

Abstract
Cyclic dinucleotides (CDNs), including cyclic di-GMP (CDG), are promising vaccine adjuvants in pre-clinical/clinical trials. The in vivo mechanisms of CDNs is not clear. Here we investigated the roles of lung DCs subsets in promoting CDG mucosal adjuvant responses in vivo. Using genetically modified mice and adoptive cell transfer, we identified lung conventional DC 2 (cDC2) as the central player in CDG mucosal responses. We further identified two functionally distinct lung cDC2 subpopulations: TNFR2+pRelB+ and TNFR2−pRelB− cDC2. The TNFR2+ cDC2 were mature and migratory upon intranasal CDG administration while the TNFR2− cDC2 were activated but not mature. Adoptive cell transfer showed that TNFR2− cDC2 mediate the antibody responses of CDG, while the TNFR2+ cDC2 generate Th1/17 responses. Mechanistically, immature TNFR2− cDC2 activate monocyte-derived DCs (moDCs), which do not take up intranasally administered CDG. moDCs promote CDG-induced generation of T follicular helper- and germinal center B-cells in the lungs. Our data revealed a previously undescribed in vivo mode of DCs action whereby an immature lung TNFR2− cDC2 subpopulation directs the non-migratory moDCs to generate CDG mucosal responses in the lung.
Introduction
Adjuvants improve vaccine safety profiles and enhance, and shape, antigen-specific immune responses. Understanding the mode of action of adjuvants is key for the development of rationally designed modern vaccines. Recently, the small molecule cyclic dinucleotides (CDNs) have emerged as a group of promising vaccine adjuvants in preclinical and clinical trials 1. CDNs include the bacterial second messengers cyclic di-GMP (CDG), cyclic di-AMP, 3’3’-cyclic GMP-AMP and the mammalian second messenger 2’3’-cyclic GMP-AMP 1. CDG is the founding member and the most studied CDNs 2. As a mucosal adjuvant, CDG does not cause acute toxicity in mice 1. Furthermore, CDG is a more potent activator of Th1 and Th2 immune response than LPS, CpG oligonucleotides (ODN) or aluminum salt-based adjuvant in mice 3. Last, CDG adjuvanted vaccines protect mice from H5N1 influenza 4, Acinetobacter baumannii 5, Staphylococcus aureus 6, Klebsiella pneumoniae 7 and Streptococcus pneumoniae 8, 9. CDG also showed cancer vaccine adjuvant activity in animal models 10, 11.
MPYS, also known as STING and MITA, is a receptor for CDNs 12 and a critical player in sensing cytosolic DNA 13, 14. MPYS−/− mice lose CDNs adjuvant activity 15. Additionally, TNF signaling, not type I IFN signaling, is essential for the adjuvant activity of CDG in vivo 15, 16. Notably, when administered intranasally, CDG not only induced lung production of TNF, IL-1β but also the anti-inflammatory cytokine IL-10 17. Consequently, CDG adjuvant does not induce exaggerated inflammation responses in the lung 17. The precise in vivo mechanism by which TNF mediates CDG adjuvant activity in vivo is unknown.
DCs orchestrate vaccine adjuvant responses 18. They consist of developmentally and functionally distinct subsets that promote either immunogenic or tolerogenic immune responses 19–22. Murine lung DCs consist of three subsets: the CD103+ conventional DC (cCD1), the CD11b+CD24+CD64− conventional DC (cDC2) and monocyte-derived CD11b+CD24−CD64+ DC (moDCs) 21. Using MPYSfl/flCD11cCre mice, we previously showed that CDG adjuvant activity depends on MPYS expression in DCs 17. The lung DC subset mediating CDG adjuvant activity is unknown.
In this report, we revealed important heterogeneity in the lung cDC2 population and identified the cDC2 subpopulation that is responsible for CDG adjuvant effect. Surprisingly, the antibody responses of CDG adjuvant depends on an activated but immature TNFR2− cDC2 subpopulation, which drive moDCs maturation to generate T follicular helper (Tfh) cells in the lung.
RESULTS
CDG directly targets lung cDC1 and cDC2
DCs mediate CDG adjuvant activity in vivo 17. There are three lung DC subsets: cDC1 (CD103+ CD24+ CD64−CD11b−), cDC2 (CD103−CD24+CD64−CD11b+) and moDCs (CD103−CD24−CD64+ CD11b+) 21, 23–27 (Fig S1). All DC subsets express the CDG receptor MPYS, which is an ER-resident protein (Fig 1A). CDG has two phosphate groups preventing it from directly passing through the cell membrane. To determine which lung DC subset took up CDG, mice were intranasally administered with FITC-conjugated CDG and FITC+ lung cells were examined after 5hrs. Among lung DCs, cDC1 and cDC2 had the highest percentage of CDG-FITC whereas moDCs had no CDG-FITC (Fig 1B).
Figure 1. cDC2 play a central role in mediating the adjuvant activity of CDG.

A. Flow cytometry analysis of MPYS expression in lung DC subsets. n>3. B. Absolute number of lung DC subsets in C57BL/6 mice administered (i.n.) with saline or 5µg FITC-CDG for 5 hours. n=3.C. Absolute number of CD86+ lung DC in mice administered (i.n.) with saline or 5µg CDG for 16 hours. n>3. D. Absolute number of CCR7+ lung DC in mice administered (i.n.) with saline or 5µg CDG for 16 hours. n>3. E-G. Flow cytometry analysis of pRelA and pRelB in cDC1 (E), cDC2 (F), and moDCs (G) from mice treated with saline or CDG for 16hrs. n>3. H. C57BL/6, Batf3−/−, and IRF4fl/flCD11ccre mice were immunized (i.n.) with two doses (14 days apart) of PspA or PspA plus CDG (5ug). Anti-PspA IgG in serum and IgA in BALF were determined by ELISA. n>3. I. IRF4fl/flCD11ccre mice were adoptively transferred (i.n.) with lung cDC2 sorted from WT mice lung and immunized (i.n.) with PspA or CDG/PspA. Serum anti-PspA IgG and BALF IgA were determined by ELISA. n=3. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
Alveolar macrophages (AM) take up CDG but are dispensable for CDG adjuvant activity.
AM (CD11c+MHC IIint) took up most of the fluorescent CDG in vivo (Fig S2A). Unlike DCs, AM did not increase expression of CD86 following CDG treatment (Fig S2B). To determine whether AM are required for CDG responses in vivo, we used the MPYSfl/flLysMcre mice 17, which deleted MPYS expression in AM (Fig S2C). The activation of lung DCs by CDG was unaltered in the MPYSfl/flLysMcre mice (Fig S2D). Importantly, MPYSfl/flLysMcre mice produced similar anti-pneumococcal surface protein A (PspA) antibody as the WT upon CDG/PspA immunization (Fig S2E).
CDG differentially activates lung DC subsets in vivo.
DCs subsets are functionally distinct. CDG increased CD86 and CCR7 expression in lung cDC1 and cDC2 (Fig 1C & 1D) 17. In addition, both cDCs migrate, bearing processed antigen, to the lung draining lymph nodes (dLNs) (Fig S3A-B). However, we found that following intranasal CDG administration, cDC1 activated RelA (Fig 1E) while cDC2 activated RelB (Fig 1F). Notably, some cDC2 have constitutively activated RelB (Fig 1F). To further demonstrate that CDG differentially activates lung DC subsets, we used the IRF4fl/flCD11ccre and Batf3−/− mice.
The development of cDC1 and cDC2 are controlled by transcriptional factors Batf3 and IRF4 respectively. IRF4fl/flCD11ccre mice lack cDC2 in the lung (Fig S1C) 28–30 while Batf3−/− mice lack cDC1 (Fig S1D) 31, 32. We found that CDG-induced lung production of TNF, IL-1β and IL-12p70 were dramatically reduced in IRF4fl/flCD11ccre mice but not in the Batf3−/− mice (Fig S3C). Conversely, CDG-induced MCP-1 production was largely absent in the Batf3−/− mice but not in the IRF4fl/flCD11ccre mice (Fig S3C).
CDG indirectly activates moDCs
Although moDCs did not take up CDG, they increased expression of CD86 in response to intranasal administration of CDG (Fig 1C). This was independent of MPYS expression in moDCs as moDCs in MPYSfl/flLysMcre mice had normal levels of CD86 expression (Fig S2D). Notably, activated moDCs did not increase CCR7 (Fig 1D) and did not migrate to dLNs (Fig S3B). Last, moDCs activated both RelA and RelB in response to CDG (Fig 1G). We concluded that CDG indirectly activate moDCs and activated lung moDCs were not migratory 25, 33.
cDC2 play a central role in mediating CDG adjuvant activity
We next asked which cDC subset mediates CDG adjuvant activity. IRF4fl/flCD11ccre mice lack cDC2 in the lung 28, 29(Fig S1C). The cDC1 and moDCs were retained in the IRF4fl/flCD11ccre mice (Fig S1C). We examined CDG adjuvant activity in the IRF4fl/flCD11ccre mice. Mice were intranasally administered with PspA alone or with CDG. PspA-specific Ab responses were examined in the blood and bronchoalveolar lavage fluid (BALF). Unlike the WT mice, CDG did not induce anti-PspA Abs in BALF and serum (Fig 1H, S3D-E) from immunized IRF4fl/flCD11ccre mice. Ex vivo recall assay in lung cells and splenocytes from immunized IRF4fl/flCD11ccre mice also did not show PspA-specific Th1, Th2 or Th17 responses (Fig S3F-G).
To further demonstrate that lung cDC2 mediate the adjuvant activity of CDG, we adoptively transfer (i.n.) WT lung cDC2 into IRF4fl/flCD11ccre mice. The recipient IRF4fl/flCD11ccre mice were then immunized with CDG/PspA. We found that adoptive transfer of WT cDC2 generated PspA-specific serum IgG and IgA in the IRF4fl/flCD11ccre mice similar to the WT mice (Fig 1I). We concluded that lung cDC2 are critical for CDG adjuvant activity.
In contrast to the IRF4fl/flCD11ccre mice, Batf3−/− mice mounted antigen-specific IgG and IgA responses in a manner comparable to the WT following CDG/PspA immunization (Fig 1H, S3D-E). We concluded that cDC2 play a central role in mediating CDG adjuvant activity. Batf3−/−mice had impaired Th1 responses following immunization (Fig S3F-G). Whether the defect is due to the lack of cDC1 remains to be determined since T cells also express Batf3.
TNFR2 is essential for CDG adjuvant activity
TNF signaling is critical for CDG adjuvant activity in vivo 15, 16. TNF signals through two TNFR1 and TNFR2. TNFR1 binds transmembrane TNF (mTNF) and soluble TNF (sTNF) while TNFR2 only binds to mTNF 34–37. The lung DC compartment is not altered by the lack of either TNFR1 or TNFR2 (Fig. S4A-B). Consistent with the previous report, we found that CDGinduced reduced humoral and cellular immune responses in TNFR1−/− mice (Fig 2A, S4C-F). Surprisingly, CDG completely lost its adjuvant activity in TNFR2−/− mice (Fig 2A, S4C-F).
Figure 2. cDC2 expression of TNFR2 is required for CDG-induced lung cDC2 maturation in vivo by activating RelB.

A. C57BL/6 (WT), TNFR1−/− and TNFR2−/− mice were immunized (i.n.) with two doses of PspA or PspA plus CDG (5ug). Serum anti-PspA IgG and BALF IgA were determined by ELISA. n=3. B. WT and TNFR2−/− mice were treated (i.n.) with saline or CDG (5µg) for 16 hours. CD86 expression in lung cDC2 were determined by Flow cytometry. n=3. C. IRF4fl/flCD11ccre mice were adoptively transferred (i.n.) with lung cDC2 sorted from WT or TNFR2−/− mice lung. The recipient mice were administered (i.n.) with saline or CDG (5µg) for 16hrs. CD86 expression in lung cDC2 were determined by Flow cytometry. n=3. D. Flow cytometry analysis of TNFR2 expression on lung cDC2 in WT mice administered (i.n) with saline or CDG for 16hrs. n>3. E. Flow cytometry analysis of TNFR2 expression on pRelB+ cDC2 from mice treated with saline or CDG. n=3. F. Flow cytometry analysis of pRelB expression on TNFR2+ cDC2 from mice treated with CDG. n=3. G. Flow cytometry analysis of pRelB expression on cDC2 from WT and TNFR2−/− mice. n=3. H. Flow cytometry analysis of CD86 expression on lung cDC2 in RelBfl/fl and RelBfl/flCD11CCre mice treated with CDG. n=3. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
TNFR2 expression on lung cDC2 is required for its maturation
cDC2 are critical for CDG adjuvant activity (Fig 1). We next examined lung cDC2 maturation in the TNFR2−/− mice. We found that CDG did not enhance CD86 or CCR7 expression in lung cDC2 of TNFR2−/− mice in vivo (Fig. 2B & S5A). In comparison, CDG-mediated CD86 and CCR7 expression on cDC1 of TNFR2−/− mice in vivo (Fig S5A-B). Furthermore, blocking TNFR2 by mAb inhibited CDG-mediated CD86 expression on cDC2 in vivo (Fig. S5C). Last, adoptively transferred TNFR2-deficient cDC2 into IRF4fl/flCD11ccre recipient mice failed to upregulate CD86 expression in response to intranasal CDG administration (Fig 2C).
TNFR2+ lung cDC2 has constitutively activated RelB
We next examine TNFR2 expression on lung cDC2. We found that a population of lung cDC2 constitutively express TNFR2 (Fig. 2D). In contrast, TNFR2 expression was not detected on steady-state lung cDC1 though CDG dramatically increased TNFR2 expression (Fig S5D). Steady-state lung cDC2 have a pRelB+ population (Fig 1F). Interestingly, all the pRelB+ cDC2 are TNFR2+ (Fig 2E, left panel). CDG further activate RelB in lung cDC2 upon CDG treatment (Fig 1F). We found that all pRelB+ cells in activated cDC2 expressed TNFR2 (Fig 2E, right panel) and all the TNFR2+ cDC2 are pRelB+ (Fig 2F). Last, cDC2 in TNFR2−/− mice lack pRelB indicating that RelB activation requires TNFR2 signaling (Fig 2G). Thus, TNFR2+ and pRelB+ lung cDC2 are the same population.
RelB in DCs is required for CDG-induced cDC2 maturation in vivo.
TNFR2 on lung cDC2 is required CDG-induced cDC2 maturation in vivo (Fig 2C). We reasoned that RelB was required for CDG-induced cDC2 maturation too. We used RelBfl/flCD11ccre mice to ablate RelB in DCs. RelBfl/flCD11ccre mice have normal DC populations, with all subsets intact (Fig S5E). In the absence of RelB in DCs, cDC2 failed to upregulate CD86 in response to CDG (Fig 2H). cDC1, which do not activate RelB, upregulated CD86 (Fig S5F). We concluded that CDG-induced lung cDC2 maturation depends on the cell-intrinsic signal of TNFR2-RelB.
TNFR2+ cDC2 are required for CDG-induced Th1 and Th17 responses but dispensable for the humoral responses
We next sought to determine if TNFR2 expression on cDC2 was needed for the adjuvant activity of CDG. Unexpectedly, even though adoptively transferred TNFR2-deficient cDC2 failed to upregulate CD86 in response to CDG (Fig 2C), they induced serum anti-PspA IgG and IgA when transferred into IRF4fl/flCD11ccre mice (Fig 3A). In fact, the anti-PspA IgG and IgA in IRF4fl/flCD11ccre mice receiving TNFR2-deficient cDC2 and WT cDC2 were comparable (Fig 3A).
Figure 3. cDC2 expression of TNFR2 and RelB is required for Th1 and Th17 responses, but dispensable for CDG-induced antibody response.

A. IRF4fl/flCD11ccre mice were adoptively transferred (i.n.) with lung cDC2 sorted from WT or TNFR2−/− mice lung and immunized (i.n.) with PspA or CDG/PspA. Serum anti-PspA IgG and BALF anti-PspA IgA were determined by ELISA. n=3. B. RelBfl/fl and RelBfl/flCD11CCre mice were immunized (i.n) with CDG/PspA or PspA alone as before. Serum anti-PspA IgG and BALF anti-PspA IgA were determined by ELISA. n=3. C. Lung cells from immunized RelBfl/fl and RelBfl/flCD11CCre mice were stimulated with 5∝g/ml PspA for 4 days in culture. Cytokines were measured in the supernatant by ELISA. n>3. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
We then examined CDG adjuvant activity in the RelBfl/flCD11ccre mice, which also lack mature cDC2. Upon immunization with PspA and CDG, RelBfl/flCD11ccre mice produced normal levels of IgG and IgA (Fig 3B). RelBfl/flCD11ccre mice failed to induce Th1 and Th17 responses in the lung (Fig 3C).
TNFR2 defines two functionally distinct subpopulations of lung cDC2
cDC2 is a heterogeneous population 21, 32, 38, 39. We showed that lung TNFR2+pRelB+ cDC2 were mature and required for the Th1/Th17 responses but not humoral responses while the TNFR2−cDC2 was not mature but mediates CDG-induced antibody response. We assessed whether TNFR2 expression could define functionally distinct lung cDC2 subpopulations.
Both cDC2 populations took up CDG in vivo (Fig 4A). When adoptively transferred into MPYS−/− mice, both produced lung TNF (Fig 4B). MPYS−/− mice themselves do not respond to CDG 15. Consistently, adoptively transferred TNFR2+ cDC2 upregulated CD86 and CCR7 in response to CDG whereas TNFR2− cDC2 failed to do so (Fig 4C). Importantly, adoptive transfer of TNFR2+ cDC2 into MPYS−/− (Fig 4D, S6A-B) and IRF4fl/flCD11ccre mice (Fig 4E) failed to rescue antibody production. Consistent with the RelBfl/flCD11ccre results (Fig 3), TNFR2+ cDC2 were able to rescue Th1/Th17 responses in the IRF4fl/flCD11ccre mice (Fig 4F). TNFR2+ cDC2 also rescued Th2 response, in contrast to RelBfl/flCD11ccre mice (Fig 4F & 3C). We speculate that the ability of TNFR2+ cDC2 to mediate CDG-induced Th2 responses maybe redundant in vivo.
Figure 4. Lung TNFR2− cDC2 mediate CDG-induced humoral response.

A. Flow cytometry analysis of TNFR2 expression on lung CDG-FITC+ cDC2 from WT mice. n=3. B. Lung TNF production in CDG treated (i.n.) MPYS−/− mice adoptively transferred with WT TNFR2+ or TNFR2− lung cDC2. n=3 C. Sorted TNFR2+ and TNFR2− lung cDC2 from WT mice were labelled with CFSE and adoptively transferred into the MPYS−/− mice. Recipient mice were treated with CDG (i.n.) for 16hrs. CD86 and CCR7 expression on CFSE positive lung cells were examined by Flow cytometry. n=3. D. Sorted TNFR2+ and TNFR2− lung cDC2 from WT mice were adoptively transferred into the MPYS−/−PspA or CDG/PspA twice. Serum anti-PspA mice. Recipient mice were immunized (i.n.) with IgG were determined by ELISA. n=3. E. Sorted TNFR2+ and TNFR2− lung cDC2 from WT mice were adoptively transferred into IRF4fl/flCD11ccre mice. Recipient mice were immunized (i.n.) with PspA or CDG/PspA. Serum anti-PspA IgG were determined by ELISA. n=3. F. Lung cells from recipient IRF4fl/flCD11ccre mice were stimulated with 5∝g/ml PspA for 4 days in culture. Cytokines were measured in the supernatant by ELISA. G. A cartoon illustrating following CDG administration, TNFR2+ cDC2 activate RelB to induce Th1, Th2 and Th17 responses while TNFR2− cDC2 mediate the antibody response. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
In contrast, adoptive transfer of TNFR2− cDC2 completely restored antibody, but not Th, responses in MPYS−/− (Fig 4D, S6A-B) and IRF4fl/flCD11ccre mice (Fig 4E-F). In fact, levels of anti-PspA IgG and IgA in IRF4fl/flCD11ccre mice receiving TNFR2− cDC2 were similar to the WT (Fig 4E). We concluded that lung cDC2 can be divided into two functionally distinct subpopulations: TNFR2+ cDC2 and TNFR2− cDC2. The TNFR2+ cDC2 are important for CDG-induced cellular immunity, while TNFR2− cDC2 are responsible for CDG-induced humoral responses (Fig 4G).
TNFR2+ and TNFR2− lung cDC2 are derived from pre-cDC2
We further characterized these steady-state lung cDC2 populations. We found that the TNFR2+ cDC2 are positive for BTLA, PDL-1, arginase 1 (Arg1). The TNFR2+ cDC2 also have mixed expression of PD-L2 and CD301b (Fig 5A). The lung TNFR2− cDC2 expressed CX3CR1 (Fig 5A). Both populations express common cDC2 markers as SIRPα, CD26, IRF4 and Zbtb46 (Fig 5A, 5B & S7A) 40, 41. Furthermore, both subpopulations of cDC2 were absent in Flt3−/− mice (Fig S7B-D). Last, TNFR2+ and TNFR2− cDC2 are negative for cDC1 markers IRF8, XCR1 and not affected in Batf3−/− mice confirming their identity as cDC2 (Fig 5B, S7C-D).
Figure 5. Lung cDC2 consist of two distinct subpopulations.

A. Flow cytometry analysis of lung TNFR2+ vs TNFR2− cDC2 at steady-state. n=3. B-C. Flow cytometry analysis of IRF4 and IRF8 (B) and Zbtb46 (C) in TNFR2+ and TNFR2− cDC2. n=3. D. Flow cytometry analysis in lungs of IRF4fl/flCD11ccre mice reconstituted with CD45.1+ pre-cDC2. n=3. pre-cDC2 were sorted from the bone marrow of B6.CD45.1 mice and transferred (i.n.) into IRF4fl/flCD11ccremice. n=3. E. Phenotypic analysis of CD45.1 TNFR2+ and TNFR2− cDC2 transferred into IRF4fl/flCD11ccre mice. n=3. F. Flow cytometry analysis of TNFR2 expression on cDC2 subpopulations transferred into MPYS−/− mice treated with CDG (i.n.) for 16hrs. n=3.
To further establish that the lung TNFR2+ and TNFR2− cDC2 subpopulations are cDC2, we adoptively transferred CD45.1 pre-cDC2 40, 42 into IRF4fl/flCD11ccre mice (Fig S8A-C). CD45.1+ cells were identified in lung five days after transfer and displayed a cDC2 phenotype (Fig 5D and S8C). Importantly, the CD45.1+ pre-cDC2 generated both TNFR2+ and TNFR2− lung cDC2 in the recipient mice (Fig 5D). The CD45.1+ TNFR2+ cDC2 expressed BTLA, PD-L1 and had mixed expression of PD-L2 and CD301b (Fig 5E), similar to the resident TNFR2+ cDC2. The CD45.1+ TNFR2− cDC2 only expressed CX3CR1 (Fig 5E). We concluded that the lung TNFR2+ and TNFR2− cDC2 arise from the cDC2 lineage and express distinct surface markers.
TNFR2+ and TNFR2− lung cDC2 do not represent different activation states
We next asked if the TNFR2+ and TNFR2− cDC2 populations represent different activation states of cDC2. We adoptively transferred CD45.1 lung TNFR2+ and TNFR2− cDC2 into MPYS−/− mice (Fig S8D). The recipient mice were then activated by CDG (i.n.). Consistent with our previous observation (Fig 3 & 4), TNFR2− cDC2 did not upregulate TNFR2 while TNFR2+ cDC2 maintained their expression of TNFR2 (Fig 5F). CDG treatment did not affect PD-L1 expression either (Fig 5F). Collectively, we concluded that lung cDC2 consist of two functionally and developmentally distinct subpopulations, TNFR2+ and TNFR2− cDC2.
CDG activates TNFR2-deficient cDC2 in vivo to produce TNF
Question remains how the immature TNFR2− cDC2 mediates CDG-induced antibody responses in vivo. We suspected that the TNFR2− cDC2, though not mature, might still be activated by CDG in vivo. CDG induces TNF production in vivo that is essential for its adjuvant activity 15–17. CDG-induced TNF production in vivo mainly depending on MPYS-expression in CD11c+ cells (Fig 6A) 17, specifically cDC2 as IRF4fl/flCD11ccre mice had dramatically decreased lung TNF (Fig S3C). We found that CDG induced lung TNF in TNFR2−/− mice (Fig 6B). Furthermore, cDC2 produced TNF in TNFR2−/− mice (Fig 6C). Deleting TBK1 in hematopoietic and endothelial lineages (TBK1fl/flVavCre) dramatically reduced CDG-induced lung TNF production (Fig 6D) suggesting TBK1 is needed for TNF production by CDG. Indeed, TBK1 was activated in the TNFR2−/− cDC2 (Fig 6E). Last, adoptive transferred TNFR2− cDC2 produced lung TNF in MPYS−/− lung (Fig 4B). We concluded that although TNFR2− cDC2 fail to mature, they were activated by CDG in vivo.
Figure 6. CDG activates TNFR2−/− cDC2 in vivo to produce TNF.

A-B. Indicated mice were administered (i.n.) with saline or CDG for 5hrs. TNF production was measured in lung homogenates by ELISA. n>3. C. WT and TNFR2−/− mice were treated with saline or CDG for 16 hours. TNF in lung cDC2 was determined by an intracellular cytokine stain. n=3. D. TBK1fl/fl and TBK1fl/flVavcre mice were administered (i.n.) with saline or CDG for 5hrs. TNF production was measured in lung homogenates by ELISA. n=3. E. Flow cytometry analysis of p-TBK1 expression in lung cDC2 from WT and TNFR2−/− mice treated with saline or CDG for 16 hours. n=3. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
Adoptive transfer of WT monocyte restored CDG adjuvant activity in TNFR2−/− mice
We next investigated how the activated, but immature, TNFR2− cDC2 mediate CDG-induced antibody responses. First, we found that moDCs failed to upregulate CD86 in IRF4fl/flCD11ccre mice following CDG treatment (Fig 7A). The CD86 expression in Batf3−/− mice was unaltered (Fig 7B). Second, adoptively transferred TNFR2−/− cDC2 induced CD86 expression on moDCs in IRF4fl/flCD11ccre mice (Fig 7C). Third, CDG induced CD86 expression on moDCs in RelBfl/flCD11ccre mice (Fig 7D). We concluded that cDC2, especially TNFR2− cDC2 promote CDG-induced moDCs maturation in vivo.
Figure 7. TNFR2 expression on moDCs is required for CDG adjuvant activity.

A-B. WT, IRF4fl/flCD11ccre (A) and Batf3−/− (B) mice were treated (i.n.) with saline or CDG (5µg) for 16 hours. CD86 expression in lung moDC were determined by Flow cytometry. n=3. C. WT and TNFR2−/− cDC2 were adoptively transferred into the IRF4fl/flCD11ccre mice. The mice were treated with CDG (i.n.) for 16hrs. CD86 expression on endogenous moDC were examined by Flow cytometry. n=3. D. Flow cytometry analysis of CD86 expression on lung moDCs in RelBfl/fl and RelBfl/flCD11cCre mice treated with CDG for 16hrs. n=3. E. Flow cytometry analysis of TNFR2 expression on lung moDCs of WT mice treated (i.n.) with saline or CDG for 16hrs.n=3. F. Flow cytometry analysis of CD86 expression on lung moDCs from WT or TNFR2−/−mice treated (i.n.) with saline or CDG for 16hrs. n=3. G. WT, TNFR2−/− or TNFR2−/− mice receiving (i.n.) WT monocytes or TNFR2−/− monoctyes were immunized with CDG/PspA. Serum anti-PspA IgG and BALF anti-PspA IgA were determined by ELISA. n=3. H. WT cDC2 adoptively transferred into TNFR2−/−mice were immunized with CDG/PspA. Serum anti-PspA IgG were determined by ELISA. n=3. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
Adoptively transferred TNFR2−/− cDC2 restored CDG responses in IRF4fl/flCD11ccre mice (Fig 3A). Yet, TNFR2−/− mice had no CDG responses (Fig 2A). We reasoned that TNFR2 expression on moDCs may be important for CDG responses in vivo. Indeed, we found that CDG induced TNFR2 on moDCs in WT mice (Fig 7E) and moDCs from TNFR2−/− mice did not upregulate CD86 in response to CDG in vivo (Fig 7F). Last, adoptive transfer of WT monocytes into TNFR2−/− mice restored CDG-induced IgG and IgA responses (Fig 7G). Notably, adoptive transfer WT cDC2 into TNFR2−/− mice did not restore CDG-induced antibody responses (Fig 7H). We concluded that moDCs expression of TNFR2 is critical for its maturation and subsequent induction of CDG adjuvant response.
CDG induces mTNF expression on TNFR2− cDC2 in vivo
Our data so far indicates that moDCs are matured by activated TNFR2− cDC2. Furthermore, moDC maturation requires cell intrinsic TNFR2 expression. Only mTNF can efficiently engage TNFR2 36. Both TNFR2+ and TNFR2− cDC2 produced TNF upon intranasal CDG treatment (Fig 4B). We asked if the lung TNFR2− cDC2 specifically expressed mTNF.
We intranasally administered CDG to WT mice for 16 hours and detected cell surface mTNF expression using TNFR2-Fc recombinant protein (Fig 8A). We found that the majority of lung mTNF+ DCs were cDC2 (Fig 8B). Remarkably, TNFR2− cDC2 were the main mTNF+ cDC2 cells in vivo while TNFR2+ cDC2 expressed little mTNF (Fig 8C).
Figure 8. TNFR2− cDC2 express mTNF but have few processed antigen.
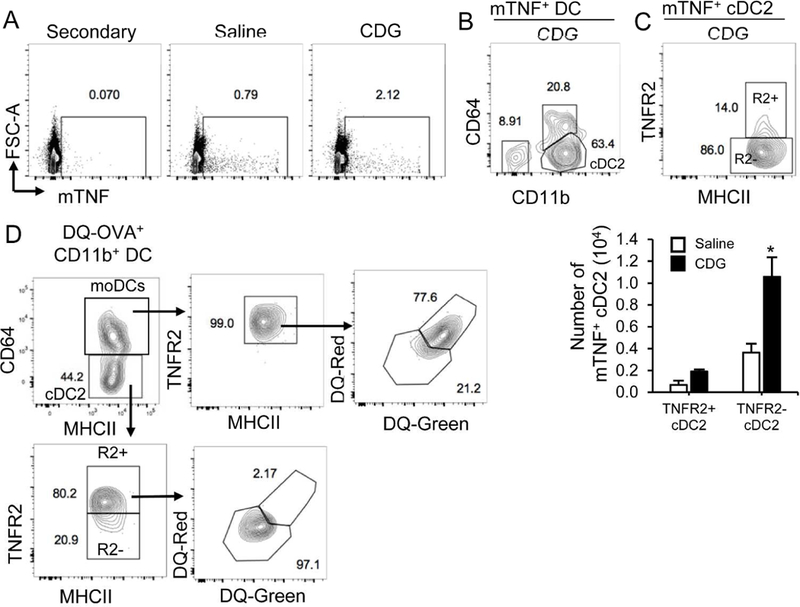
A. WT mice were administered with saline or CDG (5µg) for 16hrs. mTNF expression was determined by Flow cytometry using mouse TNFR2-Fc recombinant protein. n=3. B-C. Flow cytometry analysis of mTNF expression in lung DCs (B) and cDC2 (C) n=3. D. Flow cytometry analysis of antigen uptake and processing in lung CD11b+ DC from WT mice treated (i.n.) with DQ-OVA (20ug) and CDG (5ug) for 16 hours. n=3. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
moDCs, not TNFR2− cDC2, are very efficient in antigen processing in vivo
We next examined antigen processing in TNFR2− cDC2 in vivo. WT mice were intranasally administered with CDG/DQ™-OVA 17. DQ+ cells were examined in lung cDC2 and moDCs (Fig 8D). DQ™-OVA is a self-quenched conjugate of OVA exhibiting bright green fluorescence upon proteolytic degradation (DQ-Green). Furthermore, high concentration of digested fragments of DQ™-OVA accumulating in organelles form excimers that exhibits bright red fluorescence (DQ-Red). We found that DQ+ moDCs are mostly DQ-Red indicating a high concentration of processed antigens in moDCs. Conversely, DQ+ cDC2 were DQ-Green (Fig 8D). Strikingly, comparing to the TNFR2+ cDC2 subpopulation, very few TNFR2− cDC2 subpopulation were DQ+ (Fig 8D) suggesting that the TNFR2− cDC2 either did not take up antigen or were not efficient at antigen processing. In comparison, all DQ+ moDCs were TNFR2+ cells (Fig 8D) indicating TNFR2+ moDCs were indeed mature DCs.
moDCs promote CDG-induced Tfh and GC B cells generation in the lung
We next assessed how non-migratory moDCs (Fig 1D & S3B)25,33 promote CDG-induced antibody responses. moDCs were efficient at antigen processing (Fig 8D). We first asked if they presented antigen on cell surface. We intranasally administered C57BL/6 mice with CDG and Eα-OVA, and detected I-Ab/Eα+ cells with the YAE mAb. Indeed, CDG increased YAE+ moDCs in vivo (Fig 9A). Furthermore, the majority of YAE+ moDCs upregulated CD86 (Fig 9B), indicating their potential to activate CD4+ T cells.
Figure 9. moDCs promote CDG-induced Tfh and GC B cell generation in the lung.
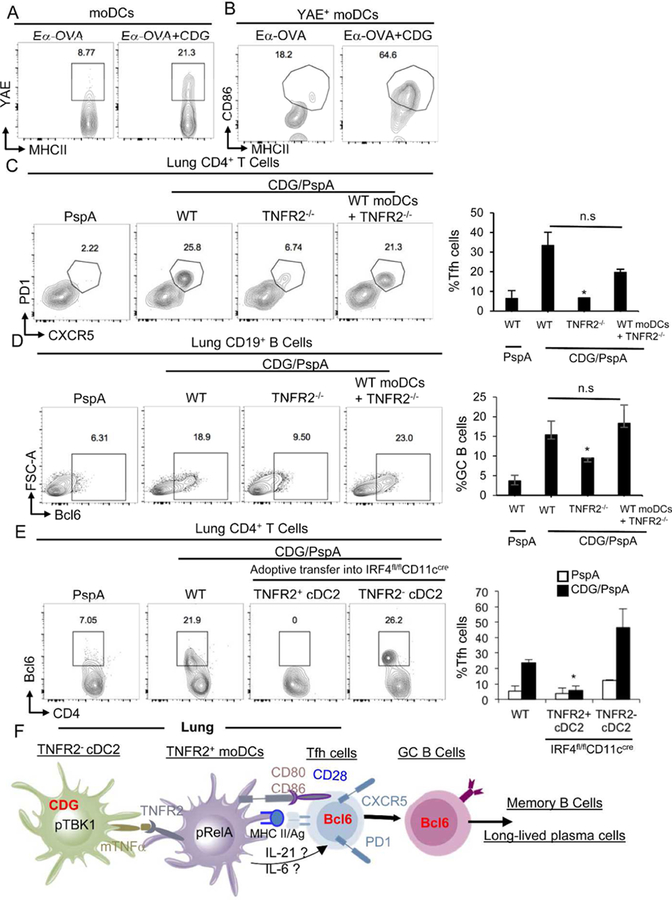
A-B. WT mice were administered with Ea-OVA (10µg) or Ea-OVA/CDG (5µg) for 16hrs. YAE+ moDCs (A) and CD86+YAE+ moDCs were determined by Flow cytometry. n=3. C-D. WT, TNFR2−/− or TNFR2−/− mice receiving (i.n.) WT monocytes were immunized with CDG/PspA. At Day 14, CD4+PD1+CXCR5+ Tfh (C) and CD19+ Bcl6+ B cells (D) were determine in lung by flow cytometry. n=3. E. TNFR2+ and TNFR2− cDC2 were adoptively transferred into IRF4fl/flCD11ccre mice and immunized with CDG/PspA. At Day 14, CD4+ Bcl6+ Tfh were determine by flow cytometry. n=3. F. Model: following CDG administration, TNFR2− cDC2 produce mTNF to activate moDCs, which will generate Tfh to mediate the antibody response. Graphs represent means ± standard error from three independent experiments. The significance is represented by and asterisk (*) where p<0.05 (unpaired Student’s t test).
Tfh cells and GC B cells play central roles in promoting humoral responses. We found that 14 days after CDG/PspA immunization (i.n.), lungs from the WT mice had PD1+CXCR5+Bcl6+ Tfh cells and Bcl6+ GC B cells (Fig 9C–9E). In contrast, TNFR2−/− mice were unable to generate lung Tfh or GC B cells (Fig 9C–9D). Importantly, adoptive transfer of WT monocytes into TNFR2−/− mice restored the generation of Tfh and GC B cells in the lung (Fig 9C–9D). We concluded that moDCs promote CDG-induced Tfh and GC B cells generation in the lung.
moDCs are activated by TNFR2− cDC2. Thus restoring TNFR2− cDC2 in the IRF4fl/flCD11ccre mice should restore Tfh cells. Indeed, we found that adoptive transfer of TNFR2− cDC2, but not TNFR2+ cDC2, into IRF4fl/flCD11ccre mice generated Tfh cells (Fig 9E). Together, we propose that CDG activates TNFR2− cDC2 that matures moDCs to generate Tfh and GC B cells promoting CDG-induced antibody responses in vivo (Fig 9F).
Discussion
In this report, we examined the mechanism by which lung DCs subsets mediate the mucosal adjuvant activity of CDG. The most exciting finding in this report is the identification of new lung cDC2 subpopulations and their unusual mode of action. cDC2 is a heterogeneous population 21, 32, 38, 39. We found that steady-state lung cDC2 have two distinct subpopulations TNFR2+pRelB+CX3CR1− and TNFR2−pRelB−CX3CR1+. Functionally, these two cDC2 subpopulations mediate the cellular and humoral immune responses to CDG adjuvant respectively. Developmentally, they derived from pre-cDC2 and do not represent different activation states of cDC2 in vivo.
Lung DC subsets are likely influenced by lung microenvironment 43. In a large scale of phenotypic and transcriptional profiling of human tissues DC subtypes, Heidkamp et. al., found that the phenotype of DCs is predominantly determined by ontogeny in the lymphoid organs whereas the phenotype of DCs is heavily influenced by the microenvironment in barrier tissues 43. The lung TNFR2− cDC2 express CX3CR1. Nakano H., et.al., showed that CX3CR1 promote pre-cDC migration to the lung at steady state 44. The lung TNFR2+pRelB+ cDC2 have not been described before. They have constitutively activated TNFR2-RelB signaling and lack mTNF themselves. We speculate that they react to modulatory signals (e.g. mTNF) from lung microenvironment that constitutively activates TNFR2-RelB. Tussiwand, R. T. et al., previously identified a Klf4-dependent SIRP-α+CD24+Mgl2+ cDC2 subpopulation that is required for Th2 response 39. A subpopulation of TNFR2+pRelB+ cDC2 have high expression of Mgl2/CD301b. More studies are needed to determine if Klf4 is required for their development
Intranasal administration of CDG leads to the maturation of the TNFR2+ cDC2 subpopulation, not the TNFR2− cDC2 subpopulation. Unexpectedly, TNFR2− cDC2 mediates the antibody responses of CDG. Why did not CDG activation mature lung TNFR2− cDC2 in vivo? NF-κB activation is essential for DC maturation. Different from cDC1 or TNFR2+ cDC2, CDG did not activate RelA or RelB in lung TNFR2− cDC2 in vivo. CDG did induce p-TBK1 in lung TNFR2− cDC2. TBK1 is critical for IRF3 activation and IFNβ production 45. However, it does not play a major role in NF-κB activation in vivo 46, 47. Previous studies, mostly done in vitro, showed that STING/MPYS pathway activates NF-κB, in particular, RelA 15, 48. Our results here indicated that the ability of STING/MPYS to engage NF-κB pathway is cell-type specific. In this lung resident TNFR2− cDC2, in vivo stimulation of STING/MPYS by CDG do not activate RelA or RelB.
How does the immature TNFR2− cDC2 promote CDG adjuvant responses in vivo? Our monocyte adoptive transfer experiment in TNFR2−/− mice showed that moDCs are critical for CDG adjuvant responses. moDCs do not directly take up intranasally administered CDG and STING expression in moDCs is dispensable. Instead, moDCs maturation requires TNFR2− cDC2 and the expression of TNFR2 on moDC. Though we can not rule out the possibility that mTNF on other cells interacts with TNFR2 on moDCs, we favor the model that mTNF on TNFR2−cDC2 engages TNFR2 on moDC to induce its maturation and subsequent CDG antibody responses.
Lung moDCs are non-migratory 25, 33. We showed that moDCs promote the generation of Tfh and GC B cells in lung suggesting the formation of the inducible bronchus-associated lymphoid tissue (iBALT). DCs are required for the formation of iBALT 49, 50. The exact lung DC subset for iBALT induction is unknown. moDCs presented antigen, expressed co-stimulator and activated RelA/RelB, which likely facilitate cytokine productions. We proposed that lung moDCs induce iBALT formation and promote CDG humoral responses.
In summary, we illustrated a previously unknown in vivo mode of action for CDG mucosal adjuvant whereby a new lung TNFR2− cDC2 subpopulation activated by CDG promoting the maturation of moDCs for the generation of Tfh cells. These findings will facilitate future mucosal vaccine development and DC research.
Methods
Mice
Eight to sixteen-week old mice, both males, and females, were used for experiments. All mice are on a C57BL/6 background. A detailed description of the lines can be found in the Supplemental Methods. Mice were housed and bred in the Animal Research Facility at the University of Florida. All experiments with mice were performed by the regulations and approval of the Institutional Animal Care and Use Committee from the University of Florida.
Intranasal CDG Immunization
A detailed description of intranasal vaccination and reagents can be found in the Supplemental Methods. Sera were collected 14 days after the last immunization. The PspA-specific Abs were determined by ELISA. To determine Ag-specific Th response, splenocytes and lung cells from PspA or CDG + PspA immunized mice were stimulated with 5µg/ml PspA for four days in culture. Th1, Th2, and Th17 cytokines were measured in the supernatant by ELISA.
Detection of Lung Cytokine Production
Mice were intranasally administered 5µg CDG, then sacrificed after 5hrs by CO2 asphyxiation 17. Lungs were harvested and lung cytokines was determined in lung homogenates. A detailed description of lung homogenates preparation can be found in the Supplemental Methods.
Isolation of lung cells
Mice were intranasally administered with or without CDG (5µg, vaccine-grade). After 20hrs, the lungs were lavaged, perfused with ice-cold PBS and harvested. A detailed description of lung cell isolation can be found in the Supplemental Methods.
In vivo Ag Uptake and Processing
Mice were intranasally administered 20µg DQ™-Ovalbumin (DQ-OVA) (Life technologies, D12053) with, or without CDG (5µg, vaccine-grade). After 20hrs, the lungs were lavaged, perfused and harvested.
Flow Cytometry and cell sorting
A detailed description of Flow antibodies used can be found in the Supplemental Methods. Cell sorting was performed on the BD FACSAriaIII Flow Cytometer and Cell Sorter. After sorting, dendritic cells were CFSE labeled, according to the protocol from the manufacturer (Invitrogen).
Intracellular staining
The intracellular cytokine staining was performed using the Cytofix/Cytoperm™ kit from BD Biosciences (cat#555028). Briefly, mice were intranasally administered saline or cyclic di-GMP (5µg, vaccine-grade). The single lung cell suspension was fixed in Cytofix/perm buffer (BD Biosciences) in the dark for 20min at RT. Fixed cells were then washed and kept in Perm/wash buffer at 4°C. Golgi-plug was present during every step before fixation.
Mouse cDC2 and monocyte purification
Primary mouse cDC2 (cat#18970A, Stemcell Technologies; cat# 480097, Biolegend) were purified from lungs of naïve mice following the protocol according to the manufacturer. Mouse monocytes (cat#19861, Stemcell Technologies) were purified from the bone marrow of naïve mice following the protocol according to the manufacturer.
Adoptive transfer
Lung TNFR2+ and TNFR2− cDC2 were sorted from the lungs of naïve donor mice with a FACSAriaIII flow cytometer. After sorting, dendritic cells were CFSE labeled, according to the protocol from the manufacturer (Invitrogen). Cells were administered intranasally into recipient mice. 24 hours later of transfer, recipient mice were intranasally vaccinated with CDG (5µg, Invivogen, cat# vac-cdg) adjuvanted PspA (2µg, BEI Resources) or PspA alone 17. Recipient mice received two doses of transferred cells and were immunized at 14 days interval.
For in vivo reconstitution of cDC2, pre-cDC2 were sorted from the bone-marrow of B6.CD45.1 naïve mice with a FACSAriaIII. pre-cDC2 were identified as Lin−MHCII−SiglecH−CD3−CD19−NK1.1−Ter119−CD11c+CD26+CD135+Ly6c+ 42. 250,000 cells were administered intranasally into recipient mice. DCs in the lung were analyzed on day 5 post transfer.
Statistical Analysis
All data are expressed as means ± SEM. Statistical significance was evaluated using Prism 5.0 software to perform a Student’s t-test (unpaired, two-tailed) for comparison between mean values.
Animals
MPYS−/− mice (Tmem173<tm1Camb>) have been described previously 1,2. The following strains were obtained from The Jackson Laboratory: B6-CD45.1 (#002014), Irf4fl (#009380)3, Batf3− (#013755)4, Tnfr1− (#002818)5, Tnfr2− (#002620)6, RelBfl (#028719)7, CD11Ccre (#008068)8. Vavcre-TBK1fl/fl mice were from Dr. Fitzgerald’s lab. FLT3−/− mice were from Dr. Miriam Merad’s lab.
Reagent
The following reagent was obtained through BEI Resources, NIAID, NIH: Streptococcus pneumoniae Family 1, Clade 2 Pneumococcal Surface Protein A (PspA UAB055) with C-Terminal Histidine Tag, Recombinant from Escherichia coli, NR-33178.
The following Abs from Biolegend were used in the flow cytometry: CD80 (16–10A1), CD86 (GL1), CD11B (M1/70), CD11C (N418), FcεRIa (MAR-1), MHC II (M5/114.15.2), CD103(2E7), CD24 (m1/69), CD64 (x54–5/7.1), CCR7 (4B12), TNF (MP6-XT22), TNFR2 (TR75–89), PD-L1 (10F.9G2), BTLA (8F4), PD-L2 (TY25), CD301b (URA-1), CX3CR1 (SA011F11), CD172a (P84), CD44 (IM7), Siglec-H (551), CD3 (145–2c11), CD19 (1D3/CD19), NK1.1 (PK136), Ter119 (TER-119), CD26 (H194–112), CD135 (A2F10), IRF4 (IRF4.3E4), PD1 (29F.1A12), CXCR5 (L138D7), CD4 (GK1.5), BCL6 (7D1). The following antibodies were from eBioscience: Ly6c (HK1.4) and YAE – Ea52–68 peptide bound to I-Ab (eBioY-Ae). IRF8 (V3GYWCH) antibody was from Thermofisher. The following Abs were from Cell Signaling: p-TBK1 (ser172, d52c2), p-RelB (ser552, d41b9), p-RelA (ser536, cat#4887S). The following reagents were from Sino Biological: TNFR2 Ab (cat# 50128-R112-A), TNFR2-Fc mouse TNFR2 extracellular domain, Met 1-Gly 258, was fused with the Fc region of human IgG1 at the C-terminus, cat# 50128-M02H), human IgG1 Fc (cat# 10702-HNAH). Alexa Fluor®488 mouse anti-human IgG1 Fc was from Invitrogen (cat# A-10631). FITC-CDG (2’-Fluo-AHC-CDG ) was from Biolog (cat# F009). Eα-OVA (ASFEAQGALANIAVDKA-OVA) from Genecust.
Lung digestion
The lungs were lavaged, perfused, and harvested at 5hr post-treatment. Excised lungs were washed in PBS and digested in DMEM containing 200µg/ml DNase I (Roche, 10104159001), 25µg/ml Liberase TM (Roche, 05401119001), at 37°C for 3hrs. Red blood cells were then lysed and a single cell suspension was prepared and analyzed by BD™ LSR II and FACScan flow cytometry.
Measure cytokines in lung homogenates
Lungs were perfused with cold PBS. The harvested lungs were washed with PBS once, then stored in 0.7ml Tissue protein extraction reagent (T-PER) (Thermo Scientific, cat#78510) containing protease inhibitors (Roche, cat#11836153001) at −80°C. Later, the lung was thawed on ice and homogenized with Minilys® (Precellys, 5,000 RPM for 30sec) using Precellys lysing kit (Precellys, cat# KT03961). Lung homogenates were transferred to a 1.5ml tube and spun at 14,000g for 30min at 4°C. The supernatant was collected and analyzed for TNF production by ELISA (eBioscience, cat#88–7324).
Intranasal immunization
Groups of mice (4 per group) were intranasally vaccinated with CDG (5µg, Invivogen, cat# vac-cdg) adjuvanted PspA (2µg, BEI Resources) or PspA alone 9. Mice were immunized twice at 14 days interval. For intranasal vaccination, animals were anesthetized using isoflurane in an E-Z Anesthesia system (Euthanex Corp, Palmer, PA). PspA, with or without CDG was administered in 20µl saline. Secondary Abs used were anti-mouse IgG1-HRP (Southern Biotech, cat#1070– 05), anti-mouse IgG2C-HRP (Southern Biotech, cat#1079–05), and anti-mouse IgA-HRP (Southern Biotech, cat#1040–05).
In vivo TNFR2 neutralization
Mice were first given CDG (i.n.). Half an hour later, treated mice were administered (i.n.) with 50µg/50µl anti-TNFR2 Ab (BioLegend, TR75–32.4), or isotype control (BioLegend, HTK888). Lung was harvested 16hr later and analyzed by Flow cytometer.
Supplementary Material
Acknowledgments:
We thank Howie Seay of Center for Immunology and Transplantation at the University of Florida for helps. We thank Jessica le Berichel and Dr. Miriam Merad of Mount Sinai for the Flt3−/− lung. This work was supported by NIH grants AI110606 and AI125999 (to L.J.) and a new faculty start-up fund from University of Florida, Department of Medicine (to L.J). S.S was supported by NIH Training grant T32 AI095213.
Footnotes
Disclosure: The authors declare no conflict of interests.
References:
- 1.Libanova R, Becker PD, Guzman CA. Cyclic di-nucleotides: new era for small molecules as adjuvants. Microb Biotechnol 2012; 5(2): 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ebensen T, Schulze K, Riese P, Morr M, Guzman CA. The bacterial second messenger cdiGMP exhibits promising activity as a mucosal adjuvant. Clin Vaccine Immunol 2007; 14(8): 952–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray PM, Forrest G, Wisniewski T, Porter G, Freed DC, DeMartino JA et al. Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities. Cell Immunol 2012; 278(1–2): 113–119. [DOI] [PubMed] [Google Scholar]
- 4.Madhun AS, Haaheim LR, Nostbakken JK, Ebensen T, Chichester J, Yusibov V et al. Intranasal c-di-GMP-adjuvanted plant-derived H5 influenza vaccine induces multifunctional Th1 CD4+ cells and strong mucosal and systemic antibody responses in mice. Vaccine 2011; 29(31): 4973–4982. [DOI] [PubMed] [Google Scholar]
- 5.Zhao L, KuoLee R, Harris G, Tram K, Yan H, Chen W. c-di-GMP protects against intranasal Acinetobacter baumannii infection in mice by chemokine induction and enhanced neutrophil recruitment. Int Immunopharmacol 2010; 11(9): 1378–1383. [DOI] [PubMed] [Google Scholar]
- 6.Hu DL, Narita K, Hyodo M, Hayakawa Y, Nakane A, Karaolis DK. c-di-GMP as a vaccine adjuvant enhances protection against systemic methicillin-resistant Staphylococcus aureus (MRSA) infection. Vaccine 2009; 27(35): 4867–4873. [DOI] [PubMed] [Google Scholar]
- 7.Karaolis DK, Newstead MW, Zeng X, Hyodo M, Hayakawa Y, Bhan U et al. Cyclic di-GMP stimulates protective innate immunity in bacterial pneumonia. Infect Immun 2007; 75(10): 4942–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogunniyi AD, Paton JC, Kirby AC, McCullers JA, Cook J, Hyodo M et al. c-di-GMP is an effective immunomodulator and vaccine adjuvant against pneumococcal infection. Vaccine 2008; 26(36): 4676–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan H, KuoLee R, Tram K, Qiu H, Zhang J, Patel GB et al. 3’,5’-Cyclic diguanylic acid elicits mucosal immunity against bacterial infection. Biochem Biophys Res Commun 2009; 387(3): 581–584. [DOI] [PubMed] [Google Scholar]
- 10.Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest 2017; 127(6): 2176–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol Immunother 2015; 64(8): 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011; 478(7370): 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dempsey A, Bowie AG. Innate immune recognition of DNA: A recent history. Virology 2015; 479-480: 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 2014; 32: 461–488. [DOI] [PubMed] [Google Scholar]
- 15.Blaauboer SM, Gabrielle VD, Jin L. MPYS/STING-mediated TNF-alpha, not type I IFN, is essential for the mucosal adjuvant activity of (3’−5’)-cyclic-di-guanosine-monophosphate in vivo. J Immunol 2014; 192(1): 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest 2015; 125(6): 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaauboer SM, Mansouri S, Tucker HR, Wang HL, Gabrielle VD, Jin L. The mucosal adjuvant cyclic di-GMP enhances antigen uptake and selectively activates pinocytosis-efficient cells in vivo. Elife 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2012; 30: 1–22. [DOI] [PubMed] [Google Scholar]
- 19.Baratin M, Foray C, Demaria O, Habbeddine M, Pollet E, Maurizio J et al. Homeostatic NF-kappaB Signaling in Steady-State Migratory Dendritic Cells Regulates Immune Homeostasis and Tolerance. Immunity 2015; 42(4): 627–639. [DOI] [PubMed] [Google Scholar]
- 20.Crowley M, Inaba K, Witmer-Pack M, Steinman RM. The cell surface of mouse dendritic cells: FACS analyses of dendritic cells from different tissues including thymus. Cell Immunol 1989; 118(1): 108–125. [DOI] [PubMed] [Google Scholar]
- 21.Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity 2014; 40(5): 642–656. [DOI] [PubMed] [Google Scholar]
- 22.Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L et al. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med 1992; 176(1): 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langlet C, Tamoutounour S, Henri S, Luche H, Ardouin L, Gregoire C et al. CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J Immunol 2012; 188(4): 1751–1760. [DOI] [PubMed] [Google Scholar]
- 24.Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol 2012; 42(12): 3150–3166. [DOI] [PubMed] [Google Scholar]
- 25.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013; 38(2): 322–335. [DOI] [PubMed] [Google Scholar]
- 26.Holt PG, Schon-Hegrad MA, McMenamin PG. Dendritic cells in the respiratory tract. Int Rev Immunol 1990; 6(2–3): 139–149. [DOI] [PubMed] [Google Scholar]
- 27.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 2013; 38(5): 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha-dendritic cell development. Proc Natl Acad Sci U S A 2004; 101(24): 8981–8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol 2014; 15(2): 161–167. [DOI] [PubMed] [Google Scholar]
- 30.Bajana S, Turner S, Paul J, Ainsua-Enrich E, Kovats S. IRF4 and IRF8 Act in CD11c+ Cells To Regulate Terminal Differentiation of Lung Tissue Dendritic Cells. J Immunol 2016; 196(4): 1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008; 322(5904): 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy KM. Transcriptional control of dendritic cell development. Adv Immunol 2013. 239–267. [DOI] [PubMed]
- 33.Nakano H, Burgents JE, Nakano K, Whitehead GS, Cheong C, Bortner CD et al. Migratory properties of pulmonary dendritic cells are determined by their developmental lineage. Mucosal Immunol 2013; 6(4): 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol 1999; 331–367. [DOI] [PubMed]
- 35.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ 2003; 10(1): 45–65. [DOI] [PubMed] [Google Scholar]
- 36.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995; 83(5): 793–802. [DOI] [PubMed] [Google Scholar]
- 37.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A 1998; 95(2): 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014; 343(6172): 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 2015; 42(5): 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016; 45(3): 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M et al. IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 2016; 45(3):626–640. [DOI] [PubMed] [Google Scholar]
- 42.Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 2015; 16(7): 718–728. [DOI] [PubMed] [Google Scholar]
- 43.Heidkamp GF, Sander J, Lehmann CHK, Heger L, Eissing N, Baranska A et al. Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Sci Immunol 2016; 1(6). [DOI] [PubMed] [Google Scholar]
- 44.Nakano H, Lyons-Cohen MR, Whitehead GS, Nakano K, Cook DN. Distinct functions of CXCR4, CCR2, and CX3CR1 direct dendritic cell precursors from the bone marrow to the lung. J Leukoc Biol 2017; 101(5): 1143–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 2003; 4(5): 491–496. [DOI] [PubMed] [Google Scholar]
- 46.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 2004; 199(12): 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 2007; 13(11): 460–469. [DOI] [PubMed] [Google Scholar]
- 48.Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 2014; 88(10): 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med 2009; 206(11): 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S et al. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J Exp Med 2009; 206(12): 2593–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES
- 1.Jin L et al. STING/MPYS Mediates Host Defense against Listeria monocytogenes Infection by Regulating Ly6Chi Monocyte Migration. J Immunol 190, 2835–2843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin L et al. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 187, 2595–2601, doi: 10.4049/jimmunol.1100088 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein U et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol 7, 773–782, doi: 10.1038/ni1357 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Hildner K et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100, doi: 10.1126/science.1164206 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeffer K et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73, 457–467 (1993). [DOI] [PubMed] [Google Scholar]
- 6.Erickson SL et al. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 372, 560–563, doi: 10.1038/372560a0 (1994). [DOI] [PubMed] [Google Scholar]
- 7.De Silva NS, Silva K, Anderson MM, Bhagat G & Klein U Impairment of Mature B Cell Maintenance upon Combined Deletion of the Alternative NF-kappaB Transcription Factors RELB and NF-kappaB2 in B Cells. J Immunol 196, 2591–2601, doi: 10.4049/jimmunol.1501120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caton ML, Smith-Raska MR & Reizis B Notch-RBP-J signaling controls the homeostasis of CD8-dendritic cells in the spleen. J Exp Med 204, 1653–1664, doi: 10.1084/jem.20062648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaauboer SM et al. The mucosal adjuvant cyclic di-GMP enhances antigen uptake and selectively activates pinocytosis-efficient cells in vivo. Elife 4, doi: 10.7554/eLife.06670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.