

Abstract
Pro-inflammatory cytokine TNFα antagonizes regulatory T cell (Treg) suppressive function with a measurable reduction of IL-10 protein secretion. Tregs are critical to suppress excessive immune activation, particularly within the intestine where high antigenic loads elicit chronic subclinical immune activation. Employing a TNFα-driven murine inflammatory bowel disease (IBD) model (TNFΔARE/+), which mirrors the Treg expansion and transmural ileitis seen in Crohn’s disease, we demonstrate that the TNFα-mediated loss of Treg suppressive function coincides with induction of a specific miRNA, miR-106a in both humans and mice, via NFκB promoter binding to suppress post-transcriptional regulation of IL-10 release. Elevation of miR-106a and impaired Treg function in this model recapitulate clinical data from IBD patients. MiR-106a deficiency promotes Treg induction, suppressive function and IL-10 production in vitro. MiR-106a knockout attenuated chronic murine ileitis, whereas T cell restricted deficiency of miR106a attenuated adoptive transfer colitis. In both models, attenuated inflammation coincided with suppression of both Th1 and Th17 cell subset expansion within the intestinal lamina propria. Collectively, our data demonstrate impaired Treg suppressive function in a murine IBD model consistent with human disease and support the potential for inhibition of miR-106a as a future therapeutic approach to treat chronic inflammatory conditions including IBD.
Introduction
Inflammatory Bowel Disease (IBD) currently affects approximately 2.5 million Europeans, though the incidence continues to increase 1. The current standard treatment, anti-tumor necrosis factor (TNFα) antibody therapy induces remission in less than 50% of IBD patients 2,3 leaving an unmet need for newer therapeutics. Clinical and experimental data indicate that IBD persistence is primarily CD4+ T cellmediated 4.
Regulatory T cells (Treg), defined by forkhead box P3 (FoxP3) expression represent a subset of immunosuppressive T cells 5. While Tregs utilize a variety of suppressive mechanisms including cAMP and adenosine, secretion of the anti-inflammatory cytokine IL-10 is considered one of the primary mechanisms for limiting T cell proliferation (for review 6). The importance of IL-10 in preventing IBD is highlighted by spontaneous enterocolitis occurring in mice lacking IL-10 7 and IL-10 receptor polymorphisms correlating with early onset IBD 8. Paradoxically, numerous studies have demonstrated increased CD4+FoxP3+ T cells in the intestinal lamina propria of IBD patients which fail to attenuate disease, potentially due to impaired Treg suppressive function 9. Interestingly, the impairment in Treg function can be reversed by anti-TNFα treatment, however, it is unclear if this is a direct consequence of TNFα blockade or due to the attenuation of intestinal inflammation 10. Nevertheless, impaired Treg function is believed to contribute to the pathogenesis of IBD.
To assess Treg suppressive function in a murine IBD model, we have employed the TNFΔARE/+ mouse model of Crohn’s disease (CD), a model in which CD4+ effector T cells play a central role 11. These mice express a single copy of a TNFα mutation which lacks the AU-rich element (ΔARE) controlling mRNA stability, leading to over-stabilized TNFα mRNA and systemic elevation of TNFα protein 12. This results in the spontaneous development of chronic transmural ileitis reminiscent of human CD in its histological features and highlights the pivotal role played by TNFα in IBD pathogenesis 11,12. We hypothesize that the ability of TNFα to transiently impair Treg function, as has been described in both Crohn’s disease and ulcerative colitis (UC) 13, may be mediated through induction of selective microRNA (miRNA). Rather than on/off regulation of genes, miRNA mediate subtle up and downregulation allowing for fine control of key biologic functions including IL-10 release 14. Despite limited clinical success 15, miRNA remain attractive therapeutic targets to modulate immune function in a more specific manner than current broad spectrum immunosuppressive therapies.
Here we examine the role of a select miRNA in regulating Treg suppressive function in the TNFΔARE/+ model. We hypothesize that disease induction and maintenance is promoted by impaired Treg function, given their role in preventing overactive inflammation. We first assessed Treg frequency at the peak of intestinal inflammation (between 8–12 weeks) along with their capacity to produce IL-10. We identified a candidate miRNA, miR-106a, that is elevated in IBD in response to TNFα, which targeted the IL-10 3′ untranslated region. MiR-106a has recently been found to be elevated in serum of IBD patients correlating with disease severity, suggesting it may also serve as a useful biomarker for both CD and UC16 We confirmed that miR-106a was elevated in our murine model and that selective inhibition of miR106a enhanced Treg function in vitro. Finally, we assessed the impact of miR-106a deficiency on inflammation in multiple IBD models using a miR-106a-363 knockout mouse strain.
Results
Expansion of impaired regulatory T cells during active disease.
Consistent with previously published data from 20 week-old mice 17, we demonstrated a significant increase in Treg frequency in the inflamed ileal lamina propria of 8–12 week-old TNFΔARE/+ mice relative to WT controls (8.1±0.5 to 15.5±1.9; P<0.01; Figure 1A), with similar findings in both the spleen and MLN. The increased frequency was offset by a reduction in CD4+FoxP3+IL-10+ positive cells (44.4±3.1% to 32.6±3.9%; P<0.01; Figure 1B) in the inflamed LP which was not recapitulated in other tissues investigated. The increased Treg frequency seen by flow cytometry was corroborated by a 6.7±1.8 fold increase in FoxP3 mRNA (relative to CD4 expression) within the TNFΔARE/+ ileum compared to WT (Figure 1C). This increased FoxP3 expression was even greater when expressed relative to 18S given the influx of CD4+ T cells into the inflamed intestine. Thus, chronic ileal inflammation results in an increased frequency of CD4+FoxP3+ Tregs with decreased IL-10 production. This increased frequency may reflect an attempt to offset the reduction in IL-10 production. Interestingly, expression analysis of isolated Tregs of WT and TNFΔARE/+ mice indicated no difference in IL-10 mRNA (Figure 1D) suggesting that the reduced IL-10 production was regulated at a post-transcriptional level.
Figure 1. Regulatory T cell expression during ileitis.
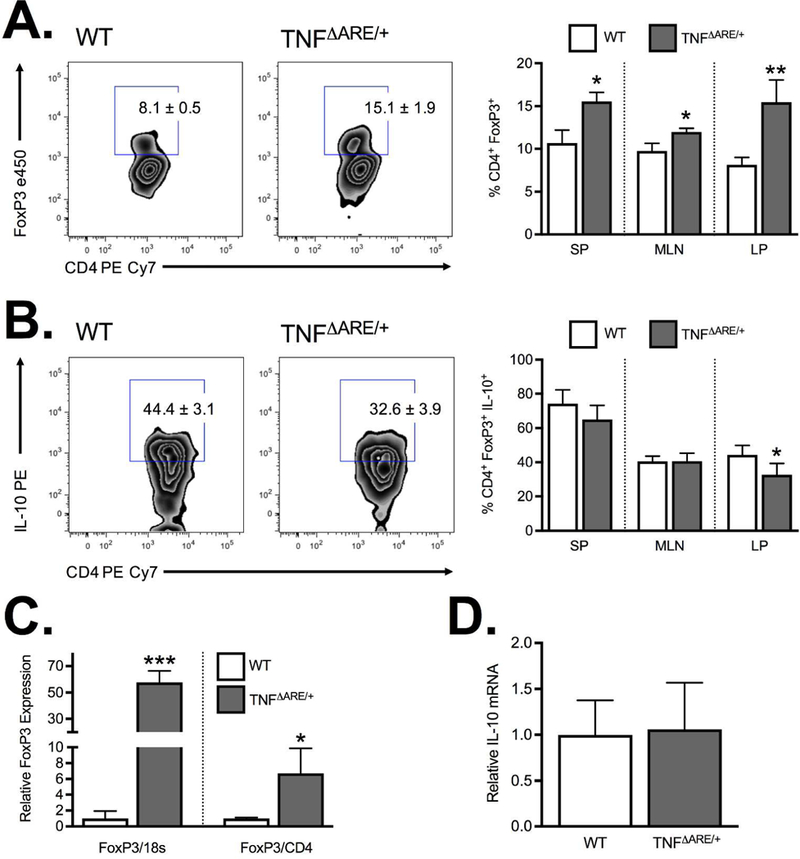
(A) Flow cytometric analysis of the frequency of CD4+FoxP3+ from the spleen (SP), mesenteric lymph node (MLN) and ileal lamina propria (LP) demonstrate a significant increase in Treg frequency at peak disease (8–12 weeks) in TNFΔARE/+ mice relative to WT. (B) The increased frequency was offset by a reduction in IL-10 producing CD4+FoxP3+ Tregs in ilea of 8wk TNFΔARE/+ mice relative to WT littermates. (C) Treg accumulation was confirmed by Taqman real-time PCR measurement of the relative FoxP3 mRNA expression in ileal whole tissue with results expressed relative to 18S or CD4. (D) PCR analysis of isolated CD4+CD25+ Tregs from WT and TNFΔARE/+ mice indicated no significant difference in IL-10 mRNA. Results represent mean ± SEM for three mice per group from three independent studies. *P<0.05, **P<0.01, ***P<0.01.
Decreased suppressive function of Tregs from inflamed mice.
To assess the impact of reduced IL-10 output from Tregs on their suppressive function, we performed an in vitro Treg suppression assay. Magnetically isolated CD4+CD25+ Tregs from TNFΔARE/+ mice were less suppressive compared to Tregs from WT littermates (Figure 2; P<0.001) with significant increases in the frequency of proliferating effectors seen at 1:1 (56.9±1.1% to 26.9±0.9%; P<0.001), 1:2 (75.3±1.2% to 41.7±3.0%; P<0.001) and 1:4 ratios (82.0±1.1% to 58.6±4.1%; P<0.001). Supplementation of IL-10 provided only partial recovery of TNFΔARE/+ Treg suppressive function (Supplemental Figure 1), indicating that additional suppressive mechanisms may play a role. Taken together, the reduction in IL-10 production and increased effector T cell proliferation support the hypothesis that Tregs from TNFΔARE/+ are functionally impaired, which may represent a key mechanism perpetuating chronic inflammatory disease.
Figure 2. Regulatory T cells from inflamed mice exhibit impaired suppressive function.
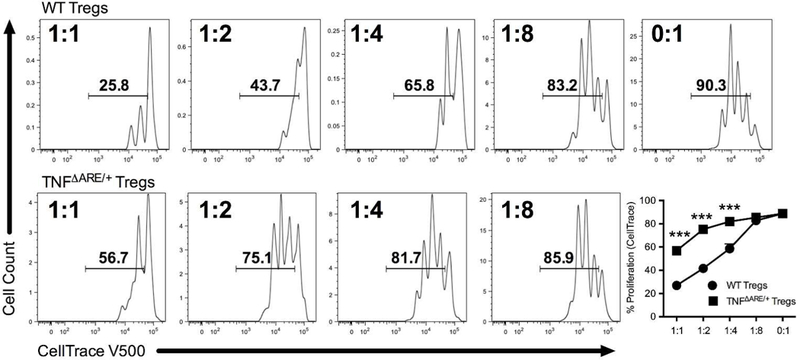
Increasing ratios of isolated CD4+CD25+ Tregs from both WT and TNFΔARE/+ mice were co-cultured with irradiated CD90Neg antigen-presenting cells and fluorescent CellTrace labeled effector cells stimulated with soluble anti-CD3 (1μg/ml) for 72 h. Proliferation of effector cells was quantified by flow cytometric analysis of the percentage of proliferating labeled lymphocytes as a surrogate marker of suppressive function. Tregs from TNFΔARE/+ displayed significantly weaker suppression of proliferation compared to WT littermates. Results represent mean ± SEM for four technical replicates from three independent studies. ***P<0.001.
TNFα suppresses IL-10 production through a post-transcriptional mechanism.
To examine the impact of recombinant TNFα on Treg IL-10 production, we employed the IL-10 IRESGFP reporter strain to allow real-time monitoring of IL-10 transcription with stoichiometric GFP production 18. This demonstrated no significant difference when recombinant TNFα was added to culture media (Figure 3A & 3B). This was further validated by harvesting RNA from isolated CD4+CD25+ Treg following 24 h incubation with TNFα and assessment of IL-10 by Taqman real-time PCR which also showed no significant difference (not shown). In contrast, ELISA of supernatants from these cultures were consistent with previous intracellular cytokine staining, showing that adding TNFα to WT Treg cultures resulted in reduced IL-10 secretion (301.8±35.7 pg/ml vs 857.4±74.3 pg/ml; P<0.00; Figure 3C). Similarly, Tregs isolated from TNFΔARE/+ mice produced significantly less IL-10 relative to WT controls (292.6±7.4 pg/ml vs 857.4±74.3 pg/ml; P<0.001; Figure 3C) yielding a similar output to that of WT Tregs challenged with TNFα in vitro. Interestingly, TNFα antibody blockade partially restored IL-10 output in TNFΔARE/+ Tregs, (518.1±26.3 pg/ml vs 292.6±7.4 pg/ml; P<0.001; Figure 3C). Taken together, these data support the ability of TNFα to modulate IL-10 protein secretion. We therefore assessed the impact of proteasomal inhibition on the secretion of IL-10 by Tregs from WT and TNFΔARE/+ mice. Treatment with the proteasomal inhibitor (MG132, 1μM) significantly increased detectable levels of IL-10 in supernatants from WT (435.0±20.1 pg/ml vs 374.5±16.5 pg/ml; P<0.05; Figure 3D) but not TNFΔARE/+ Tregs. Thus, the TNFα mediated decrease in IL-10 secretion was not altered by inhibition of the proteasome suggesting an alternative mechanism for the regulation of IL-10 production. To identify a mechanism of IL-10 posttranslational modification that may account for the TNFα -dependent changes, we turned our attention to miRNA, short sequences of ribonucleotides that act as post-transcriptional regulators by binding to complementary sequences on target mRNA, resulting in translational repression or target degradation and gene silencing 19. A candidate miRNA had to be upregulated by TNFα 20, known to downregulate IL10 14, be downregulated upon conversion to Tregs 21 and also be upregulated in human IBD 22. Based on these criteria, one particular miRNA (miR-106a) stood out as a suitable target for modifying Treg function in the TNFΔARE/+ model. Our studies validated the upregulation of miR-106a expression in isolated FoxP3-GFP+ Tregs from TNFΔARE/+ mice relative to WT littermates, which provides a rationale for targeting miR106a suppression to enhance IL-10 production (Figure 3E). Thus, TNFα appears to drive posttranscriptional suppression of IL-10 secretion independently of proteasomal degradation, implicating miR106a in this process. This may also point to an additional therapeutic mechanism of action of anti-TNFα antibodies.
Figure 3. Regulatory T cell IL-10 expression during inflammation.
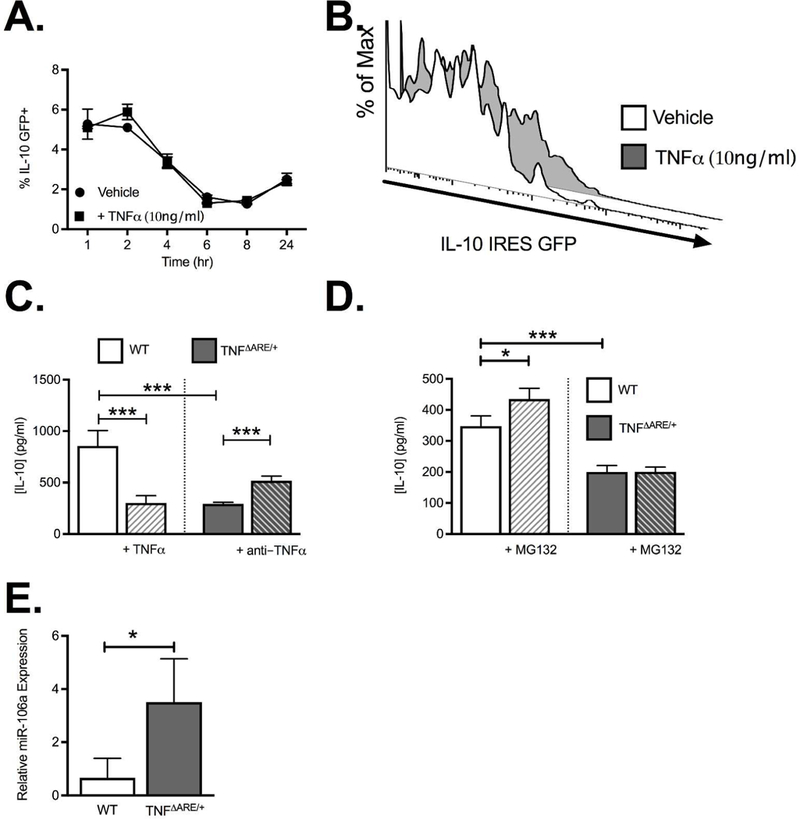
(A) Flow cytometric analysis of IL-10 mRNA in Tregs from IL-10 IRES-GFP reporter mice treated +/− TNFα (10ng/ml) demonstrates no significant difference in IL-10 mRNA transcription. (B) Representative histogram showing the GFP-positive IL-10 reporter cells. (C) ELISA quantification of IL-10 production following 24 h treatment with anti-CD3/CD28 stimulation of isolated CD4+CD25+ Tregs from WT and TNFΔARE/+ mice treated in vitro with either TNFα (10ng/ml) or anti-TNFα antibody (10μg/ml; CNTO5048). (D) ELISA quantification of IL-10 production following 24 h treatment with MG132 (1μM) demonstrated that the decreased IL-10 production seen in CD4+CD25+ Tregs from TNFΔARE/+ mice was not restored by inhibition of proteasomal degradation. (E) Real-time PCR validation of miR-106a expression by isolated CD4+CD25+ Tregs from WT and TNFΔARE/+ mice. Results represent mean ± SEM for three mice per group from three independent studies. *P<0.05, ***P<0.001.
MiR-106a promoter activity depends on NFκB binding.
To confirm TNFα was capable of regulating miR-106a expression in human T cells, Jurkat cells were treated with recombinant TNFα. We observed a concentration-dependent increase in miR-106a expression with a significant increase observed with 10ng/ml TNFα (3.3±0.2 fold induction; P<0.01; Figure 4A), consistent with murine T cell data. Interestingly, TGFβ significantly suppressed miR-106a expression in a concentration-dependent manner with the greatest suppression seen at 10ng/ml (0.18±0.01 fold expression relative to control; P<0.001; Figure 4A). This suggests that miR-106a is critical for pro-inflammatory signaling to transiently impair immunosuppressive signaling and may also have an important physiological role in Treg induction by TGFβ. To assess if TNFα and TGFβ are directly targeting miR-106a transcription we used a dual luciferase reporter assay with the human miR-106a promoter in Jurkat cells. Previous studies have identified a putative promoter region on human chromosome X for miR-106a 23 and so we aimed to confirm the ability of TNFα and TGFβ to target this miR-106a promoter. TNFα caused a concentration-dependent induction of miR-106a promoter luciferase activity relative to Renilla luciferase (4.4±0.01 RLU with 10ng/ml TNFα treatment; P<0.001; Figure 4B). In contrast, TGFβ suppressed miR-106a promoter luciferase activity in a concentration-dependent manner which was greatest at 10ng/ml of TGFβ (0.7±0.01 fold induction relative to vehicle control; P<0.01; Figure 4B). To identify potential sites on the miR-106a promoter that are responsive to TNFα signaling, we generated a number of promoter truncation mutants in arbitrary iterations of 250bp. Both the full-sized construct and the 750bp truncation mutant retained a significant TNFα responsiveness (P<0.001; Figure 4C) which was lost at 500bp, suggesting that the putative transcriptionally activity region of the promoter is between 500–1000bp. Interestingly sequence analysis of this region identified a putative NFκB binding site at position 975–984 and an AP-1 site at position 554–562 (Figure 4D). To determine the relevance of NFκB transcription factors or AP-1 transcription factors binding to the miR-106a promoter as a result of TNFα signaling, we performed luciferase assays with the full-length miR106a construct in the presence of a NFκB inhibitor, Bay 11–7082 (10μM; Sigma-Aldrich) and an AP-1 false substrate, c-JUN peptide (100μM; Tocris Bioscience) using 10ng/ml of TNFα unless otherwise stated (Figure 4E). While inhibiting AP-1 activity failed to impact TNFα-induced luciferase activity, NFκB inhibition abolished all TNFα-induced luciferase activity (0.03±0.006; P<0.0001; Figure 4E) supporting a role for NFκB binding to the miR-106a promoter as a result of TNFα signaling. This effect appears to be selective for miR-106a as expression of other miR-106 family members (Figure 4F) including miR-17, located on a paralogous cluster miR-17–92 cluster, miR-106b found on the miR106b cluster and miR-18, expressed within the same miR-106a-363 polycistronic unit, were all downregulated with TNFα treatment. However, miR-18 failed to reach statistical significance, which is consistent with some shared regulation. This phenomenon was recapitulated in isolated healthy human peripheral blood CD4+ T cells in which paired analysis demonstrated a significant increase in miR-106a in response to TNFα treatment (1.8±0.2 fold increase; P<0.05; Figure 4G). Thus, under physiological conditions, TGFβ appears to suppress miR-106a to facilitate Treg induction. In contrast, under inflammatory conditions, TNFα appears to drive upregulation of miR-106a through NFκB-dependent induction of the miR-106a promoter leading to temporary suppression of normal immune regulation.
Figure 4. MiR-106a expression and promoter activity are induced by TNFα.
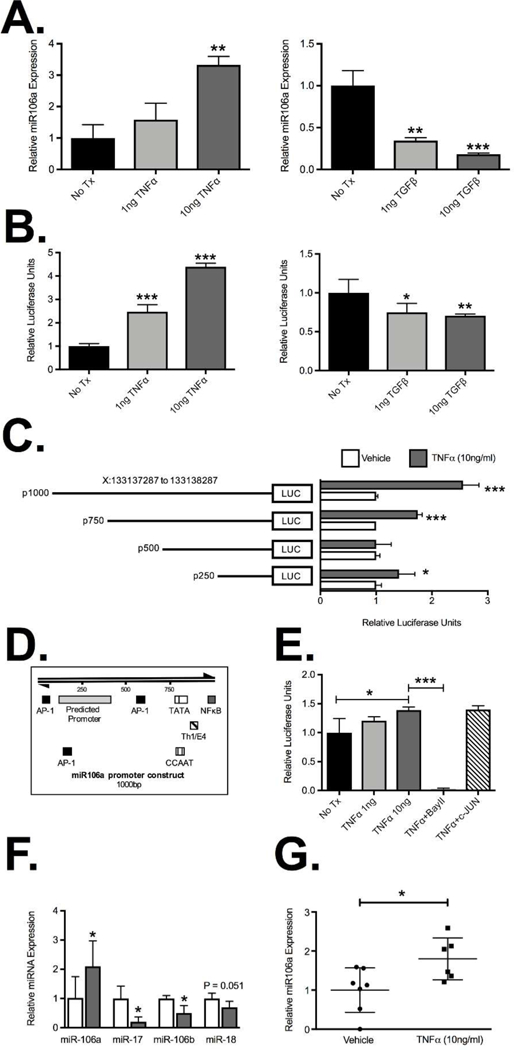
(A) Real-time PCR quantification of miR-106a expression from Jurkat T cells demonstrated induction of miR-106a (3.3±.02 fold) when treated with 10ng/ml TNFα and contrasting concentration-dependent suppression of miR-106a expression (0.18±0.01 fold) relative to control when treated with 10ng/ml of TGFβ. (B) The impact of TNFα and TGFβ treatment on miR-106a transcription was mirrored in the putative miR-106a promoter reporter luciferase activity compared to Renilla luciferase. (C) Studies using promoter truncation mutants suggested that the putative TNFα responsive target site may fall between 500bp and 1000bp on the miR-106a promoter, though results for the 250bp mutant suggested that in the absence of that region an alternative site may also exist. (D) Sequence analysis identified putative AP-1 and NFκB binding sites within the miR-106a promoter region of interest. (E) Luciferase activity from the full-length miR-106a promoter reporter was abolished by incubation with the NFκB inhibitor Bay 11–7082 (10μM) but was unaffected by incubation with c-JUN peptide (100μM). (F) Expression analysis of miR106a-associated miRNA from Jurkat T cells treated with 10ng/ml TNFα. (G) Expression of miR-106a by isolated healthy human CD4+ T cells treated for 6hr with vehicle or 10ng/ml TNFα. Results represent mean ± SEM for four technical replicates from three independent studies. Human studies were performed on seven patients per group. *P<0.05, **P<0.01, ***P<0.001.
MiR-106a deficiency enhances Treg induction and function.
Germline deletion of the miR-106a-363 cluster significantly reduced the induction of IFN+ (Figure 5A) cells and IL-17+ T cell (Figure 5B) from CD4+CD25Neg splenocytes under Th0 conditions relative to WT cells. In contrast, the frequency of CD4+FoxP3+ T cells induced under both Th0 and Tr1 conditions was significantly increased in cells from miR-106a−/− mice relative to WT controls (Figure 5C). Increased Treg induction coincided with an increased expression of IL-10 (Figure 5D). However, fold change analysis demonstrated that under Th0 conditions, the increased frequency of IL-10 producers present in miR106a−/− cells, while preferentially occurring in Tregs, was also present in non-Tregs (Figure 5E). Given the impaired Treg function seen in TNFΔARE/+ mice, we next assessed in vitro suppressive function of Tregs from miR-106a−/−TNFΔARE/+ mice relative to TNFΔARE/+ (Figure 5F). miR-106a−/−TNFΔARE/+ Tregs displayed significantly greater suppressive function. However, this enhanced suppressive function was unaffected by IL-10R blockade, suggesting that even in the absence of IL-10 signaling miR-106a deficiency improves suppression (Figure 5G). Because the miR-106a-363 knockout affects multiple miRNA, we validated the role of miR-106a in Treg suppressive function using a lentiviral approach. Lentiviral suppression of miR-106a significantly increased IL-10 secretion into cell culture supernatants measured by ELISA (192.7±21.0 pg/ml vs 130.8±9.8 pg/ml; P<0.05; Supplemental Figure 2A,B) and corresponded to an increase in IL-10 secretion from miRZIP106a-transfected CD4+ cells (10.5±0.4% vs 6.5±0.1%; P<0.001; Supplemental Figure 2C) measured by intracellular cytokine staining. While converted Tregs were less suppressive than their freshly isolated counterparts (not shown), miRZIP106a transduced Tregs demonstrated significantly improved Treg suppressive function relative to Tregs transduced with the control vector (Supplemental Figure 2E) at 1:1 Treg:Teffector ratio (24.8±2.4% vs 36.1±3.8%; P<0.05) and 1:4 Treg ratio (52.9±0.6% vs 55.3±0.7%; P<0.05). Taken together, miR-106a induction appears to be at least one mechanism by which TNFα reduces IL-10 production and impairs Treg suppressive function.
Figure 5. MiR-106a deficiency promotes anti-inflammatory T cell induction.
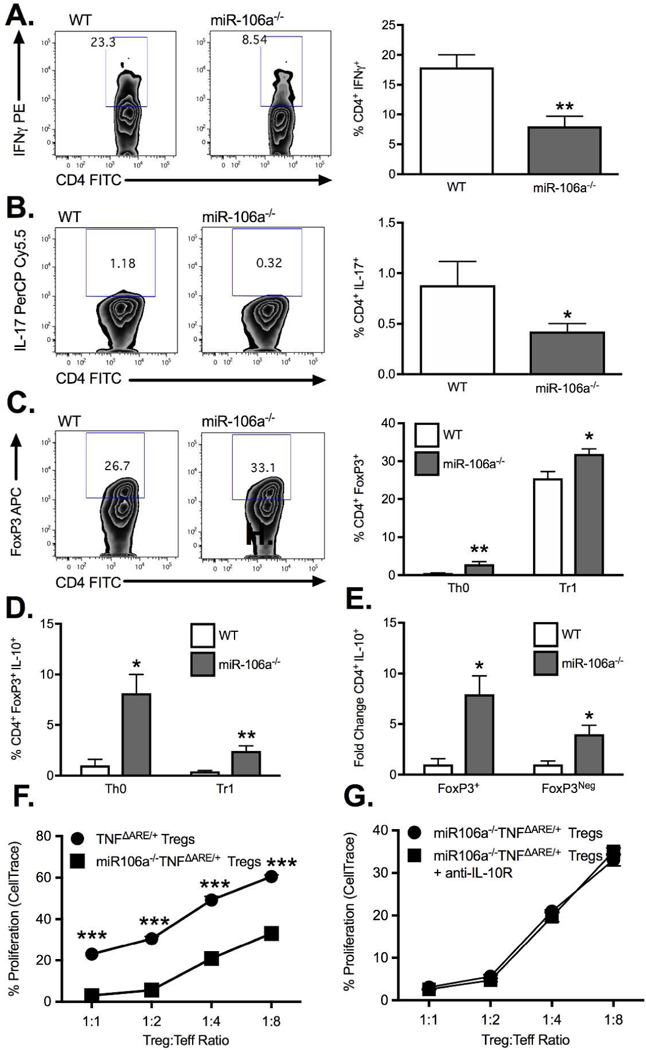
Under Th0 conditions, isolated CD4+CD25Neg cells from miR-106a−/− mice displayed a significant decrease in expression of (A) IFN and (B) IL-17 as well as an increase in (C) FoxP3 relative to WT controls. In the presence of TGFβ, miR-106a−/− CD4+CD25Neg cells underwent significantly greater Treg conversion compared to WT (C) with greater IL-10 production under both conditions (D). (E) The fold change increase in IL-10 production by miR-106a−/− cells under Th0 conditions was greater in FoxP3+ but also occurred in FoxP3Neg cells. (F) Isolated Tregs from miR-106a−/−TNFΔARE/+ mice displayed significantly greater in vitro suppressive function relative to TNFΔARE/+ alone. (G) Enhanced suppressive function was maintained with antibody blockade of IL-10R signaling. Results represent mean ± SEM for four technical replicates from three independent studies. *P<0.05, **P<0.01, ***P<0.001.
MiR-106a deficiency suppresses T cell colitogenic potential.
Employing the same miR-106a−/− strain, we demonstrated attenuated adoptive transfer colitis when naïve CD4+CD45RBHigh T cells from miR-106a−/− mice were transferred to a lymphopenic RAG1−/− host relative to WT controls (Figure 6A) as evidenced by a decrease in weight loss (Figure 6B) and a reduction in the frequency of IFN+ (Figure 6C) and IL-17+ CD4 T cell (Figure 6D) infiltration into the colonic lamina propria. Interestingly, there was a small reduction in IL-10 producing T cells within the lamina propria also though this may reflect a decreased inflammatory state (Figure 6E). Taken together, these data suggest that miR-106a−/− CD4+ T cells have reduced colitogenic potential compared with WT.
Figure 6. MiR-106a deficiency attenuates colitogenic T cell potential in vivo.
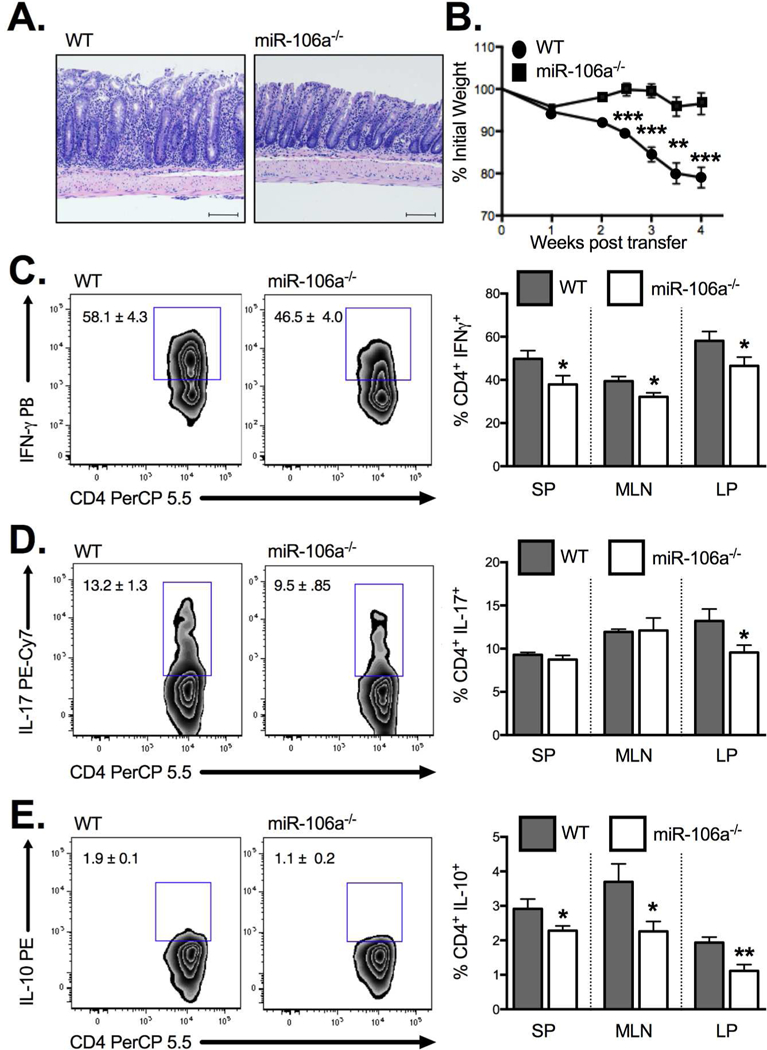
(A) Histology of the colonic lamina propria demonstrates significant villus distortion in WT CD45RBHigh T cell recipients relative to miR-106a−/− T cell recipients following adoptive transfer (B) Adoptive transfer of CD4+CD45RBHigh T cells from miR-106a−/− mice resulted in significantly attenuated weight loss, a surrogate marker of intestinal inflammation, relative to WT controls. This result coincided with a significant reduction in IFN+ CD4+ T cells (C) and IL-17+ CD4+ T cells (D) in the colonic lamina propria as well as a reduction in IL-10-producing CD4+ T cells (E). Results represent mean ± SEM for 4–7 mice per group from two or more independent studies. *P<0.05, **P<0.01, ***P<0.001.
miR-106a deficiency attenuated TNFɑ-driven intestinal inflammation in vivo.
To validate our colitis findings in a second preclinical IBD model, we crossed miR-106a−/− mice to the TNFΔARE/+ chronic murine ileitis model. We demonstrate a significant attenuation of histological indices of ileitis (Figure 7A & B) including active inflammatory index (indicative of granulocytic infiltration), chronic inflammatory index (indicative of lymphocytic infiltration), villus distortion (inflammation-associated villus blunting) and total inflammatory indices (P<0.05; n=9–14 mice/genotype) in 8–12 week-old miR-106a-/TNFΔARE/+ mice relative to TNFΔARE/+ controls. Consistent with colitis experiments, the decreased ileitis in miR-106a−/−TNFΔARE/+ mice coincided with a reduction in the frequency of IFN+ (Figure 7C) cells and IL-17+ T cell (Figure 7D) infiltration into the ileal lamina propria. Once again, we observed a reduction in IL10-producing T cells within the lamina propria likely reflecting a decreased inflammatory state (Figure 7E). This effect is unlikely to be the result of impaired gut homing of miR-106a−/− CD4+ T cells to the inflamed intestine as competitive homing assays indicated no significant difference in homing between WT and miR-106a−/− cells (Supplemental Figure 3). Taken together, these data suggest that miR-106a deficiency decreases susceptibility to spontaneous ileitis.
Figure 7. MiR-106a−/− mice display attenuated ileitis in vivo.
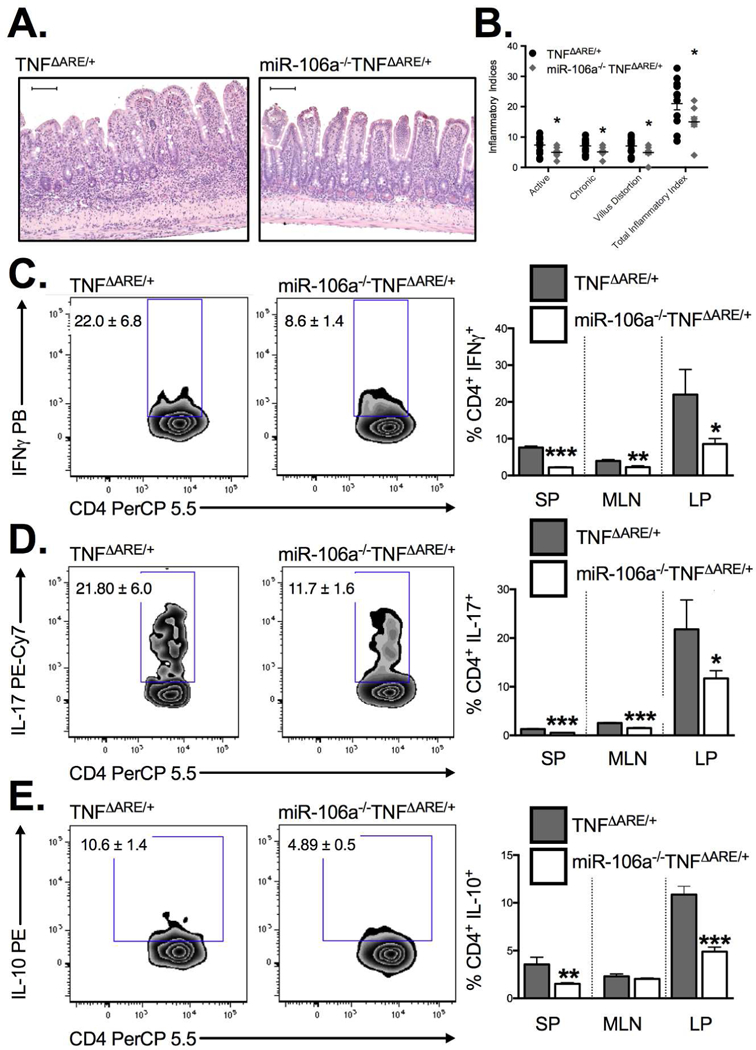
Blinded histological evaluation of intestinal inflammation in the TNFΔARE/+ ileum demonstrated a significant decrease in acute and chronic inflammatory indices in addition to a reduction in villus distortion in miR106a−/−TNFΔARE/+ relative to TNFΔARE/+ (A&B). Flow cytometry identified a concomitant reduction in (C) IFN, (D) IL-17 and (E) IL-10 producing CD4+ T the ileal lamina propria of 8–12 week-old mice. Results represent mean ± SEM for 4–7 mice per group from two or more independent studies. *P<0.05, **P<0.01, ***P<0.001.
Discussion
We have previously reported an increase in Treg frequency in the ileal lamina propria of chronically inflamed 20-week-old TNFΔARE/+ mice in the face of exacerbated inflammation 17. We now extend this finding to demonstrate that a similar induction is observed during the early stage of peak disease onset in this model at eight weeks-of-age, which is consistent with data from patients with Crohn’s disease who display a similar increase in Tregs within the inflamed ileum 9. This was counterintuitive, however, given an increase in an anti-inflammatory population would be expected to attenuate inflammation rather than facilitate it. We postulate the reason for this apparent paradox comes from understanding the impact of inflammation on Treg suppressive function. Under inflammatory conditions, pro-inflammatory cytokines acting via multiple mechanisms, including through TNFα receptors, can induce a transient resistance to suppression in effector T cells 24 that allows rapid expansion of the effector T cell population to deal with the immediate insult before undergoing activation-induced apoptosis. Thus, it is not surprising that TNFα would be able to further amplify this effect by blunting Treg suppressive function. Consistent with that hypothesis, data from human studies of Treg function in IBD patients using anti-TNFα therapy demonstrate a concomitant improvement in Treg function 10,25,26.
In our experiments, the increase in Treg frequency coincided with a reduction in IL-10 output in vivo and reduced in vitro suppressive function of Tregs from TNFΔARE/+ mice. Similar outcomes were seen if the proliferating cells were from inflamed TNFΔARE/+ mice or wildtype (not shown) suggesting a predominant defect in the TNFΔARE/+ Tregs. The reduction in IL-10 output could be directly linked to TNFα, as treatment with an anti-TNFα antibody reversed the effect. Interestingly, the decrease in IL-10 output was not mediated by downregulation of IL-10 mRNA as demonstrated using IL-10 IRES-GFP reporter mice alongside real-time PCR. IL-10 production in vivo is highly regulated with excess IL-10 increasing susceptibility to infection 27 and inadequate IL-10 signaling exacerbating IBD 8. As a result, a large number of regulatory mechanisms including AU-rich elements which control mRNA stability, stressresponse factors 28, miRNA, suppressor of cytokine signaling 3 (SOCS3), Nucleotide-binding oligomerization domain-containing protein two expression (NOD2) dysfunction of which is positively associated with IBD susceptibility 29 and IL-10 itself control IL-10 proteins levels (reviewed 30).
In the absence of transcriptional regulation of IL-10 output, we chose to assess the potential for miRNA to provide the necessary modification of Treg function based on the established alterations in miRNA associated with IBD. Our search criteria for candidate miRNA were confined to miRNA induced by TNFα, that target and suppress IL-10 translation and are induced both in human IBD and in our murine model. Based on these restrictions, one particularly suitable candidate was identified, miR-106a. MiR-106a is upregulated in both TNFΔARE/+ Tregs as well as IBD tissues 22 and known to negatively affect IL-10 production 14. Excitingly, miR-106a also represents one of the few miRNA that are actually upregulated upon T cell activation, while the majority of miRNA are downregulated 31.
Previous promoter studies have focused on a region immediately upstream of miR-106a 14, however because of how single stranded miRNA are processed, promoters of miRNA are significantly further away (as much as 90kb) from the miRNA hairpin region 23. We validated the predicted miR-106a promoter region 23 and demonstrated it was not only induced by TNFα in an NFκB-dependent manner as previously reported but that it was also inversely affected by TGFβ, consistent with a physiological role for miR-106a in Treg development. In addition, we demonstrated in vitro that miR-106a deficiency suppressed IFN+ Th1 and IL-17+ Th17 induction, while promoting FoxP3+ Treg induction, further supporting a role for TGFβ-mediated miR-106a suppression in Treg development. This increased Treg conversion coincided with increasing IL-10 production in both Tregs and non-Tregs. However, whether or not increased IL-10 output is responsible for improved Treg induction remains unclear. MiR-106a deficiency also rescued Treg suppressive function in isolated Tregs from miR-106a−/−TNFΔARE/+ mice relative to TNFΔARE/+. However, antibody-mediated IL-10 receptor blockade failed to reverse this improvement. Similar to the conversion studies, these results suggest that additional mechanisms targeted by miR-106a remain unidentified. In addition, we know that impaired IL-10 production is not the only dysfunction of Tregs impacting disease pathogenesis. Increased apoptosis 32 and IL-17 production by Tregs are also associated with IBD 33. Thus, while data from these studies support using miRNAmodification to enhance Treg function as a valid therapeutic approach for the treatment of IBD, this approach may be more successful if combined with additional miRNA.
Based on our in vitro data, we hypothesized that targeting the miR-106a cluster would attenuate intestinal inflammation in vivo. Initially, we examined the impact of miR-106a deletion on isolated CD4+ T cell colitogenic potential. Using the well-established CD45RBHigh colitis model 34, we demonstrated for the first time that deletion of the miR-106a miRNA family in CD4+ T cells resulted in attenuation of inflammation in lymphopenic recipients. This was characterized by a decrease in Th1 and Th17 cells known to perpetuate inflammation, along with a reduction in IL-10 producing T cells, which are induced in response to inflammation to serve as a negative feedback mechanism 30. While the small but significant decrease in the percent of CD4+ IL-10+ T cells in the colonic lamina propria of mice receiving miR-106a−/− T cells appears contrary to our hypothesis and data showing that miR-106a-deficiency promotes IL-10 production, we also recognize that IL-10 can be produced by Th1 and Th17 cells and this decrease more than likely reflects the marked decrease in the frequency of those cells 30. We further validated these studies with similar findings in a second preclinical mouse model of IBD, the TNFΔARE/+ model of Crohn’s disease 12. As with adoptive transfer colitis, this model is heavily CD4+ T cell dependent 11. Once again, miR-106a deficiency was associated with attenuation of intestinal inflammation. Based on recent evidence for a role for miR-106a-5P in renal cell migration 35, we also examined a potential deficit in lymphocyte gut homing that may have driven the attenuation in intestinal inflammation. However, our current studies showed no significant differences between miR-106a−/− and WT CD4+ T cell trafficking to the intestine.
While selectively targeting individual miRNA and delivering miRNA mimics or antagomirs remains a challenge to the adoption of miRNA targeting approaches 15, evidence from these studies clearly demonstrates for the first time the potential therapeutic use of miR-106a targeting for the treatment of inflammatory bowel disease. Unlike conventional therapies which offer only on/off type regulation, miRNA targeting may allow for more fine-tuning of the immune system.
Methods
Mice
The B6.129S-Tnftm2GKl/Jarn strain (TNFΔARE/+; MGI:3720980) was generated by continuous backcrosses between heterozygous TNFΔARE/+ on a mixed background 12 to C57BL6/J mice 11 and kept under specificpathogen-free conditions. Experimental animals were heterozygous for the ΔARE mutation (TNFΔARE/+) or homozygous wildtype (WT), which served as controls. MiR-106a-363−/− mice on the C57BL6/J background were obtained from Jackson Laboratories (#008461, Bar Harbor, ME). MiR-106a-363−/− mice were crossed with B6.129P-TnfΔARE (TNFΔARE/+) to produce miR-106a-363−/−/TNFΔARE/+ mice. WT/FoxP3-green fluorescent protein (GFP) and TNFΔARE/+/FoxP3-GFP reporter strains were kindly provided by Dr. de Zoeten. IL-10 internal ribosomal entry site (IRES)-GFP reporter mice were provided by Dr. Eric Clambey (#008379; Jackson Laboratories). Adoptive transfer colitis was induced by intraperitoneal injection of 1×106 cells/mouse of magnetically pre-enriched CD4+ T cells (EasySEP, STEMCELL Technologies, Vancouver, Ca) flow sorted into live CD4+ CD45RBHigh naïve T cells (BD FACSAria II, BD Biosciences, Franklin Lakes, NJ). Fecal samples were negative for Helicobacter, protozoa and helminths. All animals were handled according to procedures approved by the institutional committee for animal use.
Lymphocyte isolation
Cells from indicated organs were isolated as previously described 36. Briefly, spleens and mesenteric lymph node (MLN) cells were pressed through a 70μm filter to release leukocytes. Intestinal tissues were rinsed with phosphate-buffered saline and agitated in 1mM EDTA solution to disperse intestinal epithelia, followed by collagenase digestion of the remaining lamina propria cells to facilitate release of intestinal leukocytes.
Flow cytometry and intracellular cytokine staining
For detection of intracellular cytokine producing cells, cells were incubated with phorbol-12-myristate-13acetate (PMA, 50μg/ml; #P8139, Sigma-Aldrich, St. Louis, MO), calcium ionomycin (1mg/ml; #I0634, Sigma-Aldrich) and brefeldin-A (10μg/mL; #B7651, Sigma-Aldrich) for 4 h at 37oC. Cells were stained with antibodies against CD4 (RM4–5), CD25 (PC61.5) and fixable Live/Dead dye (Invitrogen, Carlsbad, CA) prior to permeabilization (BioLegend, San Diego, CA). Following permeabilization, cells were stained for IL-10 (JES5–16E3), IL-17 (TC11–18H10.1), IFN (XMG1.2) and FoxP3 (FJK16S). FoxP3 staining was performed using the FoxP3 FIX/PERM buffer (BioLegend). Cells were washed and fixed with 2% paraformaldehyde and analyzed using the FACS® CantoII system (BD Biosciences). Post-analyzes were performed using FLOWJo software (Tree Star Inc, Ashland, OR).
RNA isolation, cDNA synthesis and real-time PCR
Ileal tissue segments were snap frozen in liquid nitrogen prior to homogenization and RNA isolation using the RNeasy isolation kit (Qiagen, Valencia, CA). Freshly isolated CD4+CD25+ from WT and TNFΔARE/+ mice were purified by magnetic selection prior to RNA isolation. Synthesis of cDNA was performed using Superscript III cDNA synthesis kit (Invitrogen). Expression analysis was performed using Taqman (ThermoFisher, Waltham, MA) assays for FoxP3 (Mm00475162_m1), CD4 (Mm00442754_m1) and IL-10 (Mm00439614_m1) multiplexed with the endogenous 18S control (Eukaryotic 18S rRNA). Jurkat T cells were harvested in RLT buffer and RNA extracted using RNeasy columns (Qiagen) prior to cDNA synthesis using miScript II RT Kit (Qiagen). MiRNA quantification for miR-106a, RNU6–2 and SNORD68 were performed using miScript Primer Assays (Qiagen) with analysis performed on the AB7300 real-time PCR system (Applied Biosystems, Foster City, CA). For multiplex miRNA analysis, isolated Tregs from WT/FoxP3-GFP and TNFΔARE/+/FoxP3-GFP reporter mice were obtained by preenrichment using an EasySep CD4 negative selection kit (STEMCELL) followed by flow sorting for CD4+FoxP3-GFP+ cells.
T cell suppression assay
Treg-mediated suppression was measured as previously described 37. Briefly, CD4+CD25+ Tregs were isolated by negative selection of CD4+ T cells, followed by positive selection of CD25+ cells using the EasySep Treg isolation kit (STEMCELL). Treg frequency was previously confirmed by flow cytometry with approximately 85% of enriched CD4+CD25+ expressing FoxP3 38. CD4+CD25Neg effector T cells were labeled with CellTrace Violet (Invitrogen) according to manufacturer’s instructions prior to stimulation with anti-CD3 mAb (1μg/ml; 145–2C11, BioLegend) in the presence of irradiated syngeneic APC to allow for fluorescent monitoring of cell proliferation. Proliferating cells were co-cultured with varying ratios of converted CD4+CD25+ Treg from WT or TNFΔARE/+ which had been transduced with either miRZIP000 or miRZIP106a to determine the impact of selective inhibition of miR-106a on Treg suppressive function. Anti-IL-10R (10μg/ml; 1B1.3a, BioLegend) or recombinant IL-10 (10ng/ml; Peprotech, Rocky Hill, NJ) were added to culture conditions as indicated.
Cytokine production assays
Cytokine detection was performed on isolated cells by stimulating cells for 24 h with anti-CD3/CD28 antibodies (1μg/ml; 37.51, BioLegend) and snap freezing cell supernatants for subsequent analysis. Antiinflammatory cytokine IL-10 concentration was measured using the Mouse IL-10 ELISA Ready-SET-Go! Kit (eBioscience, San Diego, CA).
T cell conversion assays
Magnetically separated CD4+CD25Neg splenocytes were cultured for 72 h in 96 well round-bottom plates stimulated with plate-bound anti-CD3 (1μg/ml), soluble anti-CD28 (1μg/ml), recombinant murine IL-2 (10ng/ml; Peprotech) and without (Th0 conditions) or with (Tr1 conditions) recombinant human TGFβ (10ng/ml; R&D systems, Minneapolis, MN). Cells were then restimulated for 4 h with PMA/Ionomycin/brefeldin-A prior to fixation and permeabilization using the FoxP3 staining kit (eBioscience) according to manufacturer’s instructions and staining for IFN, IL-17, IL-10 and FoxP3.
Luciferase assays
Early passage Jurkat cells were plated at 0.5×106 cells/well in a 6-well format and transfected using Lipofectamine LTX (ThermoFisher) with 2.5μg of DNA/well of miR-106a promoter luciferase/reporter construct along with 50ng of TK Renilla control reporter vector. 24 h post transfection cells were replated to 2.5×105 cells/well in a 96-well plate and treated with increasing concentrations of recombinant human TNFα or TGFβ for 6 h. Cells were then centrifuged, supernatants removed, and cells lysed in 25μl of passive lysis buffer (Promega, Madison, WI) prior to measuring luciferase activity on a GloMax®-Multi Microplate Multimode Reader (Promega). The full and truncated miR-106a promoter constructs incorporating the human chromosome X:133137287 to 133138287 region were synthesized by GenScript (Piscataway, NJ) based on predicted sequences described previously 23.
Human CD4+ T cell assays
Peripheral blood mononuclear cells were obtained from healthy donors using CPT tubes (BD Biosciences) according to manufacturer’s instructions. Frozen PBMC were rapidly thawed and rested for 2 h in serum-free X-VIVO media (Lonza, Allendale, NJ) at 37oC prior to treatment with recombinant human TNFɑ (10ng/ml, eBioscience) for 6 hr. Unfractionated RNA was isolated using mirVana miRNA isolation kit (Invitrogen), followed by target specific cDNA synthesis using the TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems) and expression analysis performed using TaqMan miRNA probe sets expressed relative to the endogenous control RPL21.
Lentiviral transduction
Lentiviral antagomirs to inhibit miR-106a (miRZIP106a) or scrambled control vector (miRZIP000) were generated according to manufacturer’s instructions (System Biosciences, Palo Alto, CA). Briefly viral particles were generated in HEK293TN cells transfected with miRZIP vectors and pPackH1 packaging mix containing pVSV-G, pPACKH1-GAG and pPACK-H1-REV plasmids. Viral supernatant was concentrated using PEG-it virus precipitation solution. High titer virus was then incubated with isolated CD4+CD25Neg T cells, spinoculated for 2 h at 300g on day 0 and day 1 with polybrene (4mg/ml) and cultured for 72 h in the presence of TGFβ (10ng/ml), IL-2 (5units/ml), soluble anti-CD28 antibody (1μg/ml) and plate-bound anti-CD3 antibody (1μg/ml) to promote Treg induction based on a previously published method 39. Suppressive function of transfected cells was assayed as previously described.
Competitive homing assays
Magnetically separated CD4+ splenocytes were isolated from WT and miR-106a−/− donors and labeled with either CellTrace or Calcein Red-orange (Invitrogen) according to manufacturer’s instructions. Cells were then recounted and mixed in a 1:1 ratio prior to retro-orbital injection of 5×106 cells/mouse. Mice were euthanized after 18 h and expression of labeled cells identified by flow cytometry. Dye-swap experiments were performed to exclude an impact of either dye on gut homing. Results were expressed relative to injection fraction.
Statistics
Statistical analyzes were performed using the Student’s t test or 2-way ANOVA with Prism Data Analysis software (GraphPad Software, La Jolla, CA). Data were expressed as mean ± standard error of the mean (SEM). Statistical significance was set at a P value of less than 0.05.
Study approval
Experimental animal protocols were carried out in accordance with the AAALAC-accredited Institutional Animal Care and Use Committee (IACUC) at the University of Colorado School of Medicine and in compliance with recommendations in the Guide for the Care and Use of Laboratory Animals, Animal Welfare Act and PHS Policy. De-identified healthy human patient samples were collected and used in accordance with Colorado Multiple Institutional Review Board approval COMIRB #00–510.
Supplementary Material
Acknowledgments
Grant Support: NIDDK K01 DK099403 and 253596 CCFA to CBC.
Abbreviations used in this paper:
- IBD
inflammatory bowel disease
- miRNA
microRNA
- TNF
tumor necrosis factor
- Treg
regulatory T cell
- FoxP3
forkhead box P3
- CD
Crohn’s disease
- ΔARE
ΔAU-rich element
- SEM
standard error of the mean
- IRES
internal ribosomal entry site
- SP
spleen
- MLN
mesenteric lymph node
- LP
lamina propria
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists
References
- 1.Kaplan GG The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 12, 720–727 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Reinisch W et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut 60, 780–787 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein GR et al. Continuous therapy with certolizumab pegol maintains remission of patients with Crohn’s disease for up to 18 months. Clin. Gastroenterol. Hepatol. 8, 600–609 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Strober W, Fuss IJ & Blumberg RS The immunology of mucosal models of inflammation 1. Review of immunology (2002).at <http://www.annualreviews.org/doi/abs/10.1146/annurev.immunol.20.100301.064816> [DOI] [PubMed] [Google Scholar]
- 5.Groux H et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389, 737–742 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Vignali DAA, Collison LW & Workman CJ How regulatory T cells work. Nat. Rev. Immunol. 8, 523–532 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kühn R, Löhler J, Rennick D, Rajewsky K & Müller W Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Glocker E-O et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reikvam DH et al. Increase of regulatory T cells in ileal mucosa of untreated pediatric Crohn’s disease patients. Scand. J. Gastroenterol. 46, 550–560 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Boschetti G et al. Therapy with anti-TNFα antibody enhances number and function of Foxp3(+) regulatory T cells in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 160–170 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Collins CB et al. CD44 deficiency attenuates chronic murine ileitis. Gastroenterology 135, 1993–2002 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F & Kollias G Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Wu F et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology 135, 1624–1635.e24 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Sharma A et al. Posttranscriptional regulation of interleukin-10 expression by hsa-miR-106a. Proc. Natl. Acad. Sci. U. S. A. 106, 5761–5766 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luck ME, Muljo SA & Collins CB Prospects for Therapeutic Targeting of MicroRNAs in Human Immunological Diseases. J. Immunol. 194, 5047–5052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omidbakhsh A, Saeedi M, Khoshnia M, Marjani A & Hakimi S Micro-RNAs −106a and −362–3p in Peripheral Blood of Inflammatory Bowel Disease Patients. Open Biochem. J 12, 78–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins CB et al. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and T regulatory cells. Gastroenterology 141, 1821–1831 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamanaka M et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25, 941–952 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Li C, Feng Y, Coukos G & Zhang L Therapeutic microRNA strategies in human cancer. AAPS J. 11, 747–757 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suárez Y, Wang C, Manes TD & Pober JS Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J. Immunol. 184, 21–25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cobb BS et al. A role for Dicer in immune regulation. J. Exp. Med. 203, 2519–2527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu F et al. Identification of microRNAs associated with ileal and colonic Crohn’s disease. Inflamm. Bowel Dis. 16, 1729–1738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujita S & Iba H Putative promoter regions of miRNA genes involved in evolutionarily conserved regulatory systems among vertebrates. Bioinformatics 24, 303–308 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Mercadante ER & Lorenz UM Breaking Free of Control: How Conventional T Cells Overcome Regulatory T Cell Suppression. Front. Immunol. 7, 193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm. Bowel Dis. 16, 1299–1310 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Li Z et al. Restoration of Foxp3+ Regulatory T-cell Subsets and Foxp3- Type 1 Regulatory-like T Cells in Inflammatory Bowel Diseases During Anti-tumor Necrosis Factor Therapy. Inflamm. Bowel Dis. 21, 2418–2428 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Murray PJ, Wang L, Onufryk C, Tepper RI & Young RA T cell-derived IL-10 antagonizes macrophage function in mycobacterial infection. J. Immunol. 158, 315–321 (1997). [PubMed] [Google Scholar]
- 28.Collins CB et al. C/EBPβ deletion promotes expansion of poorly functional intestinal regulatory T cells. J. Crohns. Colitis (2018).doi: 10.1093/ecco-jcc/jjy105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hampe J et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 357, 1925–1928 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Saraiva M & O’Garra A The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10, 170–181 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Bronevetsky Y et al. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J. Exp. Med. 210, 417–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veltkamp C et al. Apoptosis of regulatory T lymphocytes is increased in chronic inflammatory bowel disease and reversed by anti-TNFα treatment. Gut 60, 1345–1353 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Hovhannisyan Z, Treatman J, Littman DR & Mayer L Characterization of interleukin-17producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 140, 957–965 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powrie F et al. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RB hi CD4+ T cells. Immunity 1, 553–562 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Pan Y-J et al. MiR-106a-5p inhibits the cell migration and invasion of renal cell carcinoma through targeting PAK5. Cell Death Dis 8, e3155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins CB et al. Alpha-1-antitrypsin therapy ameliorates acute colitis and chronic murine ileitis. Inflamm. Bowel Dis 19, 1964–1973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collins CB et al. Inhibition of N-terminal ATPase on HSP90 attenuates colitis through enhanced Treg function. Mucosal Immunol 6, 960–971 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones A et al. Cannabinoid receptor-2 ameliorates inflammation in murine model of Crohn’s disease. J. Crohns. Colitis (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geng X, Doitsh G, Yang Z, Galloway NLK & Greene WC Efficient delivery of lentiviral vectors into resting human CD4 T cells. Gene Ther 21, 444–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.