

Abstract
Urea carboxylic acids, products of aryl hydantoin hydrolysis, were recently identified as a new antischistosomal chemotype. We now describe a baseline structure-activity relationship (SAR) for this compound series. With one exception, analogs of lead urea carboxylic acid 2 were quite polar with LogD7.4 values ranging from −1.9 to 1.8, had high aqueous solubilities in the range of 25-100 μg/mL, and were metabolically stable. None of the compounds had measurable in vitro antischistosomal activity or cytotoxicity, but four of these had moderate worm burden reduction (WBR) values of 42-70% when they were administered as single 100 mg/kg oral doses to S. mansoni-infected mice. These data indicate that with the exception of the gem-dimethyl substructure and the distal nitrogen atom of the urea functional group, the rest of the structure of 2 is required for in vivo antischistosomal activity.
Keywords: urea carboxylic acid, antischistosomal, SAR
Graphical Abstract
The aryl hydantoin Ro 13-3978 is a promising antischistosomal lead compound (Figure 1) despite its weak antiandrogenic properties.1-4 Although Ro 13-3978 has little effect on cultured adult S. mansoni,3 it has high oral efficacy against S. mansoni, S. haematobium and S. japonicum in a variety of animal models.2,3 For example, Ro 13-3978 has a single oral dose ED50 of 15 mg/kg in the S. mansoni mouse model; in this same model, praziquantel has an ED50 of 172 mg/kg.3
Figure 1.

Metabolism of Ro 13-3978
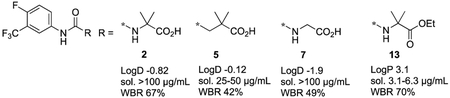
Ro 13-3978 has high metabolic stability,3,4 but when we administered a 100 mg/kg oral dose of this aryl hydantoin to mice, we observed low levels of hydroxymethyl metabolite 1 and urea carboxylic acid 2, the hydantoin hydrolysis product5 (Figure 1). Based on AUC0-24h values, the respective exposures of 1 and 2 were 3 and <1% relative to that of Ro 13-3978.4 At the same 100 mg/kg dose, metabolite 1 was inactive, whereas 2 reduced worm burden in S. mansoni-infected mice by 67%4 and thus has some promise as a new antischistosomal chemotype. We now describe physicochemical profiling, in vitro ADME, and in vivo activities against S. mansoni for a number of analogs of urea carboxylic acid 2 (3-13, Table 1) designed to increase antischistosomal efficacy and establish a baseline structure-activity relationship (SAR) for this chemotype.
Table 1.
Physicochemical properties, in vitro metabolic stability and in vivo activity against S. mansoni for 2-13.
 | |||||
|---|---|---|---|---|---|
| Compd | LogD7.4a | PSA (Å2)a |
Sol2.0/Sol6.5 (μg/mL)b |
h/m CLint (μL/min/mg protein)c |
S. mansoni WBR (%) 1 × 100 mg/kg po |
| 2 | −0.82 | 81.3 | 25-50/>100 | <7/<7 | 67d |
| 3 | −1.7 | 81.3 | >100/50-100 | <7/<7 | 20 |
| 4 | −0.8 | 63.7 | >100/>100 | <7/<7 | 14 |
| 5 | −0.12 | 69.2 | 25-50/25-50 | <7/<7 | 42 |
| 6 | −0.87 | 69.2 | 50-100/25-50 | <7/<7 | 0 |
| 7 | −1.9 | 81.3 | >100/>100 | <7/<7 | 49 |
| 8 | −1.2 | 81.3 | >100/>100 | <7/<7 | 4 |
| 9 | −0.7 | 81.3 | 50-100/>100 | 7/<7 | 21 |
| 10 | −0.7 | 72.5 | 50-100/>100 | <7/<7 | 0 |
| 11 | −0.85 | 72.5 | 50-100/>100 | <7/<7 | 0 |
| 12 | 1.8 | 84.2 | 50-100/50-100 | 19/18 | 38 |
| 13 | 3.1 | 67.4 | 3.1-6.3/3.1-6.3 | NDe | 70f |
calculated using ChemAxon JChem for Excel
Compounds in DMSO were spiked into either pH 6.5 phosphate buffer or 0.01 M HCl (approx. pH 2.0) and analyzed by nephelometry to determine a concentration range.
in vitro intrinsic clearance measured in human and mouse liver microsomes
data from Wang et al.4
Metabolic stability parameters were not determined as rapid loss of compound was noted in control microsomal incubations in the absence of co-factor
statistically different from control group (p=0.006)
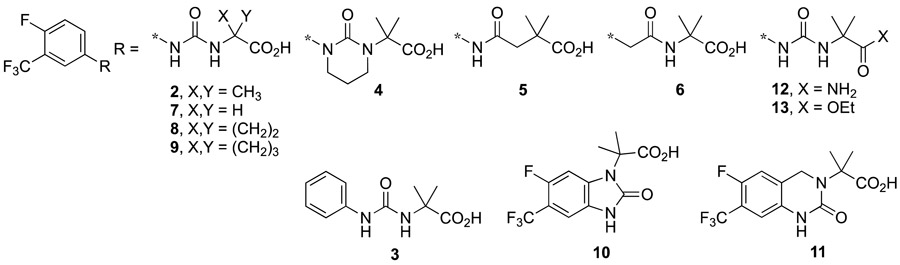
Target compounds 4-13 were prepared (Supporting Information) by a variety of reactions described in Schemes 1-5. Urea carboxylic acids 7-9 were prepared in reactions between phenyl isocyanate 146 and amino acids 15-17 in low to moderate yields (Scheme 1). Similarly, urea ester 13 was prepared in a reaction between 14 and amino ester 18 in 64% yield.
Scheme 1.

Reagents and conditions: (a) 1-2 M NaOH/CH3CN, 0 °C to rt, 12 h, then pH to 3 with aq. HC1; (b) DIPEA, CH2C12, 0 °C to rt, 12 h, then aq. HC1.
Scheme 5.

Reagents and conditions: (a) 1 M DIBAL in CH2Cl2, 0 °C to rt, 24 h; (b) tert-butyl 2-amino-2-methylpropanoate, AcOH, NaCNBH3, rt, 7 days; (c) carbonyldiimidazole, dimethoxyethane, rt to reflux, 27 h; (d) TFA, CH2Cl2, rt, 24 h.
Urea amide 12 was prepared from 2 in a two-step reaction. First, 2 was converted to N-hydroxysuccimide active ester 19 which was then exposed to cone, ammonium hydroxide to form 12 in 29% overall yield (Scheme 2).
Scheme 2.

Reagents and conditions: (a) N-(3-dimethylaminopropyl)-A’-ethylcarbodiimide hydrochloride, N-hydroxysuccinimide, DMA, rt; 12 h; (b) 28% wt. NH4OH, rt, 1.5 h.
Urea carboxylic acid 4 was prepared in 42% yield by treatment of Ro 13-3978 with sodium hydride followed by 1-bromo-3-chloropropane (Scheme 3). Carboxy amide 5 was prepared by acylation of aniline 20 with the corresponding anhydride in 93% yield whereas carboxy amide 6 was prepared by acylation of gem-dimethyl glycine with acid chloride 21 in 44% yield (Scheme 3).
Scheme 3.

Reagents and conditions: (a) NaH, DMF, 0 °C to rt, 2 h then 1-bromo-3-chloropropane, rt, 48 h and pH to 3 with AcOH; (b) 3,3-dimethyldihydrofuran-2,5-dione, THF, rt, 12 h; (c) 2-amino-2-methylpropanoic acid, 2 M NaOH, 0 ° C, 2 h, then pH to 3 with aq. HCl.
The synthesis of imidazolone carboxylic acid 10 began with conversion of aniline 227 to its isocyanate and subsequent reaction with the tert-butyl ester of gem-dimethyl glycine to afford urea ester 23 in 63% yield (Scheme 4). Exposure of 23 to CuI and DBU in hot DMSO effected ring closure to 24 in 72% yield. Ester hydrolysis of 24 afforded 10 in 97% yield.
Scheme 4.

Reagents and conditions: (a) triphosgene, toluene/EtOAc, 0-80 °C, 12 h; (b) tert-butyl 2-amino-2-methylpropanoate hydrochloride, DIPEA, CH2Cl2, 0 °C to rt, 12 h; (c) CuI, DBU, DMSO, 120 °C, 0.3 h; (d) TFA, CH2Cl2, rt, 24 h.
The synthesis of dihydropyrimidinone carboxylic acid 11 began by conversion of benzonitrile 257 to benzaldehyde 26 with DIBAL in 33% yield. Reductive amination to amino ester 27 (53%) followed by ring closure with carbonyldiimidazole afforded 28 (93%). Ester hydrolysis of 28 furnished 11 in 70% yield. Known target compound 3 was prepared according to the method of Durini et al.8
We now consider the physicochemical and in vitro ADME properties of these analogs of 2 (Table 1). With the exception of amide 12 and ethyl ester 13, all compounds were quite polar with calculated LogD7.4 values below 0 and high aqueous solubilities in the range of 25-100 μg/mL. In spite of this high polarity, 2 exhibited rapid and extensive absorption following oral administration to mice at a dose of 100 mg/kg (Figure 2) suggesting that the polarity does not limit oral exposure. Consistent with their high polarity, these compounds were metabolically stable with low intrinsic clearance values (corresponding to in vitro half-lives in excess of 200 min) in human and mouse liver microsomes. The only exception to this was ethyl ester 13 which underwent non-NADPH mediated degradation, presumably by non-specific esterases present in the microsomal test systems.
Figure 2.

Plasma concentration versus time profile for 2 following oral administration to mice at a dose of 100 mg/kg. Data points represent the mean of n=2 mice with the exception of the points marked with * (n=1).
Similar to 2,4 3-13 had no activity against schistosomula (NTS) or cultured adult S. mansoni9 at a 10 μM concentration (data not shown). In contrast, praziquantel has an LC50 of 77 nM against adult S. mansoni in vitro.10 To assess host cell cytotoxicity, target compounds were tested for growth inhibition of the rat myoblast L6 cell line; these cells were unaffected at compound concentrations up to 200 μM (data not shown).
In vivo antischistosomal activity was determined by measuring worm burden reduction (WBR) values following administration of single 100 mg/kg oral doses to S. mansoni-infected mice (Table 1). As we had previously observed for 2,4 none of the compounds tested had high in vivo activity with WBR values >90%, although 5, 7, and 13 had moderate WBR values of 42, 49, and 70%, respectively, the latter being statistically significant (p=0.006). These data indicate that with the exception of the gem-dimethyl substructure and distal nitrogen atom of the urea functional group, the rest of the structure of 2 is required for antischistosomal activity. Further, 4, 10 and 11 reveal that conformational restriction is not tolerated. Even though none of these compounds had high in vivo activity, it is instructive to note that at this same 100 mg/kg dose, praziquantel reduces worm burden by only 15%10
Drug discovery starting with active metabolites11,12 can be a fruitful strategy. However, in this initial SAR investigation, we were not able identify an analog of 2 with a superior ADME and antischistosomal profile, although our data does suggest that a prodrug strategy could be useful.
Supplementary Material
Urea carboxylic acids, a new antischistosomal chemotype.
Baseline structure-activity relationship (SAR).
Analogs were quite polar, had high aqueous solubility, and were metabolically stable.
Modifications to the gem-dimethyl and urea functional group were tolerated.
Acknowledgment
We thank Monica Cal and Marcel Kaiser for technical assistance and acknowledge the U.S. National Institutes of Health (AI116723-01) and the European Research Council (ERC-2013-CoG 614739-A HERO) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernauer K, Link H, Stohler H. Antiandrogenic and schistosomicidal imidazolidine derivatives. U. S. Patent 4,234,736 1980.
- 2.Link H, Stohler HR. 3-Arylhydantoine, eine substanzklasse mit schistosomizider wirkung. Eur J Med Chem – Chim Ther. 1984;19:261–265. [Google Scholar]
- 3.Keiser J, Panic G, Vargas M, Wang C, Don Y, Gautam N, Vennerstrom JL. Aryl hydantoin Ro 13-3978, a broad spectrum antischistosomal. J Antimicrob Chemother. 2015;70:1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Zhao Q, Vargas M, Jones JO, White KL, Shackleford DM, Chen G, Saunders J, Ng ACF, Chiu FCK, Dong Y, Charman SA, Keiser J, Vennerstrom JL. Revisiting the SAR of the antischistosomal aryl hydantoin (Ro 13-3978). J Med Chem. 2016;59:10705–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen JB, Hafliger O. Determination of the dissociation constant of a weak acid using a dissolution rate method. J Pharm Sci. 1983;72:429–431. [DOI] [PubMed] [Google Scholar]
- 6.Zhan W, Li Y, Huang W, Zhao Y, Yao Z, Yu S, Yuan S, Jiang F, Yao S, Li S. Design, synthesis and antitumor activities of novel bis-aryl ureas derivatives as Raf kinase inhibitors. Bioorg Med Chem. 2012;20:4323–4329. [DOI] [PubMed] [Google Scholar]
- 7.Borchardt A, Davis R, Beauregard C, Becker D, Gamache D, Noble SA, Hellberg M, Klimko P, Qiu Z, Payne J, Yanni J. Azoloquinoxaline compounds as inhibitors of histamine receptors for the treatment of disease and their preparation. U.S. Pat. Appl. Publ 2011; US 20110257137 A1 [Google Scholar]
- 8.Durini M, Russotto E, Pignataro L, Reiser O, Piarulli U. SupraBox: Chiral supramolecular oxazoline ligands. Eur J Org Chem. 2012;5451–5461. [Google Scholar]
- 9.Keiser J. In vitro and in vivo trematode models for chemotherapeutic studies. Parasitology 2010;137:589–603. [DOI] [PubMed] [Google Scholar]
- 10.Keiser J, Manneck T, Vargas M. Interactions of mefloquine with praziquantel in the Schistosoma mansoni mouse model and in vitro. J Antimicrob Chemother. 2011;66:1791–1797. [DOI] [PubMed] [Google Scholar]
- 11.Fura A, Shu YZ, Zhu M, Hanson RL, Roongta V, Humphreys WG. Discovering drugs through biological transformation: Role of pharmacologically active metabolites in drug discovery. J Med Chem. 2004;47:4339–4351. [DOI] [PubMed] [Google Scholar]
- 12.Fura A. Role of pharmacologically active metabolites in drug discovery and development. Drug Discov Today. 2006;11:133–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.