverse, 5ʹ -GAATTCCACGCC CAGTTTGA-3ʹ; class B scavenger receptor (CD36) forward, 5ʹ -GAT GACGTGGCAAAGAACAG-3ʹ and reverse, 50 -TCCTCGGGGTCCTGAGT TAT-30. QPCR reactions were performed using Brilliant III Ultra-Fast SYBR Green QPCR Master Mix (Agilent Technologies) and the Mx3005P Real-Time PCR System (Agilent Technologies). 36B4 mRNA was used as an internal control.
2.5. Statistical analysis
Experimental values are expressed as the mean ± standard error (SE). Statistical analyses were performed using the Student’s t-test for unpaired data. p < 0.05 was considered statistically significant.
3. Results
The LXR ligand T0901317 was administered at a concentration of 0.025% in the diet for 7 days. Serum lipid and glucose content of the mice are summarized in Table 1. The ob/ob genetic background causes severe obesity and symptoms of type 2 diabetes. Hyperglycemia in ob/ob mice was significantly suppressed by treatment with T0901317. Consistent with a previous study [10], serum cholesterol levels were increased in OB/OB-PparγWT and ob/ob-PparγWT mice. However, the T0901317-induced changes in serum glucose and cholesterol were similar to those in wild-type PPARc (PparγWT) and Pparγ∆H mice in an OB/OB or ob/ob genetic background. These results suggested that hepatic PPARγ was not involved in mediating the effects of T0901317 on serum cholesterol and glucose levels.
Table 1.
Body weight and blood parameters in mice treated by LXR ligand.
|
PparγWT |
Pparγ∆H |
|||||||
|---|---|---|---|---|---|---|---|---|
|
OB/OB |
ob/ob |
OB/OB |
ob/ob |
|||||
| Control | T0901317 | Control | T0901317 | Control | T0901317 | Control | T0901317 | |
| Body weight (g) | 22.9 ± 1.3 | 26.9 ± 0.8* | 38.7 ± 1.6 | 40.9 ± 1.9 | 21.9 ± 1.3 | 22.3 ± 0.9 | 41.6 ± 0.5 | 40.3 ± 1.0 |
| Glucose (mg/dl) | 182 ± 20 | 188 ± 7.0 | 476 ± 85 | 258 ± 42* | 214 ± 1.3 | 194 ± 8.0 | 516 ± 82 | 247 ± 40* |
| Serum TG (mg/dl) | 14.7 ± 1.5 | 12.9 ± 0.7 | 25.4 ± 2.7 | 25.9 ± 4.0 | 16.9 ± 1.0 | 22.7 ± 4.4 | 39.7 ± 5.5 | 47.1 ± 7.6 |
| Serum CHO (mg/dl) | 184 ± 8.0 | 265 ± 8.0*** | 277 ± 12 | 378 ± 15*** | 192 ± 7.0 | 301 ± 10*** | 307 ± 15 | 494 ± 39** |
Each group is from 5 to 9 mice. TG, triglyceride; CHO, total cholesterol; PparγWT, wild-type PPARγ mice; Pparγ∆H, hepatocyte-specific PPARγ null mice. Values are mean ± SE.
p < 0.05.
p < 0.01.
p < 0.001 versus control without T0901317.
The ob/ob-PparγWT mice had severe fatty liver. However, the livers of ob/ob-Pparγ∆H mice showed recovery from fatty liver disease (Fig. 1A), as reported previously [6]. Livers of T0901317-treated ob/ob mice were significantly enlarged compared to those of untreated mice and were yellow in color (Fig. 1A). Notably, T0901317-induced development of fatty liver was also observed in ob/ob-Pparγ∆H mice lacking hepatic PPARγ. Treatment with T0901317 led to a significant increase in the liver weight and hepatic triglyceride (TG) content in all mice (Fig. 1B and C). The T0901317-induced changes in liver weight and TG levels were similar to those in OB/OB-PparγWT and OB/OB-Pparγ∆H mice. The relative fold-inductions of TG by T0901317, compared to the control, were as follows: OB/OB-PparγWT, 1.8-fold; OB/OB-Pparγ∆H, 1.7-fold; ob/ob-PparγWT, 2.4-fold, and ob/ob-Pparγ∆H, 2.5-fold.
Fig. 1.
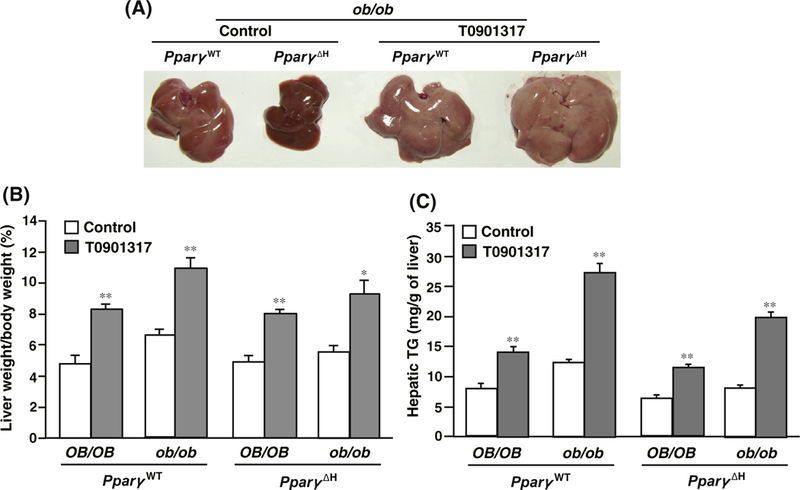
Effect of LXR ligand on hepatic triglyceride content in Pparγ∆H mice. (A) T0901317-treated ob/ob-PparγWT or ob/ob-Pparγ∆H livers. (B) Liver weight and (C) hepatic triglyceride (TG) in mice belonging to each genotype. The number of mice belonging to each genotype was as follows. Control group (untreated): OB/OB-PparγWT, n = 5; ob/ob-PparγWT, n = 9; OB/OB-Pparγ∆H, n = 9; and ob/ob-Pparγ∆H, n = 6. T0901317-treated group: OB/OB-PparγWT, n = 6; ob/ob-PparγWT, n = 8; OB/OB-Pparγ∆H, n = 8; and ob/ob-Pparγ∆H, n = 6. Values are the mean ± SE. *p < 0.01, **p < 0.001 versus the untreated control.
The fold-induction of LXRα by T0901317 treatment in all mouse genotypes was lower than that in untreated mice (Table 2). Previous studies demonstrated that LXRα is a direct target of PPARγ in macrophages [7,8]. Therefore, the level of LXRγ mRNA expressed in the livers of untreated PparγWT mice was compared with that expressed in Pparγ∆H livers. Consistent with a previous study [6], the relative ratio of LXRα mRNA in PparγWT versus Pparγ∆H mice was nearly unchanged and were as follows: OB/OB-PparγWT: OB/OB-Pparγ∆H = 1.0:0.9 and ob/ob-PparγWT: ob/ob-Pparγ∆H = 1.0:0.8. These data suggest that hepatic LXRα is not upregulated by PPARγ. Although ChREBP and SREBP-1c are also LXRα targets, the fold-inductions in OB/OB-PparγWT and OB/OB-Pparγ∆H mice were greater than those in ob/ob-PparγWT and ob/ob-Pparγ∆H mice (Table 2).
Table 2.
Effect of LXR ligand on genes involved in lipid metabolism in PPARc-deficient mouse liver.
| Gene |
PparγWT |
Pparγ∆H |
||||||
|---|---|---|---|---|---|---|---|---|
|
OB/OB |
ob/ob |
OB/OB |
ob/ob |
|||||
| Control | T0901317 | Control | T0901317 | Control | T0901317 | Control | T0901317 | |
| PPARc | 1.0 ± 0.03 | 0.96 ± 0.04 | 1.0 ± 0.04 | 0.98 ± 0.02 | N.D. | N.D. | 1.0 ± 0.13 | 1.1 ± 0.04 |
| LXRa | 1.0 ± 0.04 | 0.67 ± 0.01** | 1.0 ± 0.03 | 0.52 ± 0.01*** | 1.0 ± 0.07 | 0.84 ± 0.02* | 1.0 ± 0.05 | 0.54 ± 0.02** |
| ChREBP | 1.0 ± 0.07 | 1.3 ± 0.05* | 1.0 ± 0.03 | 1.0 ± 0.01 | 1.0 ± 0.02 | 1.6 ± 0.01*** | 1.0 ± 0.06 | 0.95 ± 0.04 |
| SREBP-1c | 1.0 ± 0.02 | 2.0 ± 0.10* | 1.0 ± 0.02 | 1.0 ± 0.03 | 1.0 ± 0.03 | 2.5 ± 0.21* | 1.0 ± 0.03 | 1.6 ± 0.05* |
| Fas | 1.0 ± 0.05 | 11 ± 0.21*** | 1.0 ± 0.09 | 3.1 ± 0.14** | 1.0 ± 0.16 | 10 ± 0.10*** | 1.0 ± 0.07 | 5.3 ± 0.17*** |
| Scd1 | 1.0 ± 0.02 | 8.2 ± 0.49*** | 1.0 ± 0.06 | 2.7 ± 0.05*** | 1.0 ± 0.02 | 8.4 ± 0.45*** | 1.0 ± 0.04 | 3.8 ± 0.11*** |
| Gpat | 1.0 ± 0.05 | 3.6 ± 0.07*** | 1.0 ± 0.03 | 2.4 ± 0.04*** | 1.0 ± 0.03 | 4.0 ± 0.03*** | 1.0 ± 0.10 | 4.3 ± 0.05*** |
| Lpk | 1.0 ± 0.04 | 0.66 ± 0.01** | 1.0 ± 0.05 | 0.89 ± 0.04 | 1.0 ± 0.07 | 1.4 ± 0.05*** | 1.0 ± 0.09 | 1.0 ± 0.03 |
| Ap2 | 1.0 ± 0.01 | 1.7 ± 0.33* | 1.0 ± 0.06 | 1.6 ± 0.04** | 1.0 ± 0.02 | 1.7 ± 0.14** | 1.0 ± 0.06 | 1.4 ± 0.04** |
| CD36 | 1.0 ± 0.08 | 5.9 ± 0.05*** | 1.0 ± 0.04 | 0.9 ± 0.10 | 1.0 ± 0.03 | 6.1 ± 0.14*** | 1.0 ± 0.00 | 1.2 ± 0.01*** |
Each group is from 3 mice. PparγWT, wild type PPARγ mice; Pparγ∆H, hepatocyte-specific PPARγ null mice ChREBP, carbohydrate response element-binding protein; SREBP-1c, sterol regulatory element-binding protein-1c; Fas, fatty acid synthase; Scd1, stearoyl-CoA desaturase 1; Lpk, liver-type pyruvate kinase; Gpat, glycerol-3-phosphate acyltransferase; Ap2, adipocyte fatty acid-binding protein; CD36, class B scavenger receptor. N.D., not detected. Values are mean ± SE.
p < 0.05.
p < 0.01.
p < 0.001 versus control without T0901317.
In a previous report, several LXRα-SREBP-1c target genes in ob/ob-Pparγ∆H mouse livers were shown to decrease [6]. To examine the potential role of hepatic PPARγ on the T0901317-mediated induction of lipogenic genes, the relative fold-induction of LXRα-SREBP-1c target genes for each genotype in the control and T0901317 groups were compared (Table 2). T0901317 treatment induced expression of Fas, Scd1, and Gpat mRNAs, known hepatic LXRα-SREBP-1c [2] and PPARγ targets [6], in PparγWT and Pparγ∆H mice with an OB/OB or ob/ob genetic background. The fold-inductions of lipogenic genes in ob/ob-PparγWT mice were nearly identical to those in ob/ob-Pparγ∆H mice as well as those with an OB/OB genetic background. Following treatment with T0901317, the ChREBP target gene Lpk [4] was induced only in the livers of OB/OB-Pparγ∆H mice. Ap2 and CD36 genes are well-known PPARγ targets. The fold-induction of Ap2 by T0901317 treatment in all mouse genotypes was slightly greater (approximately 1.5-fold) than that in untreated mice. The CD36 gene was markedly induced by T0901317 treatment in OB/OB genetic background mice, but was unchanged in the livers of ob/ob background mice. These results clearly suggested that hepatic PPARγ did not contribute to the induction of lipogenic genes and the development of fatty liver following LXRα activation.
4. Discussion
A previous study revealed that in the fatty livers of ob/ob mice, hepatic PPARγ upregulates several lipogenic genes that are LXRα targets [6]. These results suggested possible crosstalk between LXRα and PPARγ signaling pathways in the fatty livers of ob/ob mice. In the present study, using hepatocyte-specific PPARγ knockout mice, Pparγ∆H, hepatic PPARγ and LXRα were found to regulate independently the expression of the lipogenic genes SREBP-1c, Fas, Scd1, and Gpat. These results suggest that PPARγ does not function upstream or downstream of LXRα. The expression of PPARγ in the livers of mice fed a normal low-fat chow diet is very low [11]. However, PPARγ is expressed at markedly elevated levels in the fatty livers of a number of diabetes and obesity mouse models, including ob/ob mice [12,13]. Although the mechanism of induction of PPARγ in fatty liver remains unclear, a recent study showed transcriptional regulation of PPARγ by activator protein 1 (AP-1) is involved [14]. Therefore, although hepatic PPARγ and LXRα independently regulate lipogenic genes, PPARγ likely contributes to the generation of fatty liver (Fig. 2).
Fig. 2.
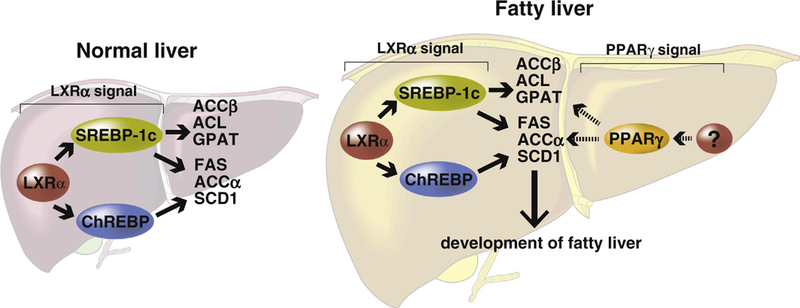
Schematic diagram illustrating the independent effects of PPARγ and LXRα in hepatic lipogenesis. The LXRα-SREBP-1c and ChREBP signals upregulate downstream lipogenic genes in normal and obesity model mouse livers [2,5]. The expression of PPARγ in normal livers is low, but is upregulated in ob/ob fatty livers [13]. The hepatic PPARγ pathway is likely more active in fatty liver rather than in normal livers. Therefore, hepatic PPARγ appears to contribute significantly to the development of fatty liver rather than LXRα. ACC, acetyl-CoA carboxylase; ACL, ATP citrate lyase; GPAT, glycerol-3-phosphate acyltransferase; FAS, fatty acid synthase; SCD1, stearoyl-CoA desaturase 1.
Previous studies have found that LXRα is a direct target for PPARγ in macrophages [7,8]. The expression of LXRα mRNA in macrophages was induced by ligands for PPARγ, but not by PPARα or PPARβ agonists. LXRα promoter activity is enhanced by PPARγ, which binds to PPAR-responsive elements in the LXRα promoter. However, in the present and a previous study, hepatic LXRα expression was nearly similar in PparγWT and Pparγ∆H mice [6]. Therefore, PPARγ does not appear to regulate directly the expression of LXRα in the liver. Indeed, LXRα is highly expressed in normal livers that express low, nearly undetectable levels of PPARγ. Therefore, PPARγ regulates LXRα in a cell- or tissue-type specific manner.
Although lipogenic genes, Ap2 and CD36 were induced in OB/OB- and ob/ob-Pparγ∆H mice by LXR ligand, the degree of induction depended on each gene. These genes appeared to be independently regulated by hepatic PPARγ and LXRα. Ap2 and CD36 genes are well known PPARγ targets; however, these genes were induced by LXR ligand. Recently, both genes were also reported as LXR targets [15,16]. Interestingly, the induction of CD36 by T0901317 was observed in mice with an OB/OB genetic background, but not in those with an ob/ob background. The mechanism remains unclear and warrants further study.
The serum TG of T0901317-treated or untreated ob/ob-Pparγ∆H mice was higher than that of ob/ob-PparγWT mice (Table 1). The details of this mechanism remain to be defined; however, the higher TG level in ob/ob-Pparγ∆H mice was also reported in our previous study [6]. The results of that study demonstrated that very-low-density lipoprotein and chylomicron in ob/ob-Pparγ∆H mice accumulate in the blood. Therefore, the elevated TG level in ob/ob-Pparγ∆H mice may be due to having impaired TG-rich lipoprotein clearance.
In summary, the present study demonstrated that PPARγ and LXRα independently regulate the expression of lipogenic genes and contribute to the development of fatty liver. Lipogenesis is known to contribute to the development of non-alcoholic fatty liver disease (NAFLD) [17]. Therefore, a complete understanding of these mechanisms might lead to the development of promising new therapeutics to control hepatic TG accumulation in NAFLD.
Acknowledgments
This work was supported by the Mochida Memorial Foundation for Medical Research and a KAKENHI Grant (No. 25460348).
Abbreviations
- PPARγ
peroxisome proliferator-activated receptor γ
- LXRα
liver X receptor α
- OB/OB
normal genetic background
- ob/ob
obese genetic background
- PparγWT
wild-type PPARγ
- Pparc∆H
hepatocyte-specific PPARγ knockout
References
- [1].Baranowski M (2008) Biological role of liver X receptors. J. Physiol. Pharmacol 59 (Suppl. 7), 31–55. [PubMed] [Google Scholar]
- [2].Cha JY and Repa JJ (2006) The liver X receptor (LXR) and hepatic lipogenesis: the carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem 282, 743–751. [DOI] [PubMed] [Google Scholar]
- [3].Uyeda K, Yamashita H and Kawaguchi T (2002) Carbohydrate responsive element-binding protein (ChREBP): a key regulator of glucose metabolism and fat storage. Biochem. Pharmacol 63, 2075–2080. [DOI] [PubMed] [Google Scholar]
- [4].Ishii S, Iizuka K, Miller BC and Uyeda K (2004) Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 101, 15597–15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chisholm JW (2003) The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J. Lipid Res 44, 2039–2048. [DOI] [PubMed] [Google Scholar]
- [6].Matsusue K, Haluzik M, Lambert G, Yim S-H, Gavrilova O, Ward JM, Brewer B, Reitman ML and Gonzalez FJ (2003) Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest 111, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee Y-H, Ricote M, Glass CK, Brewer HB and Gonzalez FJ (2002) Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol. Cell. Biol 22, 2607– 2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chawla A, Boisvert WA, Lee C-H, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM and Tontonoz P (2001) A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 7, 161–171. [DOI] [PubMed] [Google Scholar]
- [9].Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S and Gonzalez FJ (2008) Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab 7, 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schultz JR (2000) Role of LXRs in control of lipogenesis. Genes Dev 14, 2831– 2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tontonoz P, Hu E, Graves RA, Budavari AI and Spiegelman BM (1994) MPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8, 1224–1234. [DOI] [PubMed] [Google Scholar]
- [12].Bedoucha M, Atzpodien E and Boelsterli UA (2001) Diabetic KKAy mice exhibit increased hepatic PPARgamma1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J. Hepatol 35, 17–23. [DOI] [PubMed] [Google Scholar]
- [13].Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C and Feingold KR (2000) Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 141, 4021–4031. [DOI] [PubMed] [Google Scholar]
- [14].Hasenfuss SC, Bakiri L, Thomsen MK, Williams EG, Auwerx J and Wagner EF (2014) Regulation of steatohepatitis and PPARc signaling by distinct AP-1 dimers. Cell Metab 19, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Seo JB, Moon HM, Kim WS, Lee YS, Jeong HW, Yoo EJ, Ham J, Kang H, Park M-G, Steffensen KR, Stulnig TM, Gustafsson J-A, Park SD and Kim JB (2004) Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor gamma expression. Mol. Cell. Biol 24, 3430–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL and Xie W (2008) Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 134, 556–567. [DOI] [PubMed] [Google Scholar]
- [17].Shinji Tamura IS (2005) Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Invest 115, 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]