

Abstract
Although microRNAs (miRs) have been implicated in the pathogenesis of various human malignancies, limited information is available regarding mechanisms by which these noncoding RNAs contribute to initiation and progression of tobacco-induced esophageal cancers. In this study, array and quantitative RT-PCR (qRT-PCR) techniques were used to examine miR expression in immortalized esophageal epithelia (IEE) and esophageal adenocarcinoma (EAC) cells cultured in normal media (NM) with or without cigarette smoke condensate (CSC). Under relevant exposure conditions, CSC significantly decreased miR-217 expression in these cells. Endogenous levels of miR-217 expression in cultured EAC cells (EACC)/ primary EACs were significantly lower than those observed in IEE/ paired normal esophageal tissues. RNA cross-link immunoprecipitation, qRT-PCR and immunoblot experiments demonstrated direct interaction of miR-217 with kallikrein 7 (KLK7), encoding a putative oncogene not previously implicated in EAC. Repression of miR-217 correlated with increased levels of KLK7 in primary EACs, particularly those from smokers. Chromatin and methylated DNA immunoprecipitation experiments demonstrated that CSC-mediated repression of miR-217 coincided with DNMT3b-dependent hypermethylation and decreased occupancy of nuclear factor 1 (NF-1) within the miR-217 genomic locus. Deoxyazacytidine induced miR-217 expression, and down-regulated KLK7 in EACC; deoxyazacytidine also attenuated CSC-mediated miR-217 repression and up-regulation of KLK7 in IEE and EACC. Over-expression of miR-217 significantly decreased, whereas over-expression of KLK7 increased proliferation, invasion and tumorigenicity of EACC. Collectively, these data demonstrate that epigenetic repression of miR-217 contributes to the pathogenesis of EAC via up-regulation of KLK7, and suggest that restoration of miR-217 expression may be a novel treatment strategy for these malignancies.
Keywords: esophageal cancer, miR-217, microRNA, epigenetics, cigarette smoke, KLK7, TGF-β1
Introduction
Esophageal carcinomas rank among the top ten causes of cancer-related deaths worldwide. These highly lethal neoplasms exhibit striking geographic variations in prevalence rates, as well as histologic and molecular phenotypes (1;2). Squamous cell carcinomas predominate in high prevalence regions throughout Asia, Africa, Middle East and South America. In contrast, adenocarcinomas are the dominant histologies observed in the United States and Western Europe (3). Although global numbers of esophageal squamous cell carcinomas (ESCC) have remained relatively stable, incidence rates for esophageal adenocarcinomas (EAC) have risen steadily during the past thirty years (3). EACs typically arise in the context of Barrett’s esophagus – a chronic condition in which the squamous epithelium of the distal esophagus is replaced by proliferating gastric- or intestinal-type columnar epithelium, in association with gastroesophageal reflux and obesity (4). Recently, cigarette smoking have been implicated in the pathogenesis of EAC; however, the precise etiology of these malignancies remains obscure (3;5–7).
In addition to genetic as well as epigenetic aberrations (1;2), a variety of microRNA (miR) alterations have been identified in EAC (8;9). For instance, miR-21, miR-143–145, miR-192, miR-194, and miR-215 appear to be consistently up-regulated, whereas let7c, miR-203, miR-205 and miR-375 are down-regulated in EAC as well as Barrett’s epithelia; these alterations in microRNA expression inhibit death signaling, and promote cell cycle progression, as well as invasion and metastatic potential of cancer cells (reviewed in ref(10;11). Whereas some of these alterations appear to be associated with particular risk factors or overall prognosis of EAC patients (9;12;13), the precise mechanisms and clinical implications of miR dysregulation during esophageal adenocarcinogenesis have not been fully defined. The present study was undertaken to characterize miR alterations induced by cigarette smoke that potentially contribute to initiation and progression of EAC.
Results
Repression of miR-217 in cultured cells exposed to CSC.
Preliminary PCR-array experiments were undertaken to examine the effects of CSC on miR expression in a panel of immortalized esophageal epithelial cells (IEEC) and EAC cells (EACC). Under exposure conditions approximating 1 pack per day, cigarette smoke modulated expression of numerous miRs; representative results pertaining to Het-1A (SV40-immortalized esophageal squamous epithelial cells), as well as EsC1 and EsC2 (EACC) are depicted in Supplementary Figure S1. Notably, CSC decreased expression of miR-217, a relatively uncharacterized micro-RNA in these cell lines.
To further investigate this phenomenon, quantitative RT-PCR (qRT-PCR) experiments were performed to examine miR-217 expression in EsC1, EsC2, OE19, and OE33 EACC, as well as Het-1A, and two h-TERT-immortalized Barrett’s epithelial cell lines (CP-A and CP-C), cultured in normal media (NM) with or without CSC for 5 days. Endogenous levels of miR-217 in untreated EACC were > six fold lower than those observed in IEEC (Figure1A; p <0.01); additional analysis demonstrated that 5 day CSC exposure down-regulated miR-217 expression approximately 3.9, 6.3, and 6.2 fold in Het-1A, CP-A, and CP-C relative to controls (Figure 1B). Similar treatment decreased miR-217 expression approximately 1.5, 2.8, 9.2, and 2.9 fold in EsC1, EsC2, OE19, and OE33 cells, respectively (Figure 1B).
Figure 1.
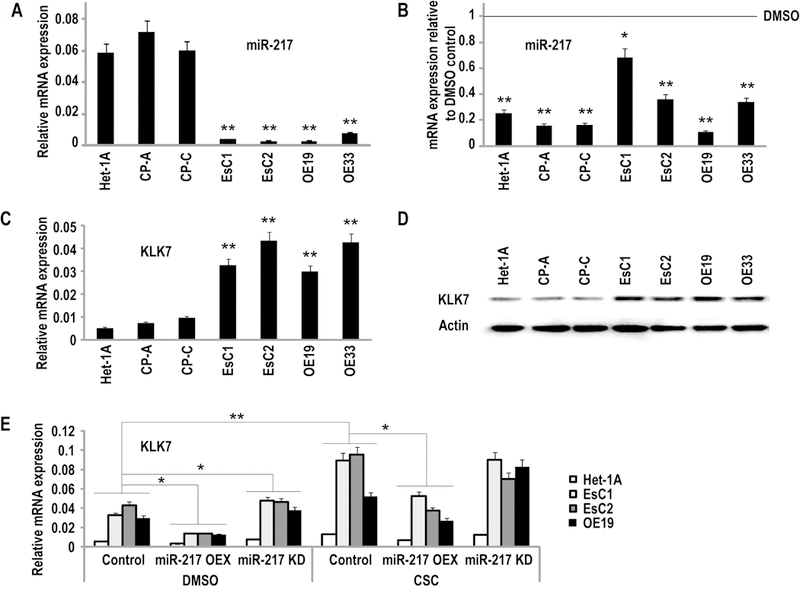
Expression of miR-217 in cultured IEEC and EACC. (*p<0.05; **p<0.01)
A: qRT-PCR analysis of endogenous miR-217 expression normalized with control miRNA (RNU44) in IEEC and EACC. Basal levels of miR-217 were significantly lower in in EACC relative to IEEC.
B: qRT-PCR analysis demonstrating repression of miR-217 in IEEC and EACC following 5 day CSC exposure.
C and D: qRT-PCR and immunoblot analysis demonstrating up-regulation of KLK7 expression in EACC relative to IEEC.
E: q-PCR analysis of KLK7 expression in Het-1A, EsC1, EsC2, and OE19 cells over-expressing precursor or antisense miR-217 constructs. CSC enhanced KLK7 expression particularly in EACC. Over-expression of miR-217 significantly attenuated CSC-mediated induction of KLK7.
Additional experiments were performed to examine if nicotine or nicotine-derived nitrosamine ketone (NNK), two major components in cigarette smoke, could recapitulate the effects of CSC on miR-217 expression in IEEC and EACC. As shown in Supplementary Figure S2, five day nicotine exposure mediated repression of miR-217 in Het-1A, EsC2 and OE19 cells at doses ranging from 50–100µg/ml, which far exceeded the concentration of nicotine in our condensates (approximately 3µg/ml; left panel). Similarly, down-regulation of miR-217 was observed in these cells following exposure to NNK at a dose of 100µg/ml, which was four fold higher than the total CSC concentration (25 µg/ml) typically used for our exposure experiments (right panel).
KLK7 is a direct target of miR-217.
Software-guided analysis suggested that miR-217 targets kallikrein 7 (KLK7), a putative oncogene, which to date has not been implicated in EAC (14;15). To further examine this issue, qRT-PCR and immunoblot experiments were performed to examine KLK7 expression levels in IEEC and EACC. As shown in Figures 1C and 1D (corresponding densitometry analysis summarized in Supplementary Table S1), KLK-7 mRNA and proteins levels were higher in EACC compared to IEEC.
Additional experiments were undertaken to ascertain if modulation of miR-217 affected expression of KLK7 in IEEC and EACC. Briefly, Het-1A, EsC1, EsC2, OE19, and OE33 cells were transfected with miR-217 precursor constructs or siRNA targeting miR-217, or respective control vectors; CP-A and CP-C cells were not used for transfection experiments due to propagation costs. qRT-PCR analysis demonstrated 7.6–33 fold increases and 18.9–26.4 fold decreases in miR-217 levels in the aforementioned cells exhibiting over-expression or knockdown of miR-217, respectively (Supplementary Figure S3). Additional analysis demonstrated that over-expression of miR-217 decreased KLK7 mRNA levels 1.7, 2.4, 3.2, and 2.4 fold, respectively, in the four cell lines relative to vector controls (Figure 1E). Repression of KLK7 by miR-217 appeared somewhat more pronounced in EACC compared to Het-1A, possibly due to lower levels of endogenous miR-217, and higher levels of KLK7 in the cancer cells. Furthermore, over-expression of miR-217 significantly attenuated CSC-mediated induction of KLK7 expression in IECC and EACC. Knock-down of miR-217 only modestly increased KLK7 mRNA levels in IEEC and EACC cultured in control media, and did not appear to significantly enhance induction of KLK7 by cigarette smoke, suggesting that the effects of miR-217 knockdown and CSC exposure on KLK-7 expression were mediated at least in part by redundant mechanisms. Modulation of miR-217 did not affect expression of KLK6, a related kallikrein family member, which is not a predicted target of miR-217 and is not modulated by CSC (Supplementary Figure S4).
Ago-RNA cross-link immunoprecipitation (CLIP) techniques (16) were used to examine if miR-217 directly interacts with KLK7 mRNA. Figure 2A depicts a single predicted miR-217 binding site within the 3’ UTR of KLK7. RT-PCR analysis revealed that the AGO family antibody precipitated KLK7 transcripts in the cytoplasm of untreated Het-1A, EsC1, and EsC2 cells (Figure 2B). Increased PCR products corresponding to a sequence of the KLK7 3' UTR containing the miR-217 binding site were observed in Het-1A, EsC1, and EsC2 cells over-expressing miR-217; this phenomenon was not observed in miR-217-depleted cells. For these experiments, β-actin served as the negative control, since its 3′ UTR contains no sequences targeted by miR-217. Immunoblot experiments demonstrated that over-expression of miR-217 decreased, whereas knock-down of miR-217 increased KLK7-but not KLK6 protein levels, respectively, in Het-1A, EsC1, and EsC2 cells (Figure 2C; corresponding densitometry analysis summarized in Supplementary Table S2). Collectively, these experiments strongly suggest that miR-217 modulates KLK7 via post-transcriptional mechanisms in IEEC and EACC.
Figure 2:

miR-217 targets KLK7. (**p<0.01)
A: The putative targeting site of miR-217 within the KLK7 3′UTR.
B: Ago-CLIP analysis of miR-217 interactions with KLK7 in Het-1A, EsC1, and EsC2 cells. Over-expression or knockdown of miR-217 significantly increased or decreased precipitation of KLK7, respectively, consistent with direct interaction of miR-217 with the KLK7 3′UTR. Actin served as the negative control.
C: Immunoblot analysis demonstrating that over-expression of miR-217 depletes KLK7, whereas knockdown of miR-217 enhances KLK7 expression in Het-1A, EsC1 and EsC2 cells.
D: qRT-PCR analysis of miR-217 and KLK7 expression levels in human esophageal cancers relative to paired adjacent normal esophageal tissues. miR-217 levels were lower whereas KLK7 levels were higher in tumors compared to adjacent normal esophageal tissues. Furthermore, miR-217 levels were lower and KLK7 levels were higher in tumors as well as normal esophageal tissues from smokers/former smokers relative to nonsmokers.
E: Higher KLK7 levels in both normal tissue (p=0.025 unadjusted; p=0.074 adjusted) and tumor tissue (p=0.057 unadjusted; p=0.17 adjusted) were associated with decreased survival of EAC patients.
Expression of miR-217 and KLK7 in primary EACs and adjacent normal esophageal tissues.
qRT-PCR experiments were performed to examine miR-217 and KLK7 expression levels in a randomly selected panel of 84 primary esophageal adenocarcinomas and adjacent histologically normal esophageal tissue samples. As shown in Figure 2D, miR-217 expression was significantly decreased (mean −6.4 fold; range −0.6–29 fold) in EACs relative to paired normal esophageal tissues (p<0.01). Additionally, miR-217 levels were significantly lower in EACs as well as histologically normal esophageal tissues from active or former smokers relative to never-smokers; in fact, miR-217 levels were lower in histologically normal esophageal tissues from smokers than EACs from never-smokers (P< 0.01). The opposite phenomenon was observed regarding KLK7 expression. Specifically, KLK7 levels were higher in primary EAC specimens compared to paired adjacent normal esophageal tissues (P< 0.01). Levels of KLK7 expression in EACs and normal esophagus were significantly higher in smokers/former smokers compared to never-smokers. KLK7 expression in tumor was inversely, moderately-well correlated with miR-217 expression in tumor (r = −0.51) and normal tissue (r = −0.53) while KLK7 expression in normal tissue was also inversely but slightly less well correlated with miR-217 expression in tumor (r = −0.45) and normal tissue (r = −0.49).
Additional studies were performed to ascertain if miR-217 and KLK7 levels in EACC were associated with survival of esophageal cancer patients. Briefly, patient outcome data were available for 65 of the 84 EACC specimens used for the aforementioned analysis. A total of 33 deaths had occurred in the cohort, but five were not due to cancer, and hence these 5 patients were censored and cancer-related survival was determined. Although not statistically significant, univariate analysis demonstrated that lower KLK7 levels in EACC (p = 0.057 unadjusted; p = 0.17 adjusted) and adjacent normal esophageal tissues (p = 0.025 unadjusted; p = 0.074 adjusted) were potentially marginally associated with better cancer-related survival (representative Kaplan Meier Curves are depicted in Figure 2E). Cox proportional hazards modeling revealed that KLK7 levels remained potentially associated with cancer-related survival after adjusting for the effect of pathologic stage (p = 0.10; HR = 2.72; 95% CI for HR: 0.82–9.05 for association of KLK7 in tumor after adjusting for pathologic stage, and p = 0.056; HR = 2.32; 95% CI for HR: 0.98–5.51 for KLK7 in normal tissue). miR-217 did not show any association with cancer-related survival (p>0.20 for levels in both tumor and normal tissue; data available upon request).
KLK7 functions as an oncogene in EACC.
A series of experiments were performed to examine the effects of KLK7 expression in EACC. Briefly, EsC1, EsC2, OE19, and OE33 cells were transfected with either cDNA for KLK7, shRNA targeting KLK7 or control vectors. Direct cell count experiments (Figure 3A) demonstrated that constitutive over-expression of KLK7 increased proliferation of EACC by 23–37% relative to respective controls (P< 0.05); in contrast, knock-down of KLK7 decreased proliferation of these cells by 29–40% (P < 0.05). Subsequent matrigel experiments (Figure 3B) demonstrated that overexpression of KLK7 increased, whereas knock-down of KLK7 inhibited invasion of EACC, respectively; Het-1A do not invade matrigel, hence, were not used for this experiment. Additional murine xenograft experiments were performed to examine if KLK7 expression modulated growth of EACC in vivo. As shown in Figure 3C, constitutive over-expression of KLK7 significantly increased volumes and masses of subcutaneous EsC2 and OE19 xenografts in athymic nude mice. Collectively, these findings suggest that KLK7 functions as an oncogene in EACC.
Figure 3.
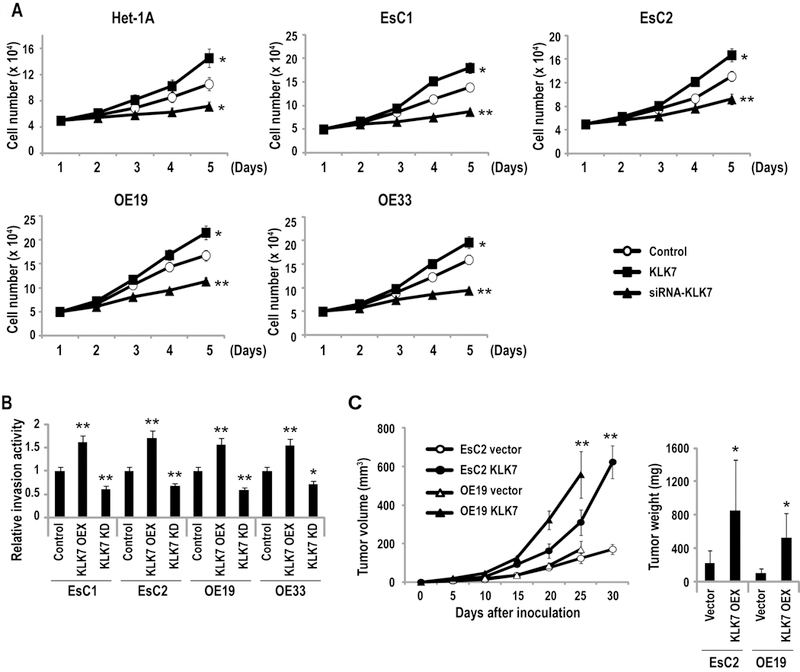
KLK7 functions as an oncogene in EAC. (*p<0.05; **p<0.01)
A: Direct cell count assays depicting in-vitro proliferation of Het-1A, EsC1, EsC2, and OE19, and OE-33 cells with over-expression or depletion of KLK7. Over-expression of miR-217 significantly enhanced, whereas knock-down of KLK7 decreased proliferation, respectively, of EACC and Het-1A cells.
B: Matrigel assays demonstrating that over-expression (OEX) of KLK7 enhances whereas knock-down (KD) of KLK7 inhibits invasion of EACC.
C: Growth of EsC2 and OE19 xenografts in nude mice. Volumes and masses of xenografts derived from EsC2 and OE19 cells over-expressing KLK7 were significantly larger than vector controls.
miR-217 is a tumor suppressor in human EACC.
Additional experiments were undertaken to examine the effects of miR-217 on the malignant phenotype of EACC. First, in-vitro growth assays were performed using Het-1A, EsC1, EsC2, OE19, and OE33 cells following over-expression or knock-down of miR-217. As shown in Figure 4A, constitutive over-expression of miR-217 decreased proliferation of EACC by ~25–45% relative to vector controls; in contrast, knock-down of miR-217 increased proliferation of Het-1A, EsC2, and OE19 cells by approximately 39.5%, 20.7%, and 21.6%, respectively, but did not appear to enhance proliferation of EsC1 or OE19 cells.
Figure 4.

miR-217 functions as a tumor suppressor in EAC. (*p<0.05; **p<0.01)
A: Direct cell count assays depicting in-vitro proliferation of Het-1A, EsC1, EsC2, OE19, and OE33 cells treated with precursor-miR-217 or anti-miR-217 constructs. Over-expression of miR-217 significantly decreased proliferation of Het-1A and EACC. Knock-down of miR-217 increased proliferation of Het-1A, but had minimal effects on proliferation of EACC, possibly due to the low endogenous levels of miR-217 in these cells.
B: Matrigel experiments depicting effects of miR-217 on invasion of EACC. Over-expression of miR-217 significantly inhibited invasion of EACC, and attenuated CSC-mediated enhancement of invasion in all four lines. Knock-down of miR-217 had minimal effects on invasion, presumably due to very low endogenous levels miR-217 in these cells.
C: Growth of EsC2 and OE19 tumor xenografts in nude mice. Volumes and masses of xenografts derived from EsC2 and OE19 cells over-expressing miR-217 were significantly smaller than vector controls.
D: qRT-PCR analysis demonstrating significant reduction of KLK7 expression in EACC xenografts over-expressing miR-217.
Matrigel assays were performed to examine the effects of miR-217 on invasion potential of EACC. Constitutive over-expression of miR-217 significantly decreased invasion of EACC (representative results pertaining to EsC1, EsC2, OE19, and OE33 cells are depicted in Figure 4B). Cigarette smoke significantly increased invasion of vector control EACC, the magnitude of which was greater than that observed in these cells following knock-down of miR-217 (p<0.01), possibly due to low endogenous levels of miR-217 in EACC prior to knock-down of this miR. Over-expression of miR-217 significantly attenuated CSC-mediated augmentation of EACC invasion. Knock-down of miR-217 did not reproducibly increase invasion of control EAC cells or enhance invasion of CSC-treated EAC cells, suggesting that the effects of miR-217 repression and CSC treatment were mediated, at least in part, by redundant mechanisms. Additional qRT-PCR experiments demonstrated that CSC tended to increase whereas miR-217 decreased expression levels of a variety of markers of EMT in cultured EACC, although the effects were relatively modest (Supplementary Figure S5).
Additional experiments were performed to examine the effects of miR-217 expression on in vivo growth of EACC. As shown in Figure 4C, volumes and masses of subcutaneous xenografts established from EsC2 and OE19 cells constitutively expressing miR-217 were significantly smaller than tumors derived from respective vector control cells. qRT-PCR experiments demonstrated significantly decreased KLK7 expression levels in xenografts constitutively expressing miR-217 relative to vector controls (Figure 4D).
Epigenetic alterations coinciding with CSC-mediated repression of miR-217.
A series of experiments were performed to examine mechanisms mediating repression of miR-217 by cigarette smoke. miR-217 is encoded within a putative host gene (DA732292) on chromosome 2 (17;18). Preliminary qPCR experiments suggested that the genomic region encoding miR-217 (Chr.2:56209989; cat# hs03219554 cn) is not deleted in esophageal carcinomas (data not shown). As such, qRT-PCR experiments were performed to examine if CSC affected expression of DA732292. As shown in Figure 5A, under exposure conditions mediating repression of miR-217, CSC did not appear to down-regulate DA732292; furthermore, CSC did not diminish expression of miR-216a, which is upstream of miR-217 within DA732292. These findings suggested that CSC mediated repression of miR-217 by more subtle mechanisms. In silico analysis of DA732292 revealed a potential CpG island approximately 5 kb upstream of the miR-217 genomic locus (Figure 5B; upper panel), which suggested that epigenetic mechanisms regulate transcription of miR-217. Sodium bisulfite sequencing experiments demonstrated increased DNA methylation within this CpG island in EACC relative to the Het-1A, and revealed that CSC enhanced DNA methylation in this region (Figure 5B; lower panel). Methylation specific PCR (MSP) experiments confirmed increased DNA methylation within this CpG island in EACC relative to IEEC (Figure 5C). Quantitative ChIP analysis demonstrated decreased levels of H3K4me3 (activation mark) with increased H3K27me3 (repressive histone mark) within this CpG island in EACC relative to Het-1A. Cigarette smoke dramatically decreased H3K4me3 and increased H3K27me3 levels within this CpG island in all of the cell lines (Figure 5D). Consistent with these findings, CSC exposure induced recruitment of EZH2 and SUZ12 to the miR-217 regulatory region in EACC and to a lesser extent, Het-1A (Supplementary Figure S6A). Subsequent, qRT-PCR analysis (Figure 5E and Supplementary Figure S6B) revealed that deoxyazacytidine (DAC; 100 nM x 72hrs) significantly increased expression of primary miR-217 but not miR-216a in EACC, and to a lesser extent, Het-1A. DZNep, which has been shown to deplete EZH2, decrease H3K27Me3 levels and augment gene induction mediated by DNA demethylating agents in cancer cells (19;20), had no effect on miR-217 expression when administered alone (0.5µM or 5µM), yet potentiated DAC-mediated induction of miR-217 in OE19, but not in EsC1 or EsC2 cells. Methylated DNA immunoprecipitation (MeDIP) experiments (Figure 5F) demonstrated that DAC significantly diminished DNA methylation in the CpG island upstream of miR-217 in EsC1 and EsC2, but not in Het-1A cells; furthermore, DAC abrogated CpG methylation induced by CSC in Het-1A as well as EsC1 and EsC2 cells.
Figure 5.
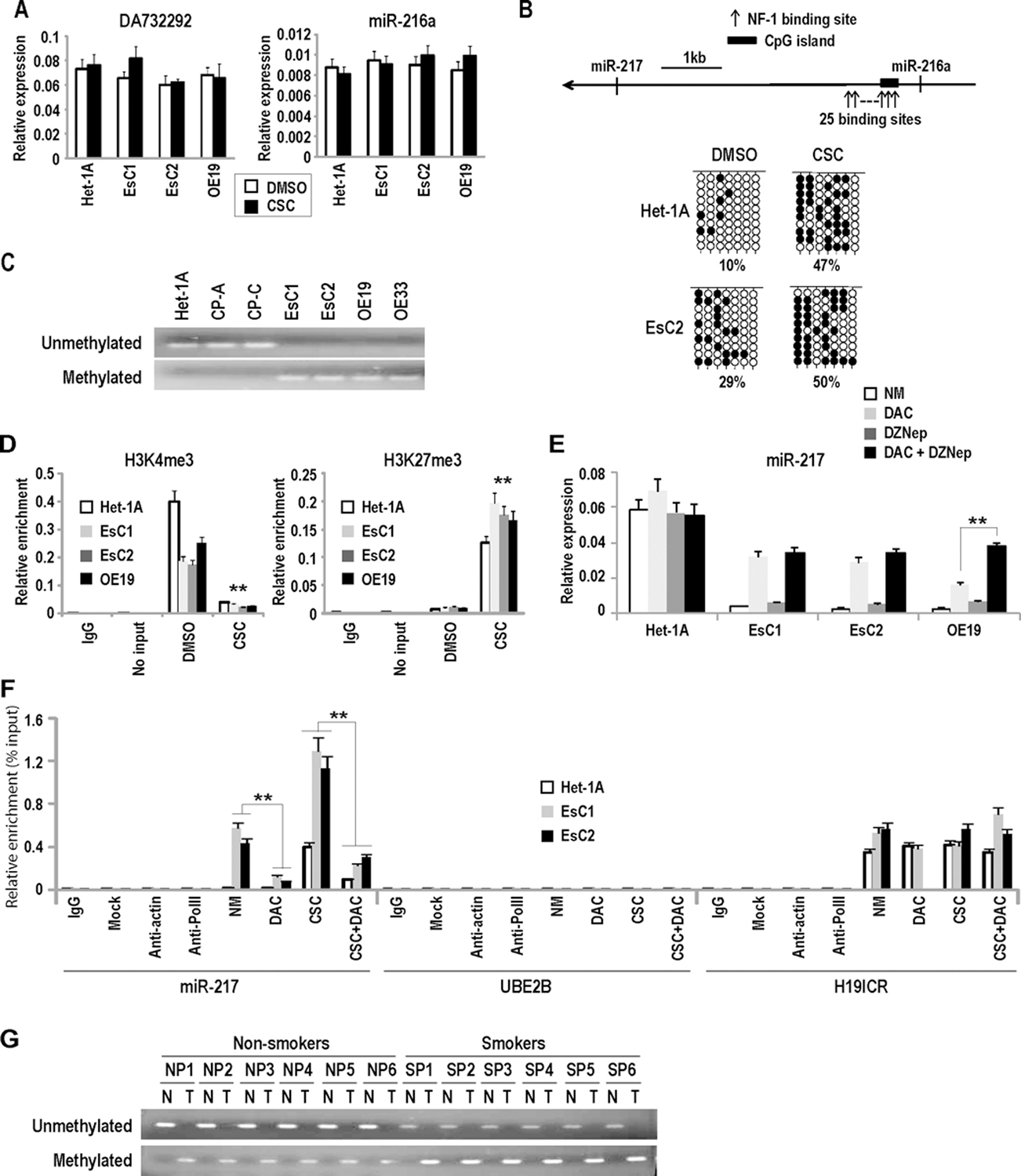
Epigenetic alterations coinciding with CSC-mediated repression of miR-217. (*p<0.05; **p<0.01)
A: qRT-PCR analysis demonstrating that CSC does not decrease DA732292 and miR-216a expression levels in Het-1A and EACC.
B: Upper panel: Schematic depiction demonstrating CpG island approximately 5 kb up-stream of the miR-217 genomic locus. 25 NF1 binding sites are densely distributed within or adjacent to this CpG island. Lower panel: results of bisulfite sequencing experiments demonstrating increased DNA methylation within the miR-217 associated CpG island in EsC2 cells relative to Het-1A. Basal levels of DNA methylation in this CpG island are higher in EACC relative to IEEC. CSC increased DNA methylation in this CpG island in Het-1A as well as EACC.
C: MSP analysis demonstrating increased methylation within the miR-217 associated CpG island in EACC relative to IEEC.
D: Quantitative ChIP analysis demonstrating that CSC decreases H3K4me3 and increases H3K27me3 levels within the miR-217 associated CpG island. These findings are consistent with results of bisulfite sequencing and MSP experiments shown above.
E: qRT-PCR analysis demonstrating that DAC (100 nM x 72h) significantly increases expression of miR-217 in EACC but not Het-1A cells. DZNep (5 uM) had minimal effects when administered alone, but augmented DAC-mediated induction of miR-217 in OE19 cells.
F: MeDIP analysis of DNA methylation profiles within the CpG island upstream of miR-217 in Het-1A, EsC1, and EsC2 cells treated with CSC and/or DAC (100 nM x72h). CSC dramatically increased methylation in this CpG island. DAC significantly abrogated CSC-mediated CpG methylation in these cells. CSC and DAC had no effect on DNA methylation levels within the UBE2B promoter (negative methylation control) or the H19 ICR (positive methylation control).
G: Representative MSP analysis of primary EAC and paired adjacent normal esophageal tissues demonstrating methylation of the miR-217 associated CpG island in EAC but not normal esophageal mucosa. Methylated PCR products were more prominent in EAC from smokers/former smokers relative to non-smokers, suggesting increased methylation of the miR-217 CpG island in patients with a history of cigarette abuse.
Additional experiments were performed to ascertain if repression of miR-217 in primary esophageal cancers was attributable to DNA methylation. MSP experiments were conducted using 16 of the samples from which the aforementioned miR-217 and KLK7 expression analyses were performed; these experiments revealed increased DNA methylation of the miR-217-associated CpG island in all of the tumors relative to matched normal esophageal tissues (Figure 5G). The magnitude of DNA methylation in this region appeared more pronounced in tumors from smokers/former smokers relative to never-smokers.
Quantitative ChIP experiments were performed to further examine epigenetic alterations within the miR-217 regulatory region in primary EAC. Consistent with the aforementioned MSP results, ChIP experiments demonstrated significantly higher levels of DNMT3a and DNMT3b, but not DNMT1 within the miR-217 regulatory region in primary EAC compared to paired normal esophageal parenchyma (Figure 6). Levels of DNMT3b in this region were significantly higher in EAC as well as normal adjacent esophageal tissues from smokers relative to non-smokers. This phenomenon was less evident for DNMT3a, although esophageal specimens from smokers tended to have higher levels of this DNMT compared to non-smokers. Additional ChIP experiments demonstrated increased EZH2 and SUZ12 levels within the miR-217 regulatory region in EAC compared to adjacent normal esophageal tissues. Consistent with these observations, H3K4Me3 levels were lower, whereas H3K27Me3 levels were higher in the miR-217 regulatory region in EAC compared to adjacent normal esophageal tissues. Similar to what was observed for DNMT3b, levels of EZH2, SUZ12 and H3K27Me3 within the miR-217 regulatory region were higher in EAC as well as normal esophageal mucosa from smokers relative to non-smokers.
Figure 6.

Quantitative ChIP analysis of DNMT1, DNMT3a, DNMT3b, EZH2, SUZ12, H3K4me3 and H3K27me3 levels within the miR-217 regulatory region in primary EAC and adjacent normal esophageal mucosa from 16 patients [six non-smokers (N), and 10 smokers (S)]. P< 0.013 for all relative enrichment values for IgG and no input controls. DNMT3b, EZH2, SUZ12 and H3K27me3 levels were higher in EAC compared to adjacent normal mucosa. PN: never-smokers vs. smokers in normal tissue; PT: never-smokers vs. smokers in tumor tissue. See text for further details.
Role of Nuclear Factor-1 in CSC-mediated regulation of miR-217.
Additional experiments were performed to further examine mechanisms by which CSC mediates repression of miR-217. Preliminary software analysis revealed 25 potential binding sites for nuclear factor-1 (NF-1) within 750 bp spanning the CpG island upstream of miR-217 (Figure 5B). qRT-PCR experiments demonstrated that basal levels of NF-1 were somewhat higher in control Het-1A relative to EsC1 and EsC2 cells (Figure 7A); CSC exposure did not alter NF-1 expression in these cells. Knock-down of NF-1 significantly diminished miR-217 expression in Het-1A, EsC1, EsC2 and OE19 cells (Figure 7B). Quantitative ChIP experiments (Figure 7C) demonstrated relatively lower NF-1 levels within the miR-217 CpG island in EACC relative to Het-1A, CP-A, and CP-C; cigarette smoke significantly decreased occupancy of NF-1 within the miR-217 CpG island in IEEC and EACC.
Figure 7.
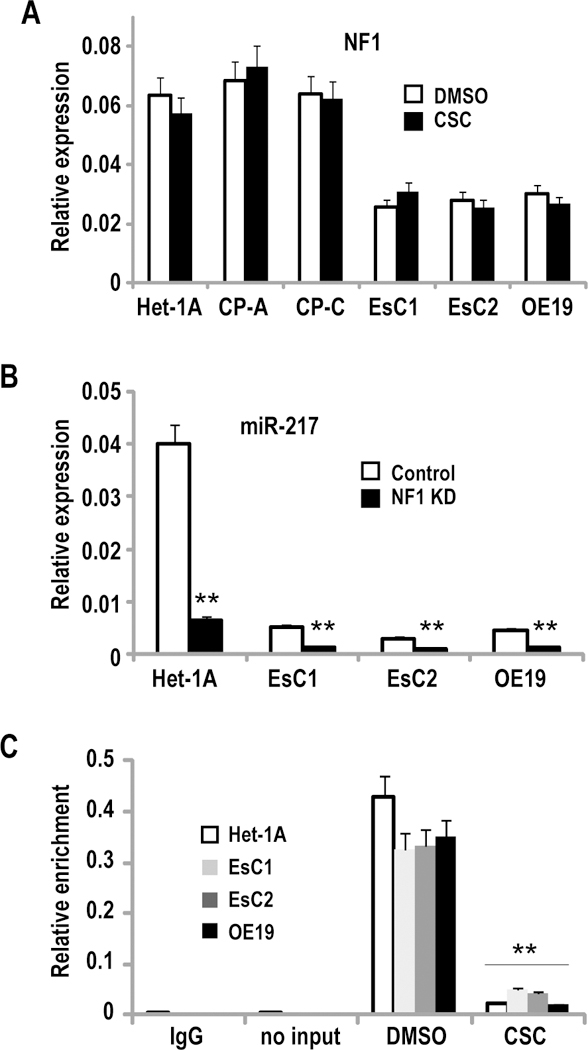
NF1 mediates CSC-induced repression of miR-217. (**p<0.01)
A: q-PCR analysis of NF1 expression in Het-1A, CP-A, CP-C, EsC1, EsC2, and OE19 cells. NF1 levels were lower in EACC relative to Het-1A, CP-A, and CP-C. CSC treatment had no significant effect on NF-1 expression in these cells.
B: q-PCR analysis demonstrating that siRNA-mediated knock-down of NF1 significantly diminishes miR-217 expression in Het-1A, and to a lesser extent in EsC1, EsC2, and OE19 cells.
C: Quantitative ChIP analysis of NF1 occupancy within the miR-217 associated CpG island in Het-1A, EsC1, EsC2, and OE19 cells cultured with or without CSC. Cigarette smoke markedly decreases occupancy of NF1 within this CpG island.
TGF-β1 mediates repression of miR-217 via de novo DNA methylation.
Recent studies have demonstrated that TGF-β1 mediates reversible DNA methylation of miR-487b loci during tobacco-induced pulmonary carcinogenesis (16). As such, additional experiments were performed to examine if TGF-β1 contributes to repression of miR-217 in tobacco associated esophageal cancers. As shown in Figure 8A, qRT-PCR analysis demonstrated that CSC enhanced TGF-β1 expression in Het-1A as well as EsC1, EsC2, and OE19 cells. Recombinant TGF-β1 repressed miR-217 in Het-1A as well as EACC (Figure 8B). A neutralizing antibody against TGF-β1 increased endogenous levels of miR-217 but not miR-216a in EACC (Figure 8C). MeDIP experiments (Figure 8D) demonstrated that recombinant TGF-β1 increased DNA methylation within the miR-217 CpG island in IEEC and EACC. A somewhat higher TGF-β1 dose (10ng/ml vs. 5ng/ml) was required to increase DNA methylation within the CPG island in EACC relative to Het-1A, possibly due to the relatively higher DNA methylation content in EACC. In contrast, TGF-β1 did not appear to increase DNA methylation levels within the methylated H19 imprinting control region or the unmethylated UBE2B locus.
Figure 8.
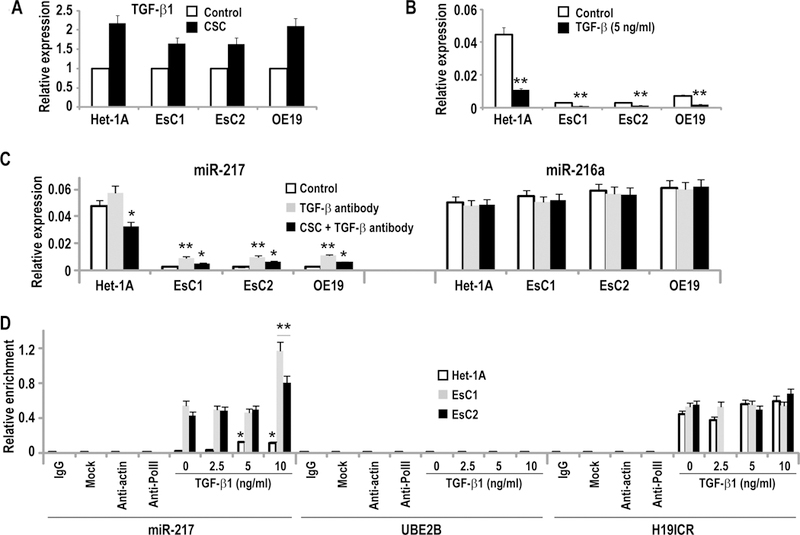
TGF-β1 mediates repression of miR-217 via de novo methylation. (*p<0.05; **p<0.01)
A: qRT-PCR analysis demonstrating that CSC exposure (48h) increased TGF-β1 expression in Het-1A, EsC1, EsC2, and OE19 cells.
B: qRT-PCR analysis demonstrating that 48h exposure to recombinant TGF-β1 decreased miR-217 expression in Het-1A and EACC.
C: qRT-PCR analysis demonstrating that a neutralizing TGF-β1 antibody modestly increased miR-217, but not miR-216a expression in Het-1A, EsC1, EsC2, and OE19 cells.
D: MeDIP analysis demonstrating that recombinant TGF-β1 increased DNA methylation levels in Het-1A cells at 5ng/ml, and in EsC1 and EsC2 cells at 10ng/ml level. This neutralizing antibody had no effect on DNA methylation within UBE2B or H19 ICR.
DNMT3b mediates CSC-induced DNA methylation of miR-217.
Additional experiments were performed to identify which DNA methyltransferase mediates epigenetic repression of miR-217 by cigarette smoke. Briefly, parental HCT116 cells, as well as HCT116 cells with knock-out of DNMT1 or DNMT3b, or combined DNMT1/DNMT3b knock-out (HCT116, DNMT1−/−, DNMT3b −/−, and DKO cells, respectively), were cultured with or without CSC for 5 days. qRT-PCR experiments demonstrated increased endogenous levels of miR-217 in DNMT3b −/− and DKO cells relative to DNMT1 −/− or parental HCT116 cells (Figure 9A). CSC decreased miR-217 expression in parental as well as DNMT1 −/− cells, but not in DNMT3b −/− or DKO cells (Figure 9A). Quantitative ChIP experiments demonstrated that CSC induced recruitment of DNMT3b, but not DNMT1 to the CpG island upstream of the miR-217 genomic locus in parental HCT116 and DNMT1−/−cells (Figure 9B). Loss of DNMT3b occupancy without appreciable change in low-level occupancy of DNMT1 within the miR-217 CpG island in DNMT3b −/− cells coincided with increased basal levels of miR-217 and complete abolishment of miR-217 repression by CSC. Additional knock-out of DNMT1 (DKO cells) only modestly augmented the effects of DNMT3b knock-out regarding endogenous levels of miR-217 or abrogation of CSC-mediated repression of this miR. Additional quantitative ChIP experiments (Figures 9C and 9D) demonstrated that TGF-β1 induced recruitment of DNMT3b, but not DNMT1, to the CpG island near the miR-217 genomic region in Het-1A, as well as EsC2, OE19 and OE33 cells; this phenomenon was not observed in EsC1 cells. With the exception of EsC2 cells, the magnitude of DNMT3b recruitment mediated by TGF-β1 signaling was not as great as that seen following CSC exposure, suggesting that TGF-β1 is not the sole mediator of CSC-induced repression of miR-217. The mechanisms and potential effects of miR-217 repression by CSC are summarized in Figure 10.
Figure 9:
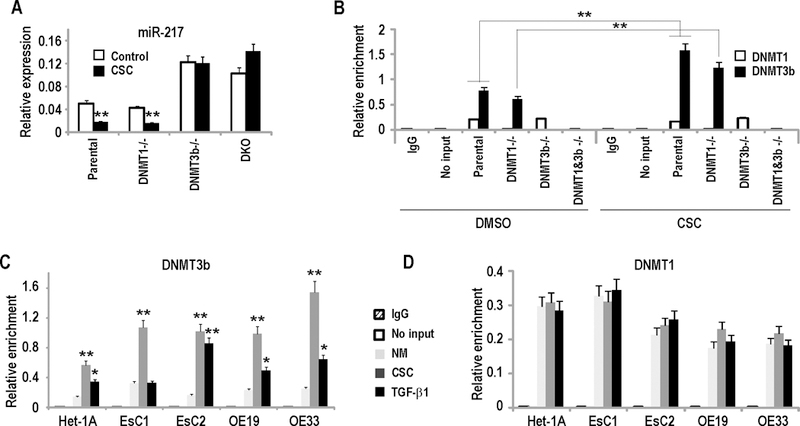
DNMT3b mediates CSC-induced repression of miR-217. (*p<0.05; **p<0.01)
A: qRT-PCR analysis of mir-217 expression in parental HCT116, DNMT1−/−, DNMT3b−/− and DKO cells cultured in the presence or absence of CSC. Loss of DNMT3b increased endogenous miR-217 levels, and abolished CSC-mediated repression of miR-217.
B: Quantitative ChIP analysis of DNMT1 and DNMT3B occupancy in the CpG island near miR-217 in parental HCT116, DNMT1−/−, DNMT3b−/−, and DKO cells cultured with or without CSC. CSC induced recruitment of DNMT3B, but not DNMT1 to this CpG island.
C/D: Quantitative ChIP analysis of DNMT3b (C) or DNMT1 (D) occupancy within the CpG island near the miR-217 genomic locus in Het-1A, EsC1, EsC2, OE19, and OE33 cells. CSC as well as recombinant TGF-β1 specifically induced recruitment of DNMT3B, but not DNMT1 to this CpG island.
Figure 10:
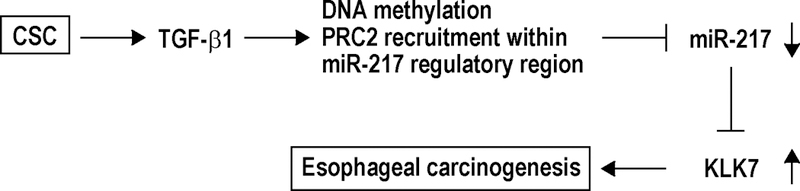
Proposed mechanism for CSC-induced esophageal carcinogenesis. Enhancement of TGF-β1 due to CSC exposure increases de novo CpG methylation and recruitment of PRC2 proteins within miR-217 regulatory region, leading to repression of miR-217 and its consequent derepression of KLK7, which promotes esophageal carcinogenesis.
Discussion
Although alterations in micro-RNA expression have been implicated in the pathogenesis of a variety of human cancers (21;22), the mechanisms by which these non-coding RNAs mediate initiation and progression of esophageal cancers have not been fully delineated. In the present study we sought to characterize micro-RNAs in esophageal epithelia and esophageal adenocarcinoma cells, which are modulated by cigarette smoke - a well-established risk factor for ESCC that only recently has been implicated in esophageal adenocarcinogenesis (23–25). Our analysis demonstrated that cigarette smoke mediates repression of miR-217- a relatively uncharacterized intragenic micro-RNA in cultured IEEC and EACC. Interestingly, cigarette smoke did not appear to repress DA732292- the host gene for miR-217, nor diminish expression of miR-216, which is upstream of miR-217 within the same host gene. Instead, CSC induced DNA hypermethylation and recruitment of polycomb repressor complexes within a CpG island between miR-216 and miR-217; heterochromatin structure coincided with decreased occupancy of NF-1 in this CpG island and repression of this microRNA. miR-217 levels were significantly lower in primary EACs- particularly those from smokers relative to paired normal esophageal mucosa. Constitutive over-expression of miR-217 inhibited, whereas knockdown of miR-217 increased growth of EACC in vitro and in vivo. To the best of our knowledge, these experiments are the first to implicate epigenetic repression of miR-217 in the pathogenesis of EAC.
Whereas our studies clearly demonstrated that CSC-mediated repression of miR-217 coincided with recruitment of DNMT3b to the miR-217 regulatory region, the mechanisms contributing to this phenomenon have not been fully delineated. Cigarette smoke contains thousands of chemicals including several hundred carcinogens; as such, identifying which components mediate repression of miR-217 may be quite difficult. Nicotine and NNK- two prototypic tobacco components, repressed miR-217 only at supra-physiologic concentrations; these observations suggest that nicotine and NNK are not the primary mediators of CSC-induced repression of miR-217, and/or that down-regulation of this miR is not primarily due to aryl hydrocarbon receptor signaling. Additional comprehensive studies are necessary to delineate associations, if any, between cigarette smoking and other environmental and genetic/epigenetic factors on miR-217 expression and risk as well as prognosis of EAC (26–28).
miR-217 has been reported to function as a tumor suppressor in pancreatic ductal carcinomas by regulating KRAS (29), and to modulate maturation of osteoblasts and chondrocytes by targeting RUNX2 and TRPS1 (30;31). In addition, miR-217 has been shown to inhibit proliferation and invasion of hepatocellular carcinoma cells via down-regulation of E2F3 (32). More recently, miR-217 has been shown to deplete DNMT3a, and to sensitize Ph+ leukemia cells to BCR/ABL tyrosine kinase inhibitors (33).
In the present study we observed that miR-217 directly targets KLK7, one of 15 human kallikrein-related serine proteases, which exhibit differential expression in normal and cancerous tissues (34). KLK7 is expressed in normal esophagus, heart, liver and skin, and contributes to desquamation of epithelial cells (34). In malignancy, KLK7 is believed to enhance invasion and metastasis by proteolytic degradation of extracellular matrix. For example, KLK7 cleaves E-cadherin to generate pro-invasive soluble E-cadherin (35;36), and increases shedding of soluble urokinase-type plasminogen activator receptor (37). In addition, KLK7 catalyzes formation of an active, truncated MMP9 variant that is not produced by other proteases (38). Over-expression of KLK7 significantly enhances invasion of glioma as well as prostate, pancreas and ovarian cancer cells (35;39–41). Consistent with these findings, we observed that KLK7 over-expression increased invasion of EACC; more importantly, KLK7 increased growth of EACC in vivo. These latter findings strongly suggest that KLK7 functions as an oncogene in EACC. The fact that inhibition of EAC xenograft growth by miR-217 coincided with significantly decreased KLK7 expression strongly suggests that the growth inhibitory effects of miR-217 in EACC are mediated, at least in part, by repression of KLK7. However our present findings do not exclude the possibility that some of the inhibitory effects of miR-217 in EACC are mediated by down-regulation of KRAS and E2F3 (29;32), or subtle epigenetic alterations due to down-regulation of DNNMT3a (33).
Over-expression of KLK7 has been associated with decreased disease free survival and overall survival in patients with colorectal carcinoma (42), as well as chemoresistance and shorter survival in patients with ovarian cancer (15). Furthermore, over-expression of KLK7 correlates with a nearly three-fold increase in mortality rates in patients with unresectable pancreas cancer (43). In our study, we were unable to demonstrate that KLK7 over-expression is a statistically significant prognostic factor in cancer-related survival in EAC patients after adjusting for pathologic stage. However, trends towards an association were identified, even after adjusting for stage. Similarly, although repression of miR-217 has been associated with decreased survival in patients with renal carcinoma (44), our analysis did not demonstrate an association between miR-217 levels and cancer-related survival of EAC patients. Particularly with respect to KLK7 expression levels, additional studies should be performed using a much larger cohort with a complete clinical database to ascertain if this parameter is associated with disease burden and overall survival in EAC patients. Furthermore, given the magnitude of miR-217 repression and up-regulation of KLK7 in EAC-particularly those from smokers, additional studies should be undertaken to examine miR-217 and KLK7 expression levels in ESCC, which are far more prevalent and associated with heavy tobacco exposure.
Materials and Methods
Cell lines and treatment conditions.
NCI-SB-EsC1 (EsC1) and NCI-SB-EsC2 (EsC2) were established in our lab from two esophageal cancer patients following chemo-radiation and definitive surgery for Barrett’s associated adenocarcinoma. OE19 and OE-33 esophageal adenocarcinoma lines were purchased from Sigma. Het-1A cells from normal esophageal epithelium immortalized with SV-40, as well as CP-A cells from non-dysplastic Barrett’s metaplasia and CP-C cells from high-grade Barrett’s dysplasia immortalized with hTERT were purchased from ATCC and were cultured as instructed. Full details of culture and smoke exposure conditions are submitted as Supplementary Materials.
Human tissues.
Primary esophageal cancer specimens and adjacent histologically normal esophageal parenchyma were harvested intra-operatively from patients undergoing potentially curative resections at the National Cancer Institute (Bethesda, MD), University of Michigan (Ann Arbor, MI), Dalhousie University (Halifax, Canada), or Weill-Cornell University Medical Center (NY, NY) under IRB-approved protocols. Full details are submitted as Supplementary Materials.
miRNA over-expression and inhibition.
Submitted as Supplementary Materials.
Quantitative RT-PCR.
Submitted as Supplementary Materials.
Chromatin immunoprecipitation (ChIP).
Submitted as Supplementary Materials.
siRNA and shRNA knockdown.
Submitted as Supplementary Materials.
MicroRNA PCR array analysis.
Submitted as Supplementary Materials.
Western blot analysis.
Submitted as Supplementary Materials.
Ago Cross-link immunoprecipitation (CLIP).
Cells were harvested and CLIP experiments were performed using either anti-AGO family or elF2C antibodies as described (45). Full details are submitted as Supplementary Materials.
Methylated DNA immunoprecipitation (MeDIP) assays.
Submitted as Supplementary Materials.
In vitro invasion assays.
Submitted as Supplementary Materials
Murine xenograft experiments.
Submitted as Supplementary Materials.
Statistical analysis.
Data are presented as means ± SEM. Differences between matched samples from the same esophageal cancer patients, such as tumor and normal tissue were tested with the signed-rank test. Groups were compared with the Wilcoxon–Mann-Whitney test. Confidence intervals for the differences in medians between two groups were computed by the bias-corrected and accelerated bootstrapping methods. Differences in tumor volumes between opposing flanks of tumor-bearing mice were tested with the Wilcoxon-signed rank test at 5-day intervals. Confidence intervals for the median of flank differences were computed as exact non-parametric two-tailed confidence intervals based on the Wilcoxon signed-rank test. P values were adjusted for multiple testing by complete resampling. P < 0.05 was considered significant.
The probability of cancer-related survival as a function of time was determined by the Kaplan-Meier method, and the statistical significance of a set of Kaplan-Meier curves was determined by the log-rank test. In some cases, univariate analyses demonstrated that combining patients into two groups, who were originally grouped into four categories on the basis of pathologic stage of mirR-217 or KLK7 levels, would result in improved association with cancer-related survival. In these cases, the adjusted p-value is equal to three times the unadjusted p-value to account for the number of implicit tests performed in order to arrive at the final grouping. The Cox proportional hazards model was used to demonstrate the joint statistical effect of factors found to have potential statistical significance in univariate analyses. The correlation between miR-217 levels and KLK7 levels was determined by Spearman rank correlation analyses.
Supplementary Material
Acknowledgements
The authors wish to express their gratitude to Ms. Jan Pappas for assistance with manuscript preparation. This study was supported by NIH intramural funding, and the Stephen J. Solarz Memorial Fund.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov 2012;2:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dulak AM, Schumacher SE, van LJ, Imamura Y, Fox C, Shim B, et al. Gastrointestinal Adenocarcinomas of the Esophagus, Stomach, and Colon Exhibit Distinct Patterns of Genome Instability and Oncogenesis. Cancer Res 2012;72:4383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Napier KJ, Scheerer M, Misra S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol 2014;6:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Estores D, Velanovich V. Barrett esophagus: epidemiology, pathogenesis, diagnosis, and management. Curr Probl Surg 2013;50:192–226. [DOI] [PubMed] [Google Scholar]
- (5).Spechler SJ. Barrett esophagus and risk of esophageal cancer: a clinical review. JAMA 2013;310:627–36. [DOI] [PubMed] [Google Scholar]
- (6).Ek WE, Levine DM, D’Amato M, Pedersen NL, Magnusson PK, Bresso F, et al. Germline genetic contributions to risk for esophageal adenocarcinoma, Barrett’s esophagus, and gastroesophageal reflux. J Natl Cancer Inst 2013;105:1711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Carr JS, Zafar SF, Saba N, Khuri FR, El-Rayes BF. Risk factors for rising incidence of esophageal and gastric cardia adenocarcinoma. J Gastrointest Cancer 2013;44:143–51. [DOI] [PubMed] [Google Scholar]
- (8).David S, Meltzer SJ. MicroRNA involvement in esophageal carcinogenesis. Curr Opin Pharmacol 2011;11:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mathe EA, Nguyen GH, Bowman ED, Zhao Y, Budhu A, Schetter AJ, et al. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res 2009;15:6192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Song JH, Meltzer SJ. MicroRNAs in pathogenesis, diagnosis, and treatment of gastroesophageal cancers. Gastroenterology 2012;143:35–47. [DOI] [PubMed] [Google Scholar]
- (11).Mayne GC, Hussey DJ, Watson DI. MicroRNAs and esophageal cancer--implications for pathogenesis and therapy. Curr Pharm Des 2013;19:1211–26. [DOI] [PubMed] [Google Scholar]
- (12).Fassan M, Volinia S, Palatini J, Pizzi M, Baffa R, De BM, et al. MicroRNA expression profiling in human Barrett’s carcinogenesis. Int J Cancer 2011;129:1661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Leidner RS, Ravi L, Leahy P, Chen Y, Bednarchik B, Streppel M, et al. The microRNAs, MiR-31 and MiR-375, as candidate markers in Barrett’s esophageal carcinogenesis. Genes Chromosomes Cancer 2012;51:473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Devetzi M, Trangas T, Scorilas A, Xynopoulos D, Talieri M. Parallel overexpression and clinical significance of kallikrein-related peptidases 7 and 14 (KLK7KLK14) in colon cancer. Thromb Haemost 2013;109:716–25. [DOI] [PubMed] [Google Scholar]
- (15).Dong Y, Tan OL, Loessner D, Stephens C, Walpole C, Boyle GM, et al. Kallikrein-related peptidase 7 promotes multicellular aggregation via the alpha(5)beta(1) integrin pathway and paclitaxel chemoresistance in serous epithelial ovarian carcinoma. Cancer Res 2010;70:2624–33. [DOI] [PubMed] [Google Scholar]
- (16).Xi S, Xu H, Shan J, Tao Y, Hong JA, Inchauste S, et al. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J Clin Invest 2013;123:1241–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res 2002;12:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol 2009;11:881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kemp CD, Rao M, Xi S, Inchauste S, Mani H, Fetsch P, et al. Polycomb Repressor Complex-2 is a Novel Target for Mesothelioma Therapy. Clin Cancer Res 2012;18:77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S, et al. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res 2011;71:4192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Diaz-Lopez A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res 2014;6:205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gu J, Xuan Z. Inferring the perturbed microRNA regulatory networks in cancer using hierarchical gene co-expression signatures. PLoS One 2013;8:e81032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hardikar S, Onstad L, Blount PL, Odze RD, Reid BJ, Vaughan TL. The role of tobacco, alcohol, and obesity in neoplastic progression to esophageal adenocarcinoma: a prospective study of Barrett’s esophagus. PLoS One 2013;8:e52192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tramacere I, La VC, Negri E. Tobacco smoking and esophageal and gastric cardia adenocarcinoma: a meta-analysis. Epidemiology 2011;22:344–9. [DOI] [PubMed] [Google Scholar]
- (25).Cook MB, Kamangar F, Whiteman DC, Freedman ND, Gammon MD, Bernstein L, et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J Natl Cancer Inst 2010;102:1344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ek WE, Levine DM, D’Amato M, Pedersen NL, Magnusson PK, Bresso F, et al. Germline genetic contributions to risk for esophageal adenocarcinoma, Barrett’s esophagus, and gastroesophageal reflux. J Natl Cancer Inst 2013;105:1711–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xu E, Gu J, Hawk ET, Wang KK, Lai M, Huang M, et al. Genome-wide methylation analysis shows similar patterns in Barrett’s esophagus and esophageal adenocarcinoma. Carcinogenesis 2013;34:2750–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Frankel A, Armour N, Nancarrow D, Krause L, Hayward N, Lampe G, et al. Genome-wide analysis of esophageal adenocarcinoma yields specific copy number aberrations that correlate with prognosis. Genes Chromosomes Cancer 2014;53:324–38. [DOI] [PubMed] [Google Scholar]
- (29).Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis 2010;31:1726–33. [DOI] [PubMed] [Google Scholar]
- (30).Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ, et al. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci U S A 2011;108:9863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang Y, Xie RL, Gordon J, LeBlanc K, Stein JL, Lian JB, et al. Control of mesenchymal lineage progression by microRNAs targeting skeletal gene regulators Trps1 and Runx2. J Biol Chem 2012;287:21926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Su J, Wang Q, Liu Y, Zhong M. miR-217 inhibits invasion of hepatocellular carcinoma cells through direct suppression of E2F3. Mol Cell Biochem 2014;392:289–96. [DOI] [PubMed] [Google Scholar]
- (33).Nishioka C, Ikezoe T, Yang J, Nobumoto A, Tsuda M, Yokoyama A. Downregulation of miR-217 correlates with resistance of ph leukemia cells to ABL tyrosine kinase inhibitors. Cancer Sci 2014;105:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shaw JL, Diamandis EP. Distribution of 15 human kallikreins in tissues and biological fluids. Clin Chem 2007;53:1423–32. [DOI] [PubMed] [Google Scholar]
- (35).Johnson SK, Ramani VC, Hennings L, Haun RS. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer 2007;109:1811–20. [DOI] [PubMed] [Google Scholar]
- (36).Grabowska MM, Day ML. Soluble E-cadherin: more than a symptom of disease. Front Biosci (Landmark Ed) 2012;17:1948–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ramani VC, Haun RS. Expression of kallikrein 7 diminishes pancreatic cancer cell adhesion to vitronectin and enhances urokinase-type plasminogen activator receptor shedding. Pancreas 2008;37:399–404. [DOI] [PubMed] [Google Scholar]
- (38).Ramani VC, Kaushal GP, Haun RS. Proteolytic action of kallikrein-related peptidase 7 produces unique active matrix metalloproteinase-9 lacking the C-terminal hemopexin domains. Biochim Biophys Acta 2011;1813:1525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Prezas P, Scorilas A, Yfanti C, Viktorov P, Agnanti N, Diamandis E, et al. The role of human tissue kallikreins 7 and 8 in intracranial malignancies. Biol Chem 2006;387:1607–12. [DOI] [PubMed] [Google Scholar]
- (40).Mo L, Zhang J, Shi J, Xuan Q, Yang X, Qin M, et al. Human kallikrein 7 induces epithelial-mesenchymal transition-like changes in prostate carcinoma cells: a role in prostate cancer invasion and progression. Anticancer Res 2010;30:3413–20. [PubMed] [Google Scholar]
- (41).Prezas P, Arlt MJ, Viktorov P, Soosaipillai A, Holzscheiter L, Schmitt M, et al. Overexpression of the human tissue kallikrein genes KLK4, 5, 6, and 7 increases the malignant phenotype of ovarian cancer cells. Biol Chem 2006;387:807–11. [DOI] [PubMed] [Google Scholar]
- (42).Talieri M, Mathioudaki K, Prezas P, Alexopoulou DK, Diamandis EP, Xynopoulos D, et al. Clinical significance of kallikrein-related peptidase 7 (KLK7) in colorectal cancer. Thromb Haemost 2009;101:741–7. [PubMed] [Google Scholar]
- (43).Iakovlev V, Siegel ER, Tsao MS, Haun RS. Expression of kallikrein-related peptidase 7 predicts poor prognosis in patients with unresectable pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2012;21:1135–42. [DOI] [PubMed] [Google Scholar]
- (44).Li H, Zhao J, Zhang JW, Huang QY, Huang JZ, Chi LS, et al. MicroRNA-217, down-regulated in clear cell renal cell carcinoma and associated with lower survival, suppresses cell proliferation and migration. Neoplasma 2013;60:511–5. [DOI] [PubMed] [Google Scholar]
- (45).Xi S, Yang M, Tao Y, Xu H, Shan J, Inchauste S, et al. Cigarette smoke induces C/EBP-beta-mediated activation of miR-31 in normal human respiratory epithelia and lung cancer cells. PLoS One 2010;5:e13764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.