

Abstract
Patients with chronic kidney disease have an increased cardiovascular morbidity and mortality. It has been recognized that the traditional cardiovascular risk factors could only partially explain the increased cardiovascular morbidity and mortality in patients with chronic kidney disease. Asymmetric dimethylarginine (ADMA) and N-monomethy L-arginine (L-NMMA) are endogenous inhibitors of nitric oxide synthases that attenuate nitric oxide production and enhance reactive oxidative specie generation. Increased plasma ADMA and/or L-NMMA are strong and independent risk factor for chronic kidney disease, and various cardiovascular diseases such as hypertension, coronary artery disease, atherosclerosis, diabetes, and heart failure. Both ADMA and L-NMMA are also eliminated from the body through either degradation by dimethylarginine dimethylaminohydrolase-1 (DDAH1) or urine excretion. This short review will exam the literatures of ADMA and L-NMMA degradation and urine excretion, and the role of chronic kidney diseases in ADMA and L-NMMA accumulation and the increased cardiovascular disease risk. Based on all available data, it appears that the increased cardiovascular morbidity in chronic kidney disease may relate to the dramatic increase of systemic ADMA and L-NMMA after kidney failure.
Keywords: Nitric oxide, kidney failure, asymmetric dimethylarginine, dimethylarginine dimethylaminohydrolase-1, heart failure
Introduction:
Chronic kidney disease (CKD) is generally described as the presence of kidney damage and reduced kidney function over 3 months. Cardiovascular diseases (CVD) are the leading cause of death in North America (28,32,37), and patents with CKD show an increase of prevalence of CVD such as hypertension, peripheral vascular disease, and congestive heart failure (CHF) (40,67,71). In addition, the cardiovascular morbidity and mortality are markedly increased in patients with CKD (67,71). It is reported that up 25 to 47% patients are with CVD such as the CHF, ischemia cardiomyopathy, or ventricular hypertrophy in patients with severe CKD (40,67,71). However, the mechanism of the increased cardiovascular risk in CKD is not very clear.
Asymmetric dimethylarginine (ADMA) and N-monomethy L-arginine (L-NMMA) are endogenous nitric oxide synthase inhibitors. ADMA and/or L-NMMA are recognized as a strong and independent risk factor(s) for various cardiovascular diseases such as hypertension, coronary artery disease, atherosclerosis, pulmonary hypertension, atrial fibrillation, stroke, peripheral vascular diseases, diabetes and CHF (2,3,4,8,15, 55,76,94). ADMA and L-NMMA attenuate nitric oxide (NO) production by inhibition of nitric oxide synthase (NOS) activity (10,11,12,41,62) (Figure 1). ADMA and L-NMMA also enhance NOS uncoupling to produce reactive oxidative species (ROS) such as superoxide anion (O2-) and peroxynitrite (ONOO–), which could further reduce the cardiovascular NO bioavailability. ADMA and L-NMMA are eliminated from the body by either DDAH1 degradation (58,61) or renal excretion (1,47). Unlike other cardiovascular disease conditions, CKD often causes a dramatic increase of circulating ADMA and/or L-NMMA (48,63,59,66,92,93). NMMA metabolism, their renal elimination, their effect on NO bioavailability and ROS production, and their important roles in promoting cardiovascular diseases. Based on the dramatic increase of circulating ADMA and/or L-NMMA in CKD patients (48,63,59,66,93), and the role of ADMA and L-NMMA in promoting cardiovascular diseases (2,3,4,8,54,73,74), the dramatic elevation of plasma ADMA and L-NMMA levels might be the major culprits for the increased cardiovascular morbidity and mortality in patients with CKD (Figure 2).
Figure 1.
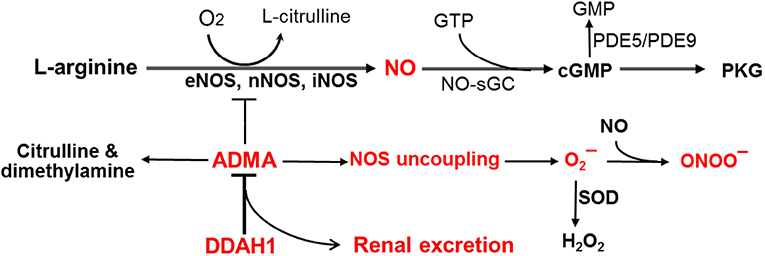
Regulation of ADMA NO production through kidney and dimethylarginine dimethylaminohydrolase-1 (DDAH1).
Figure 2.
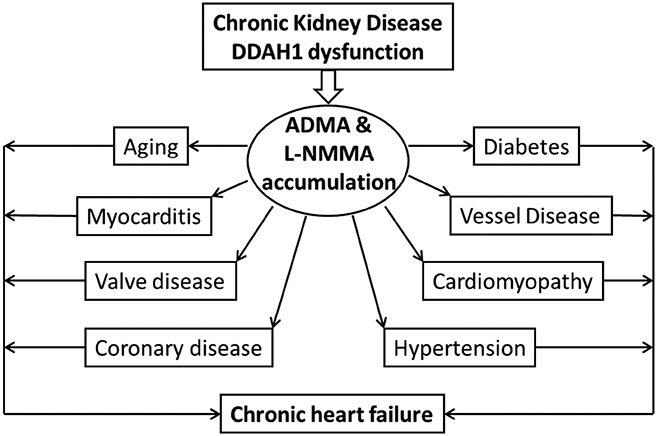
The potential role and mechanism of chronic kidney disease and dimethylarginine dimethylaminohydrolase-1 (DDAH1) dysfunction in the increased cardiovascular risks through ADMA.
Basic role and mechanism of NO in regulating cardiovascular function.
NO plays an important role in regulating various cardiovascular function (14,38,46). NO is produced by NOS by using L-arginine as a substrate. There are 3 well-recognized enzymes for NO production are neuronal nitric oxide synthase (nNOS or NOS1) and inducible nitric oxide synthase (iNOS or NOS2), and endothelial nitric oxide synthase (eNOS or NOS3) (14,38,39) (Figure 1). eNOS is mainly expressed in vascular endothelial cells, while nNOS is mainly expressed in neuronal cells. Since both eNOS and nNOS are constitutively expressed at lower levels in many other cell types (17,38), they are also called as constitutive NOS (cNOS). Under normal unstressed conditions, iNOS is mainly expressed in leukocytes such as macrophages and T cells, but it is also greatly induced in various cell types in response to inflammation or other stress conditions (38,39,96). NO production is regulated by L-arginine availability, NOS protein content, NOS activity, NOS cofactor tetrahydrobiopterin (BH4), and endogenous NOS inhibitors ADMA and L-NMMA etc. (6,14,22,45,49, 90).
NO stimulates soluble guanylate cyclase to produce cGMP, and cGMP will cause activation of cGMP dependent Protein Kinase G (PKG) (30,38). PKG activation results in smooth muscle cell relaxation, reduction of vascular resistance, and consequently increase of blood perfusion in tissues (14,29,39,78). After development of chronic heart failure, coronary artery dilatation in response to agonists or shear stress are attenuated, in part due to the reduced vascular NO cGMP and PKG signaling (7,17,19). Reduced NO-cGMP-PKG signaling can also impair angiogenesis, vascular endothelial cell growth and proliferation, platelet disaggregation, myocardial oxygen consumption, and vessel injury repair etc. (14,19,29,38,39). In addition to the well-established role of NO-cGMP-PKG signaling in maintaining normal vascular endothelial function, NO also protects against cardiac remodeling and dysfunction under stress conditions such as aging (53), myocardial infarction, hypertension, and stroke etc. (46,55). Reduced NO-cGMP-PKG signaling may also promote various cardiovascular diseases such as hypertension, coronary disease, atherosclerosis, diabetes, stroke, pulmonary hypertension, chronic heart failure, and chronic renal failure. The protective effects of eNOS and nNOS are largely attributed to NO-cGMP-PKG signaling, which also targets several proteins involved in cardiac contractility, and cardiac remodeling (7,14, 29,38,39).
Besides promoting cGMP-PKG signaling, NO also regulates cardiovascular function through post-translational modification of proteins via S-nitrosylation. For instance, S-nitrosylation of L-type calcium channels reduces ventricular arrhythmias and mortality in mice (11), and S-nitrosylation of the ryanodine receptor can reduce diastolic calcium leak (31). S-nitrosylation of G-protein coupled receptor signaling regulates cardiac ischemic injury (33). PDE5 and PDE9 degrade cGMP to reduce cGMP-PKG signaling (14,20,50,56,78), S-nitrosylation of PDE5 reduces its stability to enhance cGMP-PKG signaling (87). Since NO production is regulated by ADMA and L-NMMA (14,22,45,49, 90), factors that regulate ADMA and L-NMMA biological levels can directly regulate NO bioavailability and cardiovascular endothelial function.
Because NO signaling is often impaired in cardiovascular diseases, strategies or methods to enhance NO-cGMP-PKG signaling (such as administration of NO donor, pharmacological activation of guanylate cyclase, and blocking cGMP degradation using PDE5 and PDE9 inhibitors) have being explored as the potential treatments for various cardiovascular diseases such as pulmonary hypertension, hypertension, erectile dysfunction, CHF, and stroke etc. (24,30,50,54,56,78).
ADMA and L-NMMA production, transportation and elimination.
Protein methylation plays an important role in many cellular functions and occurs constitutively in various cells under both normal control conditions or after stresses. The production of ADMA and L-NMMA is the result of proteolysis of proteins containing methylated arginines (57,65,72). L-NMMA is formed when protein-incorporated L-arginine is methylated by the enzymes protein arginine methyltransferases type-I (PRMT-I) or type-II (PRMT-II) (57,65). PRMT-I can further methylate L-NMMA, resulting in the formation of ADMA, whereas PRMT-II can methylate L-NMMA into symmetric dimethylarginine (SDMA) (57,65). L-arginine, ADMA, L-NMMA and SDMA can be transported into circulation as well as be taken up by other cells using cationic amino acid transporter (CAT). Both ADMA and L-NMMA directly compete with L-arginine for the active site of eNOS, nNOS and iNOS to attenuate NO production. While SDMA does not directly inhibit NOS activity, it could attenuate NOS function indirectly by inhibition of the CAT (21). ADMA and L-NMMA are eliminated from the body through either DDAH1 degradation (58,61) or renal excretion (1,47). The important roles of DDAH1 and kidney in regulating ADMA and L-NMMA will be further described separately.
ADMA and L-NMMA attenuate NO-cGMP-PKG signaling and cardiovascular endothelial function.
ADMA and L-NMMA regulate cardiovascular function through attenuating NO production and increase NOS-derived superoxide anion production (Figure 1, Figure 2). As ADMA is more abundant than L-NMMA, most of the studies have focused on the physiological or pathological effects of ADMA in various biological or clinical conditions. ADMA and L-NMMA not only attenuate NO production, but also promote NOS-derived superoxide anion production (13,89,90). The superoxide anion can further bind NO to generate peroxynitrite, a far more toxic free radical. Many experimental studies have demonstrated that ADMA and/or L-NMMA attenuate vascular endothelial NO production (36), vascular endothelial cell growth, proliferation and migration (97). ADMA also attenuates vascular endothelial cell tube formation (97), and the vascular endothelial cell outgrowing capacity in isolated vessel rings (95,97). Moreover, chronic ADMA and L-NMMA accumulation in vascular endothelial specific DDAH1 knockout mice resulted in hypertension, decreased vascular NO production (36), and a reduced vascular endothelial injury repair similar to eNOS deficient mice (95). We demonstrated that global DDAH1 knockout attenuated angiogenesis or micro-vessel growth into the gel plugs in mice (95,97), while another group demonstrated that global DDAH1 knockout attenuated the hind limb angiogenesis in mice after permanent femoral artery ligation in mice (26). Chronic ADMA and L-NMMA accumulation in global DDAH1 knockout mice caused moderate hypertension and aging related cardiac hypertrophy without decreasing left ventricular function (91). However, selective deletion of DDAH1 in cardiomyocytes had no detectable effect on hypertension and aging-dependent cardiac hypertrophy, but exacerbated aortic banding induced left ventricular hypertrophy and heart failure (91), suggesting an important role of compartmentalized DDAH1 in protecting heart against hypertrophy and dysfunction. Furthermore, acute infusion of ADMA caused increase of blood pressure and decrease of endothelium-dependent vasodilation in both human subjects (1) and experimental animals (23,36). In addition, chronic ADMA infusion resulted in increase vascular angiotensin-converting enzyme, increase of oxidative stress, as well as vascular and perivascular fibrosis (75). These studies demonstrate that ADMA and/or L-NMMA can cause cardiovascular endothelial dysfunction that may contribute to the development of various cardiovascular diseases such as CHF and hypertension.
Effect of ADMA and L-NMMA on NOS uncoupling:
Studies have also demonstrated that endogenous NOS inhibitors ADMA and L-NMMA can also enhance NOS-derived O2- production and ONOO– production through a process called NOS uncoupling (Figure 1). NOS uncoupling generally occurs when NOS is exposed to oxidant stress (including peroxynitrite), when it is deficient of cofactor BH4 (22,49), or when it is deprived of its substrate L-arginine (90). Since BH4 stabilizes the dimeric forms of eNOS, nNOS and iNOS (6,23), oxidation of BH4 to BH2, or BH4 depletion can exacerbate NOS-derived O2- production (6,22). L-arginine deficiency can also exacerbate eNOS or nNOS to generate O2– and ONOO– (90, 98). Moreover, studies have demonstrated that addition of ADMA or L-NMMA also exacerbated O2- generation in purified NOS protein (13,54,62), in cultured human endothelial cells (13), isolated rat arterioles (82), and in murine lung epithelial cell line LA-4 (89). Reduced NO production or increased ROS generation is also known to increase PDE5 activity (56) through attenuating PDE5 nitrosylation (87), suggesting a further indirect reduction of cGMP signaling by NOS derived ROS. Thus, ADMA and L-NMMA also attenuate NO-cGMP bioavailability through promote ROS production and PDE5 activity.
However, unlike eNOS and nNOS, iNOS expression can be detrimental to cardiovascular system (14,32,55,96), likely due to the unmitigated production of NO and superoxide by iNOS uncoupling in the inflammatory setting of cardiovascular damage (32,96). Thus, genetic deletion or selective inhibition of iNOS often exert protective effects against the development of cardiovascular diseases (39,55,96). A recent study showed that genetic and pharmacological inhibition of DDAH1 protected mice against endotoxic shock (60).
Effect of ADMA and/or L-NMMA on cardiovascular diseases.
ADMA and/or L-NMMA accumulation also occurs in hypertension (74), atherosclerosis (43,73), cardiac valve disease (2), idiopathic cardiomyopathy (3), renal failure (44), diabetes (4,54), aging, atrial fibrillation, and CHF (27, 81) (15,16), a group of diseases cause CHF (Figure 2). Elevated plasma ADMA and/or L-NMMA level is associated with an increased risk for developing angina pectoris, myocardial infarction or cardiac death (9,10). Plasma ADMA and/or L-NMMA level is a strong and independent predictor of all-cause mortality in the community (9). Moreover, ADMA and/or L-NMMA level is strongly associated with CHF development (27, 55,81). The common clinical symptoms of CHF include shortness of breath, profound reduced exercise capacity and swelling (edema) in legs and feet etc. (28,37). Chronic CHF also causes WHO class-2 pulmonary hypertension, and severe lung vascular remodeling and inflammation (16). Interestingly, except in chronic kidney disease, plasma ADMA and/or L-NMMA levels are generally increased less than 20% in patients various cardiovascular diseases such as coronary disease (8,73), idiopathic cardiomyopathy (3), diabetes (4,54), and CHF (27, 55,81).
Experimental studies showed that infusion of exogenous ADMA resulted in reduced vascular endothelium-dependent coronary vasodilation in pacing-induced CHF dogs (20), a decrease of cardiac output in normal human subjects (1), and an increase of blood pressure in normal mice (36,52). In addition, chronic infusion of ADMA resulted in increased vascular angiotensin-converting enzyme, oxidative stress, and vessel lesions, suggesting that ADMA can cause vessel injury at least partially through modulating oxidative stress (75). Interestingly, we recently found that systolic pressure overload significantly exacerbated cardiac fibrosis and dysfunction in cardiomyocyte specific DDAH1 KO mice, a strain showed only moderate increase of myocardial ADMA content without affecting plasma ADMA content (91). Thus, chronic systemic ADMA or local myocardial ADMA accumulation can exacerbate CHF development by enhancing cardiovascular risk factors such as hypertension, diabetes, atherosclerosis, and coronary disease as summarized in Figure 2.
The essential role of DDAH1 in degrading ADMA and L-NMMA.
As ADMA was first isolated from human urine by Kakimoto and Akazawa in 1970 (47), renal excretion was initially recognized as the major route for ADMA elimination in human subjects. However, a study from McDermott subsequently showed that the urinary recoveries of L-NMMA and ADMA following intravenous injection in normal rabbits were 0.14% and 5.1%, respectively, indicating that both L-NMMA and ADMA undergo extensive metabolism in healthy animals (58). It was then reported that less than 17% of ADMA was excreted via urine in normal human subjects (1). Ogawa et al further identified the enzyme DDAH1 that catalyzes hydrolysis of L-NMMA and ADMA into L-citrulline and mono- or dimethylamine (61). A second DDAH isoform (DDAH2) was reported in 1999 (51). The initial study demonstrated that DDAH1 and DDAH2 have comparable activities for degrading ADMA and L-NMMA (51). Since DDAH1 is minimally expressed in the heart (51,83), vessels (51) and vascular endothelial cells (5), for years, it was believed that DDAH2 plays a major role in regulating ADMA and L-NMMA degradation in cardiovascular system. However, recent studies clearly indicate that DDAH1 exerts the essential role in ADMA and L-NMMA degradation, and DDAH2 exerts no detectable role in ADMA and L-NMMA metabolism (35,36,52). Thus, we observed that DDAH activity was abolished in tissues obtained from global DDAH1 knockout (KO) mice while the expression of DDAH2 was unaffected in these tissues (35). In other words, tissues obtained from global DDAH1 KO mice are unable to degrade ADMA or NMMA even though DDAH2 expression is not affected in these tissues (35). Consistent with our findings, another study demonstrated that tissue DDAH activity was reduced ~50% in heterozygous DDAH1 KO mice, in which DDAH1 was reduced 50% (52). Furthermore, we found that selective gene silencing of DDAH1 caused accumulation of ADMA and decreased NO production in cultured vascular endothelial cells, but selective gene silencing of DDAH2 did not affect ADMA and NO production in cultured vascular endothelial cells (35). Moreover, over-expression of DDAH1 decreased ADMA content in cultured vascular endothelial cells, over-expression of DDAH2 had no effect on ADMA and NO production in cultured vascular endothelial cells (95). These findings clearly indicate that DDAH1 is the enzyme for ADMA and L-NMMA degradation, while DDAH2 has no detectable role in ADMA degradation. Moreover, three DDAH2 KO strains were independently generated in different groups in past 10 years, no data has ever shown that DDAH2 KO could affect tissue DDAH activity, or systemic ADMA and/or L-NMMA in these strains. It is reasonable to believe that none of these DDAH2 KO strains affect ADMA or L-NMMA metabolism and DDAH activity. Several studies indicate that a potential role of alanine-glyoxylate aminotransferase 2 (AGXT2) in ADMA metabolism (34; 68, 70), a study using AGXT2 deficient animals could further define the significance of AGXT2 in ADMA degradation or systemic ADMA accumulation.
Global DDAH1 KO causes increases of plasma and tissue ADMA and L-NMMA, which is associated with reduced NO production, moderate hypertension, and endothelial dysfunction (35,36). DDAH1 KO also limits angiogenesis and impairs vascular injury repair (26,95,97). Conversely, over-expression of DDAH1 results in decreases of plasma and tissue ADMA levels, which was associated decrease of systemic blood pressure (25), increase of insulin sensitivity (76), increase of angiogenesis (42,95,97), reduced high fat diet induced atherosclerosis (42,88) and graft coronary artery disease (80). However, chronic ADMA and L-NMMA elevation in DDAH1 KO mice were insufficient to cause cardiac dysfunction in mice under unstressed conditions (35, 91). These findings indicate that chronic accumulation of ADMA and L-NMMA are sufficient to alter vascular tone and endothelial function, but it is insufficient to cause spontaneous cardiac dysfunction at least under control conditions. Thus, elevating DDAH1 activity to eliminate ADMA and L-NMMA could be a promising strategy for restoring NOS function and increasing NO bioavailability in cardiovascular system.
Alteration of systemic ADMA in CKD suggests an important role of kidneys in ADMA metabolism.
While two previous studies demonstrated that only small amount of ADMA and/or L-NMMA are eliminated through urine excretion in normal human subject and experimental animals (1,58), enormous evidences obtained from renal failure patients indicate that kidneys might exert an important role in elimination of ADMA and L-NMMA in clinical conditions. Thus, several studies showed that plasma ADMA and L-NMMA generally increases over 4 fold (even increased up to 10 fold in some reports) in patients with end-stage renal diseases (44,48,59,63,66,93), a group of patients with the highest burden of cardiovascular mortality (40,67,71). Patients with end-stage renal diseases or patients require hemodialysis generally have higher ADMA and/or L-NMMA contents, while kidney transplantation or hemodialysis resulted in a decreased of systemic ADMA and/or L-NMMA, and improved vascular endothelial function (66,92). While it is reasonable to believe that reduced ADMA and/or L-NMMA would contribute the beneficial effect of hemodialysis in this population, the relative impact of ADMA and/or L-NMMA reduction by hemodialysis on the overall clinical outcome could not be precisely defined. Nevertheless, plasma ADMA and L-NMMA levels are recognized as important biomarkers for CKD (44,69), and the increased cardiovascular morbidity and mortality in CKD (Figure 2).
The important role of kidneys in regulating ADMA and L-NMMA metabolism may be two folds. First, kidneys have the highest DDAH1 content and DDAH1 activity (36,83). Based on published microarray data, kidneys have the highest DDAH1 mRNA levels in mice (Figure 3), human, rats and dogs. The data in The Human Protein Atlas showed that kidney is one of the tissue with highest DDAH1 expression (https://www.proteinatlas.org/ENSG00000153904-DDAHI/tissue). Kidney is the tissue with the highest DDAH1 protein expression (Figure 4), and the tissue with the highest DDAH activity (Figure 5). It appears that tissues with higher DDAH1 protein content also have higher DDAH1 mRNA in mice (Figure 3, Figure 4), suggest that DDAH1 protein expression is regulated at the transcriptional level. Second, kidneys can excrete ADMA and L-NMMA through urine. The ADMA excretion by kidneys would be more important when DDAH1 is impaired by various pathological conditions such as oxidative stress. The dramatic increase of plasma ADMA in patients with severe CKD (44,48,59,63,66,92,93) is likely a combined outcome of the diminished renal ADMA excretion capacity and the decreased DDAH1 protein contents or activity in the damaged kidneys. While it is inappropriate to directly compare human and mouse data, the markedly increase of plasma ADMA in severe CKD patients clearly suggest that the role of kidney in controlling systemic ADMA and/or L-NMMA metabolism might have been underestimated by the ADMA field.
Figure 3.
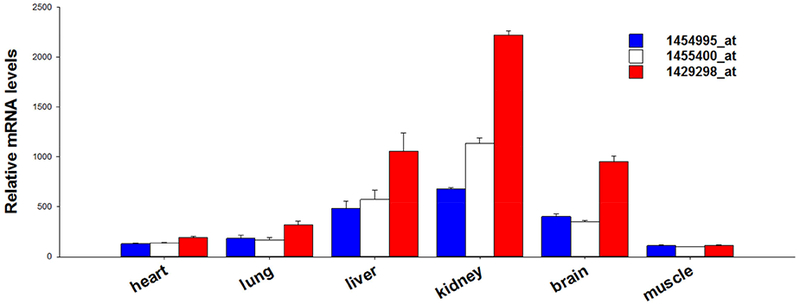
The relative mRNA content of dimethylarginine dimethylaminohydrolase-1 (DDAH1) in C57/B6J mice. The data are retrieved from GEO profile ID: 49893290 of DataSet ID:3142. The data presented here are obtained by using probe ID 1454995_at, 1455400_at, and 1429298_at, respectively. The relative DDAH1 mRNA signal intensities are varied detecting with different probes, but kidney is one of the tissues with highest DDAH1 expression by using all of these 3 probes.
Figure 4.
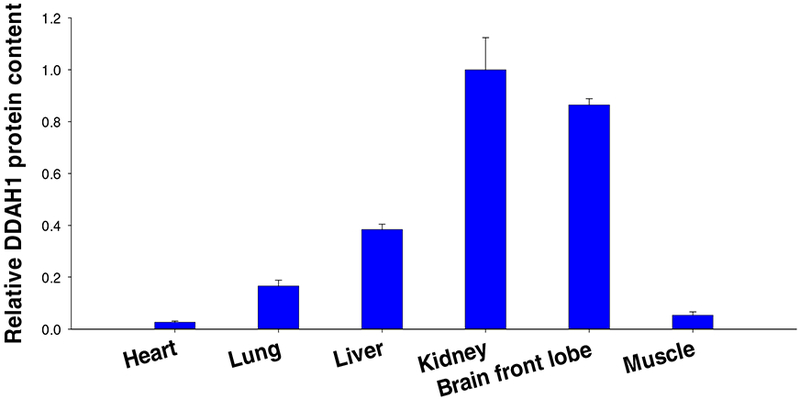
The relative dimethylarginine dimethylaminohydrolase-1 (DDAH1) protein content in normal mouse tissues. Kidney is the tissue with highest DDAH1 protein content.
Figure 5.
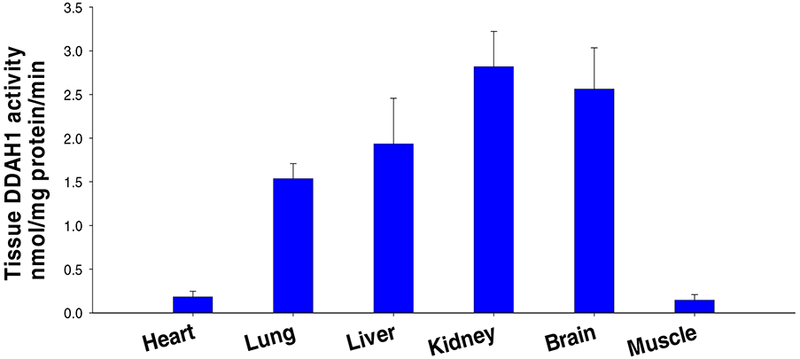
The relative dimethylarginine dimethylaminohydrolase activity in normal mouse tissues, or the tissue capacity in degrading isotope labeled ADMA.
ADMA Alteration in CKD experimental animal models.
While the clinic studies clearly show that severe CKD causes a dramatic increase of plasma ADMA contents, various experimental animal models of CKD have so far failed to recapitulate the apparent elevation of plasma ADMA observed in CKD patients. For examples, the commonly used 1/2 or 5/6 nephrectomized wild type mice or rats only caused moderate increases of plasma ADMA in mice or rats (43,64,77), suggest a better experimental animal model(s) is needed to test the causal role of chronic systemic ADMA accumulation in the exacerbated cardiovascular diseases in CKD. One potential approach may be using the 5/6 nephrectomized global DDAH1 knockout mice to create a CKD model with an increased plasma ADMA similar to the CKD patients.
The mechanism of ADMA and/or L-NMMA accumulation in cardiovascular diseases.
Although the detailed mechanism for increased ADMA and L-NMMA in cardiovascular diseases is not totally clear, in principle, the overall ADMA elevation can be a result for 3 scenario such as (i) an increased ADMA and L-NMMA production in response to stresses or increased degradation or turnover of proteins containing methylated arginine (65,72); (ii) a decreased ADMA and L-NMMA degradation by DDAH1 dysfunction; or (iii) a reduced ADMA and L-NMMA excretion by kidney dysfunction (44,48,59,66,93). Increased protein degradation by autophagy or proteasome activity during tissue remodeling and inflammation can potentially increase ADMA production (65,72). Study has demonstrated that inhibition of proteasome activity in cultured cells reduced free levels of ADMA and SDMA, while inhibition of autophagy reduced ADMA without affecting SDMA (82). Both autophagy and proteasome activity are affected in various cardiovascular diseases such as CHF (86), while protein synthesis is often increased as well. Therefore, protein degradation or protein turnover, and subsequent production of ADMA and L-NMMA may elevate in cardiovascular diseases. However, the contribution and significance of these pathways to ADMA and L-NMMA production in cardiovascular diseases are not very clear now. Nevertheless, as ADMA and L-NMMA are often increased less than 20% in most cardiovascular diseases (3,8,15,33,55,76,94), the increased ADMA and/or L-NMMA production can be largely controlled these diseases when renal function is not impaired.
In addition, there is some evidence that accumulation of ADMA and L-NMMA may result from depressed DDAH1 expression or activity(17,18), through loss-of-function polymorphisms of the DDAH1 gene (84), or increased DDAH1 degradation through post-translational modifications such as oxidation of DDAH1 protein (41,54). A previous study identified a mutation of the DDAH1 gene that is associated with high plasma ADMA levels and an increased risk for coronary heart disease and hypertension (84). DDAH activity can be depressed by oxidized LDL and TNFa (41), and high plasma glucose in diabetic rats (54). Our previous study demonstrated that chronic mechanical unloading by implant left ventricular assistant device caused significant increases of left ventricular DDAH1 mRNA and protein content in patients with severe CHF (18), suggesting that mechanical stress or pressure regulates cardiac DDAH1 protein expression in the failing heart. In the context of the critical role of DDAH1 in ADMA and L-NMMA degradation, reduced DDAH1 activity by posttranslational modification may contribute to the moderate increased systemic ADMA and/or L-NMMA in cardiovascular diseases such as CHF. Moreover, the reduced renal blood perfusion after heart failure or hypertension may also contribute to the moderate ADMA and L-NMMA elevation through reducing their renal excretion.
Potential therapeutic targets or therapies to reduce ADMA.
Since ADMA and L-NMMA are recognized risk factors for cardiovascular disease through attenuating vascular NO/cGMP signaling, effects have been undertaken to reduce ADMA and L-NMMA. As protein methylation plays an important role in many cellular functions and occurs constitutively in various cells under both normal or pathological conditions, reduction of ADMA and L-NMMA production is generally regarded as an unattractive approach. Thus, most of the effort in the ADMA field is to identify compounds to increase DDAH1 expression, or compounds to active DDAH1 activity (55). Unfortunately, no effective DDAH1 activator has been identified. Several studies have demonstrated that farnesoid X receptors (FXR) agonists such as ursodeoxycholic acid (UDCA, a major component of bile) and GW4064 increase DDAH1 expression in the liver and kidney, and decreased plasma ADMA (34,55). Currently, UDCA is approved by the FDA for treatment of primary biliary cirrhosis (79) and other liver diseases. While a clinical trial demonstrated that UDCA significantly improved peak post-ischemic blood flow in the arm (85), it is not clear whether the observed beneficial effect of UDCA was associated with alterations of DDAH1 protein expression or ADMA content. Since kidney exerts an important role in regulating systemic ADMA, methods or treatment(s) to preserve renal function should effectively attenuate systemic ADMA level.
Summary.
Cardiovascular diseases such as (hypertension, heart failure, and coronary artery disease) are generally associated mild or less than 20% increase of plasma ADMA and/or L-NMMA, and increased plasma ADMA and/or L-NMMA is a strong and independent risker factor for many cardiovascular diseases. Increased ADMA and/or L-NMMA can cause detrimental effects on vascular endothelial cell growth, abnormal angiogenesis and vessel injury repair through attenuating NO production or increase of ROS generation. Clinical studies have also unanimously demonstrated that severe CKD often causes an increase of plasma ADMA and/or L-NMMA over 4 fold (even increased ~10 fold in some reports), and the increase of plasma ADMA and/or L-NMAM appears associated with the severity of CKD. Therefore, the current literatures clearly suggest that the dramatic increased ADMA and/or L-NMMA might be at least partially responsible for the increased cardiovascular morbidity and mortality in patients with CKD.
Supplementary Material
Highlights.
Patients with chronic kidney disease have an increased cardiovascular morbidity and mortality.
ADMA is endogenous inhibitors of nitric oxide synthases, and is a strong and independent risk factor for cardiovascular diseases.
Chronic kidney disease causes profound increase of systemic ADMA that attenuates NO production.
Increased cardiovascular morbidity in chronic kidney disease may relate to the dramatic increase of systemic ADMA and L-NMMA after kidney failure.
Acknowledgements:
None.
Sources of funding: This study was supported by Grants HL098669, HL102597, R01HL105406 from the National Institutes of Health, and a grant in aid from American Heart Association. The study was also supported by the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning to Dr. Xu.
Abbreviations
- CKD
chronic kidney disease
- ADMA
Asymmetric dimethylarginine
- L-NMMA
N-monomethy L-arginine
- SDMA
symmetric dimethylarginine
- DDAH1
dimethylarginine dimethylaminohydrolase 1
- PRMT1
type-1 protein arginine N-methyltransferase
- iNOS
inducible nitric oxide synthesis
- eNOS
endothelial nitric oxide synthesis
- nNOS
neuronal nitric oxide synthesis
- ROS
reactive oxygen species
- CAT
cationic amino acid transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None.
References
- 1.Achan V, Broadhead M, Malaki M, Whitley G, Leiper J, MacAllister R, Vallance P: Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol 2003;23:1455–9. [DOI] [PubMed] [Google Scholar]
- 2.Ali OA, Chapman M, Nguyen TH, Chirkov YY, Heresztyn T, Mundisugih J, Horowitz JD. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart. 2014;100:800–5. [DOI] [PubMed] [Google Scholar]
- 3.Anderssohn M, Rosenberg M, Schwedhelm E, Zugck C, Lutz M, Lüneburg N, Frey N, Böger RH. The L-Arginine-asymmetric dimethylarginine ratio is an independent predictor of mortality in dilated cardiomyopathy. J Card Fail. 2012;18:904–11 [DOI] [PubMed] [Google Scholar]
- 4.Anderssohn M, Schwedhelm E, Lüneburg N, Vasan RS, Böger RH. Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diab Vasc Dis Res. 2010;7:105–18. [DOI] [PubMed] [Google Scholar]
- 5.Arrigoni FI, Vallance P, Haworth SG, Leiper JM. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation. 2003;107:1195–201. [DOI] [PubMed] [Google Scholar]
- 6.Baek KJ, Thiel BA, Lucas S, Stuehr DJ. Macrophage nitric oxide synthase subunits. Purification, characterization, and role of prosthetic groups and substrate in regulating their association into a dimeric enzyme. J Biol Chem. 1993;268:21120–9. [PubMed] [Google Scholar]
- 7.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–9. [DOI] [PubMed] [Google Scholar]
- 8.Boger RH, Bode-Boger SM, Tsao PS, Lin PS, Chan JR, Cooke JP. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. Journal of the American College of Cardiology. 2000;36:2287–95. [DOI] [PubMed] [Google Scholar]
- 9.Böger RH, Sullivan LM, Schwedhelm E, Wang TJ, Maas R, Benjamin EJ, Schulze F, Xanthakis V, Benndorf RA, Vasan RS. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation. 2009;119:1592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Böger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004;134:2842S–7S. [DOI] [PubMed] [Google Scholar]
- 11.Burger DE, Lu X, Lei M, Xiang FL, Hammoud L, Jiang M, Wang H, Jones DL, Sims SM, Feng Q. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation. 2009;120:1345–54 [DOI] [PubMed] [Google Scholar]
- 12.Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, Zweier JL. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem. 2007;282:879–87. [DOI] [PubMed] [Google Scholar]
- 13.Cardounel AJ, Xia Y, Zweier JL. Endogenous Methylarginine Modulate Superoxide as Well as Nitric Oxide Generation from Neuronal Nitric-oxide Synthase: Differences In The Effects Of Monomethyl-And Dimethylarginine In The Presence And Absence Of Tetrahydrobiopterin. J. Biol. Chem 2005;280:7540–9. [DOI] [PubMed] [Google Scholar]
- 14.Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal. 2013;18:1078–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cengel A, Sahinarslan A, Biberoglu G, Hasanoglu A, Tavil Y, Tulmaç M, Ozdemir M. Asymmetrical dimethylarginine level in atrial fibrillation. Acta Cardiol. 2008;63:33–7. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Guo H, Xu D, Xu X, Wang H, Hu X, Lu Z, Kwak D, Xu Y, Gunther R, Huo Y, Weir EK. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension. 2012;59:1170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Li Y, Zhang P, Traverse JH, Hou M, Xu X, Kimoto M, Bache RJ. Dimethylarginine dimethylaminohydrolase and endothelial dysfunction in failing hearts. Am J Physiol Heart Circ Physiol. 2005;289:H2212–9. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Park S, Li Y, Missov E, Hou M, Han X, Hall JL, Miller LW, Bache RJ. Alterations of gene expression in failing myocardium following left ventricular assist device support. Physiological Genomics. 2003;14:251–260. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Traverse JH, Du R, Hou M, Bache RJ. Nitric oxide modulates myocardial oxygen consumption in the failing heart. Circulation. 2002;106:273–9. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Traverse JH, Hou M, Li Y, Du R, Bache RJ. Effect of PDE5 inhibition on coronary hemodynamics in pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2003;284:H1513–20. [DOI] [PubMed] [Google Scholar]
- 21.Closs EI, Basha FZ, Habermeier A, et al. Interference of l-arginine analogues with l-arginine transport mediated by the y +carrier hCAT-2B. Nitric Oxide. 1997;1: 65–73. [DOI] [PubMed] [Google Scholar]
- 22.Cosentino F, Patton S, d’Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, Malinski T, Luscher TF. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensiverats. J Clin Invest. 1998;101:1530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–6. [DOI] [PubMed] [Google Scholar]
- 24.Dasgupta A, Bowman L, D’Arsigny CL, Archer SL. Soluble guanylate cyclase: a new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clin Pharmacol Ther. 2015;97:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dayoub H, Achan V, Adimoolam S, Jacobi J, Stuehlinger MC, Wang BY, Tsao PS, Kimoto M, Vallance P, Patterson AJ, Cooke JP. Dimethylargininedimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation. 2003;108:3042–7. [DOI] [PubMed] [Google Scholar]
- 26.Dowsett L, Piper S, Slaviero A, Dufton N, Wang Z, Boruc O, Delahaye M, Colman L, Kalk E, Tomlinson J, Birdsey G, Randi AM, Leiper J. Endothelial DDAH1 is an Important Regulator of Angiogenesis but Does Not Regulate Vascular Reactivity or Hemodynamic Homeostasis. Circulation. 2015;131:2217–25.. [DOI] [PubMed] [Google Scholar]
- 27.Dückelmann C, Mittermayer F, Haider DG, Altenberger J, Eichinger J, Wolzt M. Asymmetric dimethylarginine enhances cardiovascular risk prediction in patients with chronic heart failure. Arterioscler Thromb Vasc Biol. 2007;27:2037–42. [DOI] [PubMed] [Google Scholar]
- 28.Emanuel LL, Bonow RO. Care of patients with end-stage heart disease In: Bonow RO, Mann DL, Zipes DP, Libby P, eds. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, Pa: Saunders Elsevier; 2011. [Google Scholar]
- 29.Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93:907–16. [DOI] [PubMed] [Google Scholar]
- 30.Gheorghiade M, Marti CN, Sabbah H et al. Soluble guanylate cyclase: a potential therapeutic target for heart failure. Heart Fail Rev. 2013;18:123–34. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010; 285:28938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Habib FM, Springall DR, Davies GJ, Oakley CM, Yacoub MH, Polak JM. Tumour necrosis factor and inducible nitric oxide synthase in dilated cardiomyopathy. Lancet. 1996;347:1151–5. [DOI] [PubMed] [Google Scholar]
- 33.Huang ZM, Gao E, Fonseca FV, Hayashi H, Shang X, Hoffman NE, Chuprun JK, Tian X, Tilley DG, Madesh M, Lefer DJ, Stamler JS, Koch WJ. Convergence of G protein-coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Sci Signal. 2013;6:ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu XL, Li MP, Song PY, Tang J, Chen XP. AGXT2: An unnegligible aminotransferase in cardiovascular and urinary systems. J Mol Cell Cardiol. 2017. December;113:33–38. [DOI] [PubMed] [Google Scholar]
- 35.Hu XL, Atzler D, Xu X, Zhang P, Guo HP, Lu ZB, Fassett J, Schwedhelm E, Böger RH, Bache RJ, Chen YJ. Global Dimethylarginine Dimethylaminohydrolase-1 (DDAH1) Gene-Deficient Mice Reveal That DDAH1 Is the Critical Enzyme for Degrading the Cardiovascular Risk Factor Asymmetrical Dimethylarginine. Arterioscler Thromb Vasc Biol. 2011; 31:1540–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu XL, Xu X, Zhu GB, Atzler D, Kimoto M, Chen J, Schwedhelm E, Luneburg N, Boger RH, Zhang P, Chen YJ. Vascular Endothelial-Specific DimethylarginineDimethylaminohydrolase 1 Deficient Mice Reveal That Vascular Endothelium Plays an Important Role in Removing Asymmetric Dimethylarginine. Circulation. 2009;120:2222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunt SA, Abraham WT, Chin MH et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult. Circulation. 2005; 112: e154–235. [DOI] [PubMed] [Google Scholar]
- 38.Ignarro LJ, Napoli C, Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: an overview. Circ Res. 2002;90:21–8. [DOI] [PubMed] [Google Scholar]
- 39.Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol. 2002;53(4 Pt 1): 503–14. [PubMed] [Google Scholar]
- 40.Iimori S, Naito S, Noda Y, Nishida H, Kihira H, Yui N, Okado T, Sasaki S, Uchida S, Rai T. Anaemia management and mortality risk in newly visiting patients with chronic kidney disease in Japan: The CKD-ROUTE study. Nephrology (Carlton). 2015;20(9):601–8. [DOI] [PubMed] [Google Scholar]
- 41.Ito A, Tsao PS, Adimoolam S, Kimoto M, Ogawa T, Cooke JP. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation. 1999;99:3092–5. [DOI] [PubMed] [Google Scholar]
- 42.Jacobi J, Maas R, Cardounel AJ, Arend M, Pope AJ, Cordasic N, Heusinger-Ribeiro J, Atzler D, Strobel J, Schwedhelm E, Böger RH, Hilgers KF. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E-deficient mice by lowering asymmetric dimethylarginine. Am J Pathol. 2010;176:2559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobi J, Maas R, Arend M, Cordasic N, Hilgers KF. Effect of lowering asymmetric dimethylarginine (ADMA) on vascular pathology in atherosclerotic ApoE-deficient mice with reduced renal mass. Int J Mol Sci. 2014;15(4):5522–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobi J, Tsao PS. Asymmetrical dimethylarginine in renal disease: limits of variation or variation limits? A systematic review. Am J Nephrol. 2008;28:224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeong EM, Monasky MM, Gu L, Taglieri DM, Patel BG, Liu H, Wang Q, Greener I, Dudley SC Jr, Solaro RJ. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol. 2013;56:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones SP, Greer JJ, van Haperen R, Duncker DJ, de Crom R, Lefer DJ. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc Natl Acad Sci U S A. 2003;100:4891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kakimoto Y, Akazawa S. Isolation and identification of N-G,N-G- and N-G,N’-G-dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. J Biol Chem. 1970;245:5751–8. [PubMed] [Google Scholar]
- 48.Kielstein JT, Boger RH, Bode-Boger SM, Frolich JC, Haller H, Ritz E, Fliser D. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J Am Soc Nephrol. 2002;13:170–176. [DOI] [PubMed] [Google Scholar]
- 49.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519:472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J.1999; 343:209–214. [PMC free article] [PubMed] [Google Scholar]
- 52.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. [DOI] [PubMed] [Google Scholar]
- 53.Li W, Mital S, Ojaimi C, Csiszar A, Kaley G, Hintze TH. Premature death and age-related cardiac dysfunction in male eNOS-knockout mice. J Mol CellCardiol. 2004;37:671–80. [DOI] [PubMed] [Google Scholar]
- 54.Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation. 2002;106:987–92. [DOI] [PubMed] [Google Scholar]
- 55.Liu X, Hou L, Xu D, Chen A, Yang L, Zhuang Y, Xu Y, Fassett JT, Chen Y. Effect of asymmetric dimethylarginine (ADMA) on heart failure development. Nitric Oxide. 2016;54:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, Fassett J, Tao Y, Zhang P, Hall JL, dos Remedios C, Garry DJ, Chen Y. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation. 2010;121:1474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McBride AE, Silver PA. State of the arg: protein methylation at arginine comes of age. Cell. 2001;106:5–8 [DOI] [PubMed] [Google Scholar]
- 58.McDermott JR: Studies on the catabolism of Ng-methylarginine, Ng, Ng-dimethylarginine and Ng, Ng-dimethylarginine in the rabbit. Biochem J. 1976;154:179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mochizuki S, Ono J, Yada T, Ogasawara Y, Miyasaka T, Kimoto M, Kashihara N, Kajiya F. Systemic nitric oxide production rate during hemodialysis and its relationship with nitric oxide-related factors. Blood Purif. 2005;23:317–324. [DOI] [PubMed] [Google Scholar]
- 60.Nandi M, Kelly P, Torondel B, Wang Z, Starr A, Ma Y, Cunningham P, Stidwill R, Leiper J. Genetic and pharmacological inhibition of dimethylarginine dimethylaminohydrolase 1 is protective in endotoxic shock. Arterioscler Thromb Vasc Biol. 2012. November;32(11):2589–97. [DOI] [PubMed] [Google Scholar]
- 61.Ogawa T, Kimoto M, Sasaoka K. Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-L-arginine to L-citrulline in rats. Biochem Biophys Res Commun. 1987;148: 671–7. [DOI] [PubMed] [Google Scholar]
- 62.Olken NM, Osawa Y, Marletta MA. Characterization of the inactivation of nitric oxidesynthase by NG-methyl-L-arginine: evidence for heme loss. Biochemistry. 1994;33:14784–91. [DOI] [PubMed] [Google Scholar]
- 63.Osanai T, Fujiwara N, Saitoh M, Sasaki S, Tomita H, Nakamura M, Osawa H, Yamabe H, Okumura K. Relationship between salt intake, nitric oxide and asymmetric dimethylarginine and its relevance to patients with end-stage renal disease. Blood Purif. 2002;20:466–468. [DOI] [PubMed] [Google Scholar]
- 64.Pacurari M, Xing D, Hilgers RH, Guo YY, Yang Z, Hage FG. Endothelial cell transfusion ameliorates endothelial dysfunction in 5/6 nephrectomized rats. Am J Physiol Heart Circ Physiol. 2013; 305(8): H1256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pope AJ, Karuppiah K, Cardounel AJ. Role of the PRMT-DDAH-ADMA axis in the regulation of endothelial nitric oxide production. Pharmacol Res. 2009;60:461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raj DS, Vincent B, Simpson K, Sato E, Jones KL, Welbourne TC, Levi M, Shah V, Blandon P, Zager P, Robbins RA. Hemodynamic changes during hemodialysis: role of nitric oxide and endothelin. Kidney Int. 2002;61:697–704. [DOI] [PubMed] [Google Scholar]
- 67.Ritchie J, Rainone F, Green D, Alderson H, Chiu D, Middleton R, O’Donoghue D, Kalra PA. Extreme Elevations in Blood Pressure and All-Cause Mortality in a Referred CKD Population: Results from the CRISIS Study. Int J Hypertens. 2013;2013:597906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodionov RN, Murry DJ, Vaulman SF, Stevens JW, Lentz SR. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J Biol Chem. 2010; 285:5385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwedhelm E, Böger RH. The role of asymmetric and symmetric dimethylarginines in renal disease. Nat Rev Nephrol. 2011;7:275–85. [DOI] [PubMed] [Google Scholar]
- 70.Seppälä I, Kleber ME, Lyytikäinen LP, Hernesniemi JA, Mäkelä KM, Oksala N, Laaksonen R, Pilz S, Tomaschitz A, Silbernagel G, Boehm BO, Grammer TB, Koskinen T, Juonala M, Hutri-Kähönen N, Alfthan G, Viikari JS, Kähonen M, Raitakari OT, März W, Meinitzer A, Lehtimäki T; AtheroRemo Consortium. Genome-wide association study on dimethylarginines reveals novel AGXT2 variants associated with heart rate variability but not with overall mortality. Eur Heart J. 2014;35:524–31. [DOI] [PubMed] [Google Scholar]
- 71.Shah R, Matthews GJ, Shah RY, McLaughlin C, Chen J, Wolman M, Master SR, Chai B, Xie D, Rader DJ, Raj DS, Mehta NN, Budoff M, Fischer MJ, Go AS, Townsend RR, He J, Kusek JW, Feldman HI, Foulkes AS, Reilly MP; CRIC Study Investigators. Serum Fractalkine (CX3CL1) and Cardiovascular Outcomes and Diabetes: Findings From the Chronic Renal Insufficiency Cohort (CRIC) Study. Am J Kidney Dis. 2015;66(2):266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shirakawa T, Kako K, Shimada T, Nagashima Y, Nakamura A, Ishida J, Fukamizu A. Production of free methylarginines via the proteasome and autophagy pathways in cultured cells. Mol Med Rep. 2011;4:615–20. [DOI] [PubMed] [Google Scholar]
- 73.Sinisalo J, Vanhanen H, Pajunen P, Vapaatalo H, Nieminen MS. Ursodeoxycholic acid and endothelial-dependent, nitric oxide-independent vasodilatation of forearm resistance arteries in patients with coronary heart disease. Br J Clin Pharmacol. 1999;47:661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Surdacki A, Nowicki M, Sandmann J, Tsikas D, Boeger RH, Bode-Boeger SM, Kruszelnicka-Kwiatkowska O, Kokot F, Dubiel JS, Froelich JC. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol. 1999;33:652–8. [DOI] [PubMed] [Google Scholar]
- 75.Suda O, Tsutsui M, Morishita T, Tasaki H, Ueno S, Nakata S, Tsujimoto T, Toyohira Y, Hayashida Y, Sasaguri Y, Ueta Y, Nakashima Y, Yanagihara N. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase-deficient mice: involvement of renin-angiotensin system and oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1682–8. [DOI] [PubMed] [Google Scholar]
- 76.Sydow K, Mondon CE, Schrader J, Konishi H, Cooke JP.Dimethylargininedimethylaminohydrolase overexpression enhances insulin sensitivity. Arterioscler Thromb Vasc Biol. 2008;28:692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sydow K, Schmitz C, von Leitner EC, von Leitner R, Klinke A, Atzler D, Krebs C, Wieboldt H, Ehmke H, Schwedhelm E, Meinertz T, Blankenberg S, Böger RH, Magnus T, Baldus S, Wenzel U. Dimethylarginine dimethylaminohydrolase1 is an organ-specific mediator of end organ damage in a murine model of hypertension. PLoS One. 2012;7(10):e48150. doi: 10.1371/journal.pone.0048150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–22. [DOI] [PubMed] [Google Scholar]
- 79.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tanaka M, Sydow K, Gunawan F, Jacobi J, Tsao PS, Robbins RC, Cooke JP. Dimethylarginine dimethylaminohydrolase overexpression suppresses graft coronary artery disease. Circulation. 2005;112:1549–56. [DOI] [PubMed] [Google Scholar]
- 81.Tang WH, Tong W, Shrestha K, Wang Z, Levison BS, Delfraino B, Hu B, Troughton RW, Klein AL, Hazen SL. Differential effects of arginine methylation on diastolic dysfunction and disease progression in patients with chronic systolic heart failure. Eur Heart J. 2008;29:2506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toth J, Racz A, Kaminski PM, Wolin MS, Bagi Z, Koller A. Asymmetrical Dimethylarginine Inhibits Shear Stress-Induced Nitric Oxide Release and Dilation and Elicits Superoxide-Mediated Increase in Arteriolar Tone. Hypertension. 2007;49:563–8. [DOI] [PubMed] [Google Scholar]
- 83.Tran CT, Fox MF, Vallance P, Leiper JM. Chromosomal localization, gene structure, and expression pattern of DDAH1: comparison with DDAH2 and implications for evolutionary origins. Genomics. 2000;68:101–105. [DOI] [PubMed] [Google Scholar]
- 84.Valkonen V- P, Tuomainen T- P, Laaksonen R. DDAH gene and cardiovascular risk. Vascular Medicine. 2005;10:45–8. [DOI] [PubMed] [Google Scholar]
- 85.vonHaehling S, Schefold JC, Jankowska EA, Springer J, Vazir A, Kalra PR, Sandek A, Fauler G, Stojakovic T, Trauner M, Ponikowski P, Volk HD, Doehner W, Coats AJ, Poole-Wilson PA, Anker SD. Ursodeoxycholic acid in patients with chronic heart failure: a double-blind, randomized, placebo-controlled, crossover trial. J Am Coll Cardiol. 2012;59:585–92. [DOI] [PubMed] [Google Scholar]
- 86.Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol. 2014;71:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y, Zhang P, Xu Z, Yue W, Zhuang Y, Chen Y, Lu Z. S-nitrosylation of PDE5 increases its ubiquitin-proteasomal degradation. Free Radic Biol Med. 2015;86:343–51 [DOI] [PubMed] [Google Scholar]
- 88.Weis M, Kledal TN, Lin KY, Panchal SN, Gao SZ, Valantine HA, Mocarski ES, Cooke JP. Cytomegalovirus infection impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation. 2004;109:500–5. [DOI] [PubMed] [Google Scholar]
- 89.Wells SM, Holian A. Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am J Respir Cell Mol Biol. 2007;36:520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93:6770–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu X, Zhang P, Kwak D, Fassett J, Yue W, Atzler D, Hu X, Liu X, Wang H, Lu Z, Guo H, Schwedhelm E, Böger RH, Chen P, Chen Y. Cardiomyocyte dimethylarginine dimethylaminohydrolase-1 (DDAH1) plays an important role in attenuating ventricular hypertrophy and dysfunction. Basic Res Cardiol. 2017;112:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yilmaz MI, Saglam M, Caglar K, Cakir E, Ozgurtas T, Sonmez A, Eyileten T, Yenicesu M, Acikel C, Oguz Y, Ozcan O, Bozlar U, Erbil K, Aslan I, Vural A. Endothelial functions improve with decrease in asymmetric dimethylarginine levels after renal transplantation. T ransplantation. 2005;80:1660–1666. [DOI] [PubMed] [Google Scholar]
- 93.Yilmaz MI, Saglam M, Caglar K, Cakir E, Sonmez A, Ozgurtas T, Aydin A, Eyileten T, Ozcan O, Acikel C, Tasar M, Genctoy G, Erbil K, Vural A, Zoccali C. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am J Kidney Dis. 2006;47:42–50. [DOI] [PubMed] [Google Scholar]
- 94.Zairis MN, Patsourakos NG, Tsiaousis GZ, TheodossisGeorgilas A, Melidonis A, Makrygiannis SS, Velissaris D, Batika PC, Argyrakis KS, Tzerefos SP, Prekates AA, Foussas SG. Plasma asymmetric dimethylarginine and mortality in patients with acute decompensation of chronic heart failure. Heart. 2012;98:860–4. [DOI] [PubMed] [Google Scholar]
- 95.Zhang P, Xu X, Hu X, Wang H, Fassett J, Huo Y, Chen Y, Bache RJ. DDAH1 deficiency attenuates endothelial cell cycle progression and angiogenesis. PLoS One. 2013;8:e79444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res. 2007;100:1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang P, Hu XL, Xu X, Chen YJ, Bache RJ. Dimethylarginine dimethylaminohydrolase 1 modulates endothelial cell growth through nitric oxide and Akt. Arterioscler Thromb Vasc Biol. 2011;31:890–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.