

Abstract
Amyotrophic lateral sclerosis, the most common neurodegenerative disease affecting motor neurons, lacks an effective treatment. A small fraction of amyotrophic lateral sclerosis cases have a familial origin, related to mutations in causative genes, while the vast majority of amyotrophic lateral sclerosis cases are considered to be sporadic, resulting from the interaction between genes and environmental factors in predisposed individuals. During the past few years, dozens of drugs have been postulated as promising strategies for the disease after showing some beneficial effects in preclinical cellular and murine models. However, the translation into clinical practice has been largely unsuccessful and the compounds failed when were tested in clinical trials. This might be explained, at least partially, by the enormous complexity of the disease both from clinico-epidemiological and a pathogenic points of view. In this review, we will briefly comment on the complexity of the disease focusing on some recent findings, and we will suggest how amyotrophic lateral sclerosis research might be reoriented to foster the advance in the diagnostic and therapeutic questions.
Keywords: amyotrophic lateral sclerosis, ALS, environment, epidemiology, genes, phenotype, therapy
Amyotrophic lateral sclerosis (ALS) is the most common neurodegenerative disease affecting motor neurons (Riancho et al. 2016c; Zufiría et al. 2016). With an annual incidence that ranges from 1 to 2 cases per 100,000 habitants, it is characterised by progressive muscle wasting which usually leads to death during the first 3 years after diagnosis, commonly related to respiratory failure (Riancho et al. 2016c; Zufiría et al. 2016). More than 90 percent of cases are thought to be sporadic ALS, in which the disease likely results from the interaction between individual genetic predisposition, environment and aging (Figure 1) (Riancho et al., 2016b). Currently, riluzole and more recently edaravone, both with a very modest effect on survival, are the only approved drugs for the disease. Therefore, there is a need to find new therapies for the disease. For preparing this article, we reviewed the literature and selected relevant studies published from 2000 to 2018, prioritizing the most recent ones.
Figure 1.

Main involved components in the genesis of amyotrophic lateral sclerosis (ALS).
Each individual has a determined prenatal genetic load and during life accumulates a number of environmental exposures and some degree of age-related cell damage. Regardless the particular “weight” of each one of these components, ALS would develop when the sum of them reach a certain threshold. (A) Healthy subject, (B) sporadic ALS.
During the past few years, dozens of drugs have been postulated as promising strategies for the disease (Riancho et al., 2016a; Al Chalabi et al., 2017). However, despite the fascinating results in basic research with both neuronal cells and murine ALS models, there have existed important difficulties in the translation of these findings into patients with ALS, since all these new compounds failed when tested in clinical trials with ALS patients (Ittner et al., 2015). A potential explanation for this failure might depend on the enormous complexity of the disease.
First, from a clinical point of view, ALS is a complex syndrome with a variable phenotype. Both clinical onset patterns and survival rates markedly differ between the different forms (classic spinal onset, bulbar, flailed-arm, pseudomyopathic) of the disease (Al Chalabi et al., 2016). This, as well as the absence of useful validated biomarkers, substantially difficult the correct stratification of patients in clinical trials, therefore masking potential positive outcomes in some groups of patients (Brown and Al Chalabi, 2017). In recent years, an extensive list of potential biomarkers has been studied (Benatar et al., 2016), including biological fluid-based biomarkers, electrophysiological biomarkers and neuroimaging biomarkers. Within the first category, phosphorylated neurofilament heavy and neurofilament light seem to be the most promising ones. Based on published data, phosphorylated neurofilament heavy and neurofilament light levels in cerebrospinal fluid (and potentially in plasma) may have a role as prognostic biomarkers. Thus, they could be used in clinical trials to facilitate the stratification of study participants into treatment arms on the basis of anticipated rates of disease progression (Boylan et al., 2013). Regarding electrophysiological biomarkers, the motor unit number index (MUNIX) is gaining relevance (Nandedkar et al., 2004). MUNIX is a relatively easy technique which would theoretically allow to capture disease progression very early in the disease course, at a time when common neurophysiological parameters CMAP remained relatively stable. Unfortunately, this potential benefit is offset by the fact that repeatability of MUNIX is usually lowest early in the disease and improves only as the disease progresses into advanced stages (Felice, 1997; Bromberg, 2007). Among neuroimaging biomarkers, structural imaging analysis such as voxel based morphometry consistently show atrophy in the precentral gyrus in ALS patients compared to healthy volunteers (Menke et al., 2014). MRI tractography has also been evaluated revealing changes at corticospinal tract as disease progresses (Keil et al., 2012). However, further studies are needed to validate these results. Finally, fluorodeoxyglucose-positron emission tomography studies have revealed hypometabolism in the precentral girus and frontal regions in ALS subjects (Van Laere et al., 2014) and it may be an independent predictor of shorter survival (Van Laere et al., 2014).
It can be anticipated that a combination of validated humoral, electrophysiologic and imaging biomarkers will probably provide a dynamic understanding of the disease and its progression as well as the eventual response to a particular therapy more firmly that any single biomarker (Benatar et al., 2016).
Second, from an epidemiological point of view, there is a rapid-growing list of environmental factors that have been associated to ALS (Riancho et al., 2018). In addition to the classical ones, such as exposure to heavy metals, toxicants as paraformaldehyde or cyanotoxins, new categories of environmental factors and lifestyle characteristic are being including in that list. In recent years, some microorganisms, particularly retroviruses and gut microbiota dysregulation, have been associated with the genesis of the disease (Castanedo-Vazquez et al., 2018). Furthermore, lifestyle and other demographic parameters such as physical activity, nutrition, body mass index, cardiovascular risk factors, autoimmune diseases and cancer are being associated to the disease. Differently from most other neurodegenerative diseases, increased body mass index and metabolic syndrome have been related to an increased risk of ALS (Gallo et al., 2013). The association with autoimmunity and cancer is also intriguing: patients with previous autoimmune diseases and oncological history have been reported to have increased and decreased risk of ALS, respectively (Turner et al., 2013; Gibson et al., 2016). New research about the associations between ALS and comorbid conditions will doubtlessly provide new perspectives of the disease and will help us to better understand it (Riancho et al., 2018).
Third, from a molecular perspective, ALS should also be considered as a very complex condition. Familial forms of the disease, which represent about 10 percent cases, appear as “pure” forms, directly derived from mutations not only in the genes encoding neuron-damaging proteins, but also in genes that are involved in a variety of cellular functions, including RNA processing, autophagy, vesicular transport and energy metabolism (Zufiría et al., 2016). The mechanisms leading to disease in sporadic cases may be much more complex and at least as heterogeneous as those in familial forms. In any case, once the pathogenic process has been initiated, a plethora of metabolic and other cellular alterations precipitate a vicious cycle in ALS and other neurodegenerative diseases favouring disease progression (Zufiría et al., 2016). In other words, abnormalities in neuronal functions are often related to the combination of alterations at several levels, including alterations of gene processing (impaired DNA/RNA regulation, epigenetic aberrations), increased oxidative stress, mitochondrial dysfunction, proteostasic abnormalities, energetic imbalance, axonal transport deficits and glial cell dysfunction (Riancho et al., 2016b).
Based on this perception of the disease, several disease models, including cellular models and more complex animal models have been created by using molecular biology tools. Unfortunately, after accumulating a huge amount of data, experimental outcomes have not been translated into effective preventive or therapeutic strategies. This lack of success might respond to several reasons. Perhaps, we should consider that extrapolations and inferences may have been made in a too simplistic manner, as animal models of the disease commonly reproduce just a particular feature, rather than the whole process. In addition, we may have not paid enough attention to other factors of the internal and external environmental that influence the disease course.
If we do agree with the idea that in ALS there are several pathways involved, with multiple interactions building a complex pathogenic network, we should consider a modern perspective about causation (Rothman and Greenland, 2005). In this model, a causative factor could be unique (such as mutation in a causative gene) but more frequently could also be composed of several causal components that could be grouped in different manners constituting different sufficient causes (which may or may not share some of the component causes). When a component cause is present in all the sufficient causes it must be considered a necessary cause (Rothman and Greenland, 2005). It seems clear that according to this model, research focused on just one or two component causes will continue yielding very partial results with serious difficulties in reformulating a theory that generates valid predictive models.
These focused approaches derive, at least partially, from the structure of research funding programs. According to this new concept, research funding strategies should firmly support integrated programs of investigation considering ALS as a global neurodegenerative process rather than the result of a variety of isolated pathogenic events. These integrated projects should doubtlessly involve multi-disciplinary groups that focus together on the same problem by using different models and analytical tools, but always subordinated to a common hypothesis (Figure 2).
Figure 2.
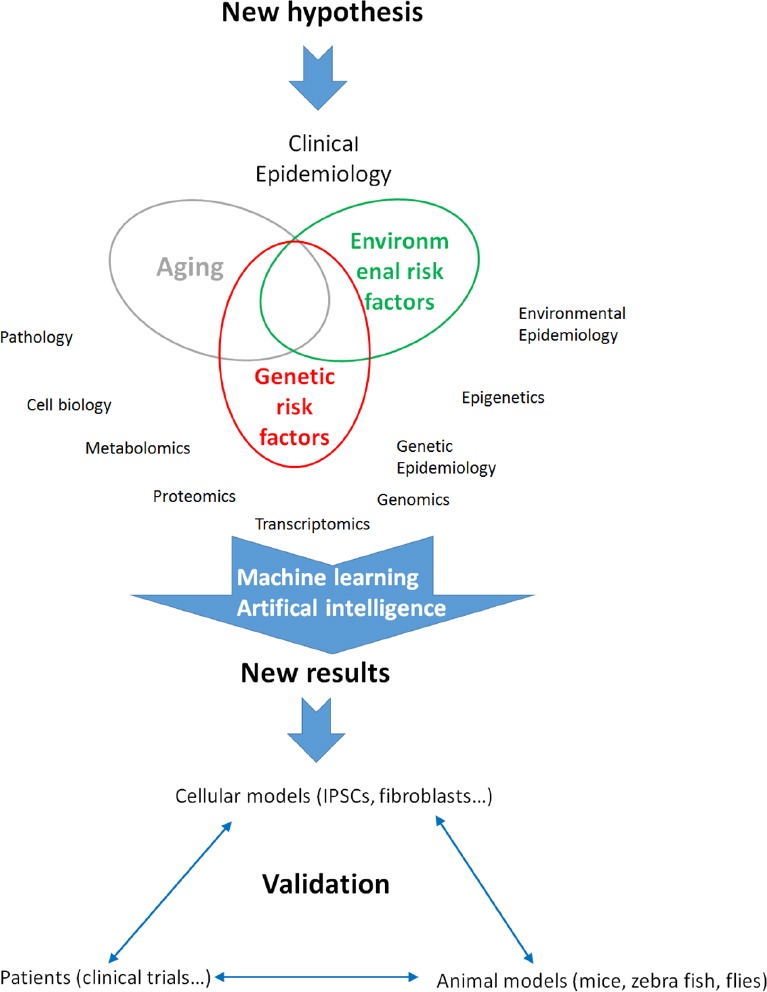
An integrated vision of amyotrophic lateral sclerosis research.
Integrated projects must include multi-disciplinary groups that focus together on the same problem by using different models and analytical tools, being always subordinated to a common hypothesis. IPSCs: Induced pluripotent stem cells.
In conclusion, we think that a multidisciplinary holistic approach to ALS-related questions is needed to better understand the pathogenesis of the disease and advance into more effective therapies. An open, honest and cooperative relationship between academia and industry, as well as between industry and health regulatory authorities will foster translational research programs leasing to effective therapies.
Footnotes
Conflicts of interest: All authors declare that they have no conflicts of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Yu J; T-Editor: Liu XL
References
- 1.Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016;15:1182–1194. doi: 10.1016/S1474-4422(16)30199-5. [DOI] [PubMed] [Google Scholar]
- 2.Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13:96–104. doi: 10.1038/nrneurol.2016.182. [DOI] [PubMed] [Google Scholar]
- 3.Benatar M, Boylan K, Jeromin A, Rutkove SB, Berry J, Atassi N, Bruijn L. ALS biomarkers for therapy development: State of the field and future directions. Muscle Nerve. 2016;53:169–182. doi: 10.1002/mus.24979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boylan KB, Glass JD, Crook JE, Yang C, Thomas CS, Desaro P, Johnston A, Overstreet K, Kelly C, Polak M, Shaw G. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as a potential prognostic biomarker in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2013;84:467–472. doi: 10.1136/jnnp-2012-303768. [DOI] [PubMed] [Google Scholar]
- 5.Bromberg MB. Updating motor unit number estimation (MUNE) Clin Neurophysiol. 2007;118:1–8. doi: 10.1016/j.clinph.2006.07.304. [DOI] [PubMed] [Google Scholar]
- 6.Brown RH, Jr, Al Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602. doi: 10.1056/NEJMc1710379. [DOI] [PubMed] [Google Scholar]
- 7.Castanedo-Vazquez D, Bosque-Varela P, Sainz-Pelayo A, Riancho J. Infectious agents and amyotrophic lateral sclerosis: another piece of the puzzle of motor neuron degeneration. J Neurol. 2018 doi: 10.1007/s00415-018-8919-3. doi: 10.1007/s00415-018-8919-3. [DOI] [PubMed] [Google Scholar]
- 8.Felice KJ. A longitudinal study comparing thenar motor unit number estimates to other quantitative tests in patients with amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:179–185. doi: 10.1002/(sici)1097-4598(199702)20:2<179::aid-mus7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 9.Gallo V, Wark PA, Jenab M, Pearce N, Brayne C, Vermeulen R, Andersen PM, Hallmans G, Kyrozis A, Vanacore N, Vahdaninia M, Grote V, Kaaks R, Mattiello A, Bueno-de-Mesquita HB, Peeters PH, Travis RC, Petersson J, Hansson O, Arriola L, et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: the EPIC cohort. Neurology. 2013;80:829–838. doi: 10.1212/WNL.0b013e3182840689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson SB, Abbott D, Farnham JM, Thai KK, McLean H, Figueroa KP, Bromberg MB, Pulst SM, Cannon-Albright L. Population-based risks for cancer in patients with ALS. Neurology. 2016;87:289–294. doi: 10.1212/WNL.0000000000002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ittner LM, Halliday GM, Kril JJ, Götz J, Hodges JR, Kiernan MC. FTD and ALS--translating mouse studies into clinical trials. Nat Rev Neurol. 2015;11:360–366. doi: 10.1038/nrneurol.2015.65. [DOI] [PubMed] [Google Scholar]
- 12.Keil C, Prell T, Peschel T, Hartung V, Dengler R, Grosskreutz J. Longitudinal diffusion tensor imaging in amyotrophic lateral sclerosis. BMC Neurosci. 2012;13:141. doi: 10.1186/1471-2202-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menke RA, Körner S, Filippini N, Douaud G, Knight S, Talbot K, Turner MR. Widespread grey matter pathology dominates the longitudinal cerebral MRI and clinical landscape of amyotrophic lateral sclerosis. Brain. 2014;137:2546–2555. doi: 10.1093/brain/awu162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nandedkar SD, Nandedkar DS, Barkhaus PE, Stalberg EV. Motor unit number index (MUNIX) IEEE Trans Biomed Eng. 2004;51:2209–2211. doi: 10.1109/TBME.2004.834281. [DOI] [PubMed] [Google Scholar]
- 15.Riancho J, Berciano MT, Ruiz-Soto M, Berciano J, Landreth G, Lafarga M. Retinoids and motor neuron disease: Potential role in amyotrophic lateral sclerosis. J Neurol Sci. 2016a;360:115–120. doi: 10.1016/j.jns.2015.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riancho J, Bosque-Varela P, Perez-Pereda S, Povedano M, de Munaín AL, Santurtun A. The increasing importance of environmental conditions in amyotrophic lateral sclerosis. Int J Biometeorol. 2018;62:1361–1374. doi: 10.1007/s00484-018-1550-2. [DOI] [PubMed] [Google Scholar]
- 17.Riancho J, Gonzalo I, Ruiz-Soto M, Berciano J. Why do motor neurons degenerate? Actualization in the pathogenesis of amyotrophic lateral sclerosis. Neurologia. 2016b doi: 10.1016/j.nrl.2015.12.001. doi: 10.1016/j.nrl.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Riancho J, Lozano-Cuesta P, Santurtún A, Sánchez-Juan P, López-Vega JM, Berciano J, Polo JM. Amyotrophic lateral sclerosis in northern spain 40 years later: what has changed? Neurodegener Dis. 2016c;16:337–341. doi: 10.1159/000445750. [DOI] [PubMed] [Google Scholar]
- 19.Rothman KJ, Greenland S. Causation and causal inference in epidemiology. Am J Public Health. 2005;95(Suppl 1):S144–S150. doi: 10.2105/AJPH.2004.059204. [DOI] [PubMed] [Google Scholar]
- 20.Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology. 2013;81:1222–1225. doi: 10.1212/WNL.0b013e3182a6cc13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, Robberecht W, Van Damme P. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. 2014;71:553–561. doi: 10.1001/jamaneurol.2014.62. [DOI] [PubMed] [Google Scholar]
- 22.Zufiría M, Gil-Bea FJ, Fernández-Torrón R, Poza JJ, Muñoz-Blanco JL, Rojas-García R, Riancho J, de Munain AL. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog Neurobiol. 2016;142:104–129. doi: 10.1016/j.pneurobio.2016.05.004. [DOI] [PubMed] [Google Scholar]