

Abstract
Oxygen is essential to the human life and life of all aerobic organisms. The complete oxidation of nutrients for the biological energy supply is one of the most important prerequisites for the formation of higher life forms. However, cells that benefit from oxidative respiration also suffer from reactive oxygen species because they adapted to oxygen as an energy source. Healthy cells balance the formation and elimination of reactive oxygen species thereby creating and keeping reactive oxygen species-homeostasis. When the concentration of free radicals exceeds a critical level and homeostasis is disturbed, oxidative stress occurs leading to damage of multiple cellular molecules and compartments. Therefore, oxidative stress plays an important role in the physiology and pathology of various diseases. Often, the antioxidant protection system becomes pathologically unbalanced in the genesis of several diseases, leading to functional losses of the organism, as in the case of amyotrophic lateral sclerosis, or cells develop metabolic mechanisms to use this system as protection against external influences, such as in the case of glioblastoma cells. Either way, understanding the underlying deregulated mechanisms of the oxidative protection system would allow the development of novel treatment strategies for various diseases. Thus, regardless of the direction in which the reactive oxygen species-homeostasis disequilibrate, the focus should be on the oxidative protection system.
Keywords: neurodegenerative disease, amyotrophic lateral sclerosis, cancer, glioblastoma, reactive oxygen species, metabolism, antioxidant protection system
Oxygen is essential to the human life and life of all aerobic organisms. The complete oxidation of nutrients for biological energy supply is one of the most important prerequisites for the formation of higher life forms. However, the cells that benefit from oxidative respiration have also been burdened by the reactive oxygen species (ROS) since adaptation to oxygen as an energy source. Oxygen radicals are continuously formed in aerobic organisms. For example, free radicals are involved in the synthesis of some hormones. Leukocytes and macrophages use free radicals as part of the respiratory burst to destroy foreign bodies such as bacteria and viruses. Furthermore, the role of ROS as a signaling and modulator molecule, as well as a transcriptional regulator, has come to the fore in recent years. Therefore, it can be assumed that ROS is part of a healthy metabolism in limited concentrations. Healthy cells balance the formation and elimination of ROS thereby creating and keeping ROS-homeostasis. When the concentration of free radicals exceeds a critical level and the homeostasis is disturbed oxidative stress occurs. Upon oxidative stress, the free radicals of oxygen and nitrogen can cause damage to cells, cell organelles, and components, such as lipids, proteins, and DNA. These damages, if unrepaired, can lead to cell death. Oxidative changes play a role in the pathogenesis of many diseases, for example in diabetes, cardiovascular and neurodegenerative diseases, such as amyotrophic lateral sclerosis, but also in chronic inflammatory diseases, amyloidosis, autoimmune processes and cancer (Cacciapuoti, 2016). Recent studies revealed also an emerging role of oxidative stress or decreased antioxidant defenses in migraine pathogenesis, which was partly associated with the mitochondrial dysfunction (Ferroni et al., 2018). To counteract this potential threat of oxidative stress, eukaryotic cells have developed protective systems that attempt to prevent the development of free radicals in parallel with the evolutionary adaptation to the oxygen-rich atmosphere by the uptake of the oxidative metabolizing mitochondria. These oxidation protection systems have in common that they can prevent or delay oxidative damage and thereby help keeping the balance between the malignant or healthy stage in various diseases. In order to fulfill this task, oxidation protection systems need to meet several requirements. We have performed a PubMed literature search of articles published in the period January 1969–September 2018 on the molecular mechanisms of oxidative stress in neurodegenerative diseases and cancer.
The requirements for a substance to have an antioxidant effect are: i) reactivity with biologically relevant oxidants and radicals, ii) availability in sufficient concentration and iii) reaction product of the substance itself is less harmful than the oxidant. The mechanisms of action consist of trapping reactive oxygen compounds, minimizing the oxygen compounds, repairing or replacing damaged target molecules. However, this different possibilities for elimination of ROS require variable antioxidant protection systems.
The antioxidant protection system of the cell can be divided into antioxidant molecules and antioxidant enzymes. The antioxidant molecules of the cell include endogenous substances such as glutathione, uric acid, ubiquinone (coenzyme Q), lipoic acid and bilirubin, or food antioxidant molecules such as ascorbic acid (vitamin C), tocopherols and tocotrienols such as α-tocopherol (vitamin E), carotenoids and flavonoids. Nevertheless, despite the variety of existing antioxidant protection systems within the cell, they all have to fulfill the above-mentioned requirements.
The tripeptide glutathione (GSH, γ-glutamylcysteinylglycine) is the cell’s most important antioxidant molecule found in mammalian cells at concentrations up to 12 mM. Mitochondria contain about 10% to 15% of the total glutathione of the cell. On the one hand GSH reacts directly in non enzymatic reactions with radicals, on the other hand GSH acts as an electron donor in the reduction of peroxides catalyzed by glutathione peroxidase. Regulation of GSH synthesis proceeds by γ-glutamylcysteine-synthetase via a feedback mechanism. Oxidized glutathione disulfide can be regenerated by glutathione reductase, which uses nicotinamide adenine dinucleotide phosphate (NADPH) as the reducing agent.
Vitamin C and Vitamin E are the most important antioxidants that can be absorbed through diet. By forming the stable ascorbyl radical, vitamin C can release an electron to radicals in the cytosol of a cell. Radicals that attack membranes are trapped by vitamin E. Vitamin E accumulates in membranes due to its lipophilic character and detoxifies lipid peroxidation products and thus constitutes a so-called chain-breaking antioxidant. Vitamin C and ubiquinone regenerate oxidized vitamin E.
An important enzymatic component of radical defense is formed by superoxide dismutase (SOD). SOD is an intracellular enzyme that catalyzes the reaction of two superoxide radicals and two protons to produce hydrogen peroxide and molecular oxygen. In this function, the SOD is an important part of the oxidation protection system. Overexpression of SOD can lead to an overproduction of H2O2, which may also lead to cell destruction. For this reason, the resulting H2O2 must be detoxified by means of catalase or glutathione peroxidase. Catalase is an intracellular, highly active enzyme found mainly in peroxisomes. This catalyzes the reaction of H2O2 to water and molecular oxygen, which traps the radical H2O2. Glutathione peroxidase also reduces H2O2 by GSH producing glutathione disulfide and lipid hydroperoxide.
If ROS formation outweighs ROS detoxification, oxidative modifications of cellular components predominate. Lipid peroxidation occurs on polyunsaturated fatty acids, which are highly concentrated in membranes of the brain and the mitochondrial membrane. The main products of lipid decomposition and biochemical markers of oxidative stress are malondialdehyde and 4-hydroxy-nonenal. Both aldehydes show cytotoxic properties, are mutagenic, attack proteins and induce oxidative conditions (Ayala et al., 2014). Lipid peroxidation products modify proteins and reactions with sugars and aldehydes cause increased protein glycosylation. Protein carbonyls that are formed in these reactions are an important marker of protein oxidation. Protein oxidation causes various modifications of the amino acid residues, e.g., cysteine and methionine side chains can react to disulfides and sulfoxides, arginine and lysine to aldehydes, aromatic side chains can be oxidized or nitrated, and aliphatic carbon atoms can be oxidized to alcohols (Davies, 2016). The elimination of modified proteins occurs by means of proteosomal and lysosomal degradation processes. The oxidation of nucleic acids is mutagenic, in which case aromatic bases, as well as the sugar backbone can be oxidized, the latter leading to DNA strand breaks. The most common product of this reaction is 8-hydroxyguanine, which is another marker of oxidative stress. Furthermore, excessive ROS-formation can damage mitochondrial DNA as well leading among others to functional limitations of the mitochondria (Nissanka and Moraes, 2018). All these modifications lead to impaired cellular processes and increased repair mechanisms. Excessive oxidative stress or disruption of pathways can induce cell death.
Numerous studies have shown that oxidative stress seems to play a major role in the pathology of amyotrophic lateral sclerosis (ALS). Patients suffering from various forms of ALS (familial ALS and sporadic ALS) show evidence of protein damage by ROS. Thus, in the spinal cord of the patients as well as in the motor cortex increased levels of protein carbonyls could be detected (Ferrante et al., 1997). At the cellular level, protein oxidation and lipid oxidation could be observed within the gray matter of the spinal cord of sporadic ALS patients in motor neurons as well as in reactive astrocytes and microglia (Shibata et al., 2001). Measurements of 8-hydroxyguanine levels have shown that oxidative DNA damage is found in the anterior horn of the cervical part of the spinal cord of ALS patients (Ferrante et al., 1997). Thus, ALS patients have the results of increased and non-physiologic ROS concentration within specific cells (Figure 1). Neuronal cells are more susceptible to oxidative stress than other cell populations, because they are post-mitotic and accumulate damage throughout life. Highly specialized cells with a large size, such as motor neurons, generally appear to be more vulnerable to stress, because they require a high energy demand and metabolic input, to maintain its functions. For this reason, additional mitochondrial functionalities are of great importance accompanied with increased production of ROS (Shaw and Eggett, 2000). In addition, neurons are more vulnerable to oxidative stress because they have a lower level of glutathione as an antioxidant than other cells of the nervous system (Makar et al., 1994; Bhattacharyya et al., 2014). Mitochondrial dysfunction was also associated with motor neuronal cell death in ALS. In vitro studies have shown that changes in the electron transport chain, the main physiological ROS producer in cells, and impaired ATP production are responsible for changes in oxidative metabolism (Mattiazzi et al., 2002; Menzies et al., 2002). Another plausible explanation for oxidative stress in ALS is provided by the findings that about 20% of familial ALS patients show different toxic gain of function mutations of the SOD1 gene with the simultaneous loss of dismutase activity (Saccon et al., 2013). Since SOD1 is an important component of the antioxidant system, it is not surprising that these cells are exposed to elevated oxidative stress levels. In ALS patients suffering from the sporadic form, this fact is not necessarily given and the cause of motor neuron degeneration is not fully understood. However, recent work on the Wobbler Mouse, an animal model of sporadic ALS, showed elevated ROS levels in the cervical part of the spinal cord (Röderer et al., 2018). In addition, the use of the unspecific ROS scavengers N-acetyl-L-cysteine has been shown to decrease the degeneration of the motor neurons in wobbler mice due to a reduction of ROS level (Henderson et al., 1996). Since 2017, a new active substance called Edaravone has been approved for the treatment of ALS in humans: Edaravone has antioxidant properties and thus prevents oxidative stress that leads to motoneuronal death in ALS patients (Ikeda and Iwasaki, 2015; Schultz, 2018). This indicates that abnormal levels of ROS may result from a non-functioning antioxidant protection system of the cell with the involvement of antioxidant molecules, increased ROS production by e.g., complexes of oxidative phosphorylation within the damaged mitochondria or altered metabolic demands or both.
Figure 1.
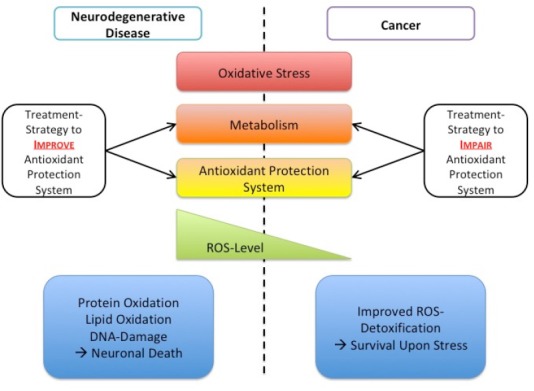
Bringing oxidative stress as common denominator into the perspective of multiple diseases.
Oxidative stress plays an important role in physiology and pathology of various diseases. Among them, neurodegenerative diseases like amyotrophic lateral sclerosis or the neoplasia of neuronal system (glioblastoma) are just a few examples. Often the concentration of reactive oxygen species (ROS) exceeds a critical level disturbing the equilibrium and thereby leading to oxidative stress. Upon oxidative stress, the free radicals of oxygen and nitrogen can cause damage to cells, cell organelles, and components, such as lipids, proteins, and DNA. Therefore, in the genesis of several diseases, antioxidant protection system often becomes pathologically unbalanced and thus leading to functional loss of the organism, as in case of amyotrophic lateral sclerosis, or cells develop metabolic mechanisms to use this system as protection against external influences, as in the case of glioblastoma cells. Either way, understanding the underlying deregulated mechanisms of the oxidative protection system would allow to develop novel treatment strategies for various diseases.
In contrast, different cancers including the neoplasia of the neuronal system (e.g., glioblastoma multiforme) adapt their antioxidant system to counteract increased ROS formation induced among others by elevated metabolism and increased metabolic demands (Matschke et al., 2016a). In particular, cancer cells rewire their metabolism at multiple levels to reduce ROS generation or improve ROS defense to maintain mitochondrial integrity and reduce ROS-dependent damage to cellular macromolecules (Levine and Puzio-Kuter, 2010; Matschke et al., 2016a; Hlouschek et al., 2018a). Therefore, cancer cells can become increasingly dependent on the uptake of additional nutrients, such as glucose, amino acids or fatty acids, for proliferation and survival in an adverse tumor microenvironment (Matschke et al., 2016b). As an example, cancer cells, including glioblastoma multiforme, may become dependent on glucose or glutamine as a carbon source to support anabolic processes that fuel proliferation and support increased production of reduction equivalents necessary for the regeneration of glutathione, the main antioxidant in the cell (Matschke et al., 2016a; Hlouschek et al., 2018a). Moreover, tumor cells may use reductive glutamine metabolism to maintain tricarboxylic acid cycle, particularly acetyl-coenzyme A production for fatty acid synthesis, rendering them independent of complex-I but dependent on fatty acid uptake (Matschke et al., 2016a) or increase the uptake of fatty acids to maintain proliferation under low oxygen conditions (Matschke et al., 2016b). However, several studies demonstrated different metabolic adaptation strategies of cancer cells to increase their cellular glutathione levels, avoid ROS-dependent damage and escape genotoxic therapies (Matschke et al., 2016a; Nakashima et al., 2017; Hlouschek et al., 2018a). Nevertheless, modulation of the antioxidant capacity in cancer cells as a consequence of metabolic adaptation seems to be a general mechanism throughout different tumor entities (Figure 1). Even the highly aggressive neoplasia of neuronal system, glioblastoma multiforme, acquires alterations in redox status during the gliomagenesis bringing therapies modulating the redox homeostasis and antioxidant capacity into the spotlight (Salazar-Ramiro et al., 2016).
In this case, the increased antioxidant capacity of tumor cells makes them cross-resistant to ROS-inducing treatments like radio- and chemotherapy and offers novel intervention strategies for treatment of radio and chemotherapy resistant tumor types, like glioblastoma multiforme, breast and non-small cell lung cancers (Matschke et al., 2016a; Hlouschek et al., 2018a, b). Generally, increased GSH levels can be targeted by using drugs interfering with the regeneration of glutathione, the provision of reduction equivalents, increased glutathione synthesis or glutathione transport and uptake (Matschke et al., 2016a; Hlouschek et al., 2018a).
In summary, the oxidative system seems to play an important role in physiology and pathology of various diseases, like amyotrophic lateral sclerosis or glioblastoma. Regardless of the direction in which the ROS-homeostasis disequilibrate - leading to functional loss of the organism, as in case of ALS, or by improving this protection system against external influences, as in the case of glioblastoma cells, the oxidative protection system should be in focus of research (Figure 1).
Acknowledgments:
We thank Prof. Dr. Verena Jendrossek [Institute of Cell Biology (Cancer Research), University of Duisburg-Essen, University Hospital Essen, Essen, Germany] for stimulating discussions and support.
Footnotes
Conflicts of interest: The authors declare no conflict of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Yu J; T-Editor: Liu XL
References
- 1.Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:1–31. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cacciapuoti F. Oxidative stress as “mother” of many human diseases at strong clinical impact. J Cardiovasc Med Cardiol. 2016;3:001–006. [Google Scholar]
- 4.Davies MJ. Protein oxidation and peroxidation. Biochem J. 2016;473:805–825. doi: 10.1042/BJ20151227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 6.Ferroni P, Barbanti P, Della-Morte D, Palmirotta R, Jirillo E, Guadagni F. Redox mechanisms in migraine: novel therapeutics and dietary interventions. Antioxid Redox Signal. 2018;28:1144–1183. doi: 10.1089/ars.2017.7260. [DOI] [PubMed] [Google Scholar]
- 7.Henderson JT, Javaheri M, Kopko S, Roder JC. Reduction of lower motor neuron degeneration in wobbler mice by N-acetyl-L-cysteine. J Neurosci. 1996;16:7574–7582. doi: 10.1523/JNEUROSCI.16-23-07574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hlouschek J, Hansel C, Jendrossek V, Matschke J. The Mitochondrial citrate carrier (SLC25A1) sustains redox homeostasis and Mitochondrial Metabolism supporting radioresistance of cancer cells With Tolerance to cycling severe hypoxia. Front Oncol. 2018a;8:14–18. doi: 10.3389/fonc.2018.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hlouschek J, Ritter V, Wirsdörfer F, Klein D, Jendrossek V, Matschke J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018b;439:24–38. doi: 10.1016/j.canlet.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda K, Iwasaki Y. Edaravone, a free radical scavenger, delayed symptomatic and pathological progression of motor neuron disease in the wobbler mouse. PLoS One. 2015;10:e0140316. doi: 10.1371/journal.pone.0140316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 12.Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem. 1994;62:45–53. doi: 10.1046/j.1471-4159.1994.62010045.x. [DOI] [PubMed] [Google Scholar]
- 13.Matschke J, Riffkin H, Klein D, Handrick R, Lüdemann L, Metzen E, Shlomi T, Stuschke M, Jendrossek V. Targeted inhibition of glutamine-dependent glutathione metabolism overcomes death resistance induced by chronic cycling hypoxia. Antioxid Redox Signal. 2016a;25:89–107. doi: 10.1089/ars.2015.6589. [DOI] [PubMed] [Google Scholar]
- 14.Matschke J, Wiebeck E, Hurst S, Rudner J, Jendrossek V. Role of SGK1 for fatty acid uptake, cell survival and radioresistance of NCI-H460 lung cancer cells exposed to acute or chronic cycling severe hypoxia. Radiat Oncol. 2016b;11:75. doi: 10.1186/s13014-016-0647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 16.Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZMA, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- 17.Nakashima R, Goto Y, Koyasu S, Kobayashi M, Morinibu A, Yoshimura M, Hiraoka M, Hammond EM, Harada H. UCHL1-HIF-1 axis-mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization. Sci Rep. 2017;7:6879. doi: 10.1038/s41598-017-06605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592:728–742. doi: 10.1002/1873-3468.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Röderer P, Klatt L, John F, Theis V, Winklhofer KF, Theiss C, Matschke V. Increased ROS level in spinal cord of Wobbler mice due to Nmnat2 downregulation. Mol Neurobiol. 2018;55:8414–8424. doi: 10.1007/s12035-018-0999-7. [DOI] [PubMed] [Google Scholar]
- 20.Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136:2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salazar-Ramiro A, Ramírez-Ortega D, Pérez de la Cruz V, Hérnandez-Pedro NY, González-Esquivel DF, Sotelo J, Pineda B. Role of redox status in development of glioblastoma. Front Immunol. 2016;7:156. doi: 10.3389/fimmu.2016.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz J. Disease-modifying treatment of amyotrophic lateral sclerosis. Am J Manag Care. 2018;24:S327–S335. [PubMed] [Google Scholar]
- 23.Shaw PJ, Eggett CJ. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol. 2000;247(Suppl 1):I17–I27. doi: 10.1007/BF03161151. [DOI] [PubMed] [Google Scholar]
- 24.Shibata N, Nagai R, Uchida K, Horiuchi S, Yamada S, Hirano A, Kawaguchi M, Yamamoto T, Sasaki S, Kobayashi M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001;917:97–104. doi: 10.1016/s0006-8993(01)02926-2. [DOI] [PubMed] [Google Scholar]